1. Introduction
It is widely known that CO oxidation is one of the most important reactions in environmental research. Extant studies [
1] indicate that transition metal oxides, including MnO
2, Fe
2O
3, Co
2O
3, NiO, and CuO, as well as noble metals, are highly active for CO oxidation. The catalytic performance of metal oxides is enhanced by increasing their surface area. Thus, metal oxide catalysts were synthesized in the forms of nanoparticles to yield high surface areas. Generally, nanostructured metal oxides are significantly active catalysts when compared with non-nanostructured metal oxides. However, it is not easy to utilize these oxides as a medium for noble metal catalysts due to their extremely small size. Among the transition metal oxides, iron oxide is synthesized in the form of spherical particles with a controlled size, which attracts research attention with respect to its utilization as support. Noble metals on Fe
2O
3 particles were indicated as appreciably active [
2]. Recently, Fe
2O
3 was frequently used as a model system in studies that focused on strong metal–support interactions [
3]. Specifically, Fe
2O
3 spherical particles with a submicron size have been rarely examined as a catalyst and support to date.
In any kinetics study, monitoring the time-dependent variation of the concentrations of reactants and/or products is considered to be crucial. It has been known that photoacoustic spectroscopy (PAS) is suitable to in situ monitor a reaction processes in its initial stage [
4]. Since signals obtained from PAS rely on the concentration of a target species and PAS detects extremely low level molecular gases with high selectivity and large signal intensity, temporal variations in concentration can be readily measured even in a short period of reaction times where rates are high. Thus, low molecular level photoacoustic results are believed to provide more accurate kinetic information for catalytic reactions, especially when the reaction occurs on a relatively clean surface of catalysis.
In this study, we prepared α-Fe2O3 spherical particles (sp) with an average diameter of approximately 200 nm by using a solvothermal method. Subsequently, α-Fe2O3 submicron powder was tested as a catalyst for CO oxidation and as a medium for a Pd metal catalyst. CO oxidation from α-Fe2O3 and Pd/α-Fe2O3 submicron powder catalysts was performed in a static reactor at a total pressure of 40 Torr. A CO2, laser-based, photoacoustic technique with a differential photoacoustic cell was employed to investigate the kinetics of CO oxidation. Rates of CO2 formation were calculated from the CO2 photoacoustic data at an early reaction stage. The rates were measured with a stoichiometric reaction mixture and aided in determining the apparent activation energies in the temperature range of 225–350 °C. The partial orders with respect to CO and O2 were calculated from the rates that were measured at various partial pressures of CO and O2 at 350 °C. The catalytic behaviors of both the α-Fe2O3 and Pd/α-Fe2O3 submicron powders for CO oxidation were compared with those of an α-Fe2O3 fine powder.
2. Results
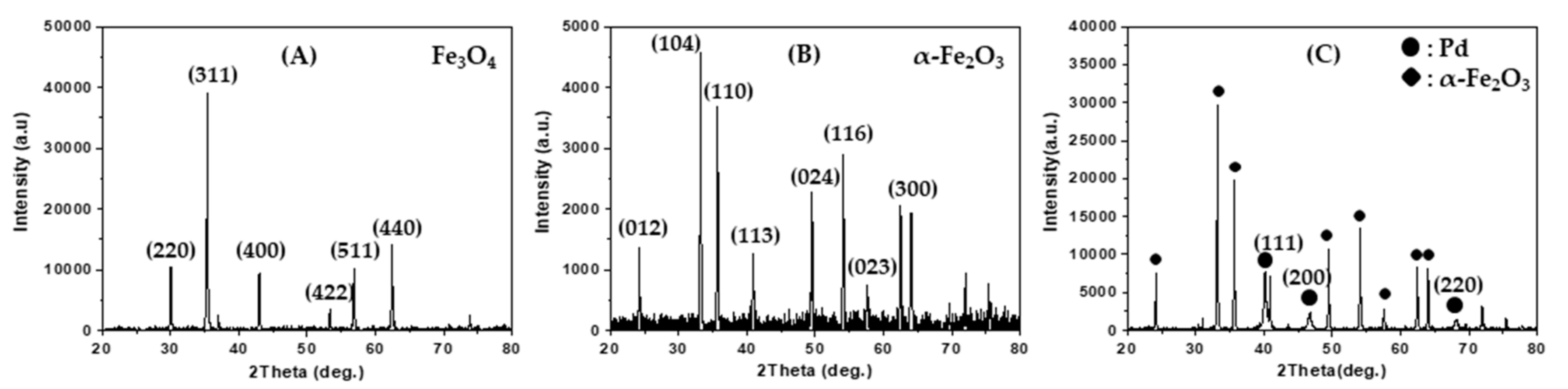
Figure 1A shows the X-ray powder diffraction (XRD) pattern of magnetite spherical submicron particles that were directly prepared by using a solvothermal method. The diffraction pattern indicates that the magnetite phase has a face-centered cubic structure (JCPDS Card No. 00-039-1346). The magnetite powder was calcined at 600 °C in a flow of O
2 (10%)/He mixture, and a red powder was obtained.
Figure 1B shows the XRD pattern of the red powder and indicates that the red powder corresponds to an α-Fe
2O
3 phase with a rhombohedral structure (JCPDS Card No. 01-089-0598). In this study, Pd-loaded α-Fe
2O
3 spherical submicron particles were prepared by using the impregnation method.
Figure 1C shows the XRD pattern of the Pd/α-Fe
2O
3 particles, which exhibits reflection peaks arising from Pd metal crystallites with a face-centered cubic structure (JCPDS Card No. 87-0641) at 2
θ = 40.1°, 46.6°, and 68.1° as denoted by the Miller indices (111), (200), and (220), respectively. By performing energy dispersive X-ray spectroscopy (EDS) on the Pd/α-Fe
2O
3 sample, the Pd/Fe atomic ratio was determined as 0.123, which corresponds to 14 wt % Pd.
Figure 2A shows a field emission-scanning electron microscopy (FE-SEM) image of the α-Fe
2O
3 spherical submicron particles and indicates that the particles possess a spherical morphology with a rough surface.
Figure 2B exhibits a high-resolution transmission electron microscopy (HR-TEM) image of the α-Fe
2O
3 spherical submicron particles.
Figure 2C displays a FE-SEM image of the Pd (14 wt %)-loaded α-Fe
2O
3 spherical submicron particles and shows that Pd nano-particles are loaded on the surface of α-Fe
2O
3 spherical submicron particles.
Figure 3A shows the particle size distribution of the α-Fe
2O
3 spherical submicron particles. From the histogram, the mean diameter of the α-Fe
2O
3 spherical submicron particles was estimated to be 196 nm. The α-Fe
2O
3 spherical submicron powder sample is denoted as α-Fe
2O
3 (sp200). N
2 adsorption measurements were performed for the α-Fe
2O
3 (sp200) sample at liquid nitrogen temperature. The N
2 adsorption–desorption isotherm is shown in
Figure 3B. An International Union of Pure and Applied Chemistry (IUPAC) classification reveals that the hysteresis loop of
Figure 3B corresponds to the H3 type [
5].
Figure 3C reveals the pore size distribution as determined by the Barret–Joyer–Halenda (BJH) method from the adsorption branch of the isotherm and shows that the pore size is distributed in the range of 3–20 nm. The Brunauer–Emmett–Teller (BET) surface area was calculated to be 19.0 m
2/g for the α-Fe
2O
3 (sp200) powder, 7.4 m
2/g for the Pd/α-Fe
2O
3 (sp200) powder, and 6.8 m
2/g for the α-Fe
2O
3 fine powder (α-Fe
2O
3 (fp)). The surface area of the Pd/α-Fe
2O
3 (sp200) is smaller than that of α-Fe
2O
3 (sp200), suggesting that the openings of some pores could be blocked by the Pd particles.
A kinetic study of CO oxidation was performed with the static method, and photoacoustic spectroscopy was employed to measure the rate of CO
2 formation in the early reaction stage of CO oxidation. Generally, the CO
2 photoacoustic signal measured with the CO
2 laser-based photoacoustic method is dependent on the excitation wavelength, the power of the light source, the chopping frequency, and the sensitivity of the microphone.
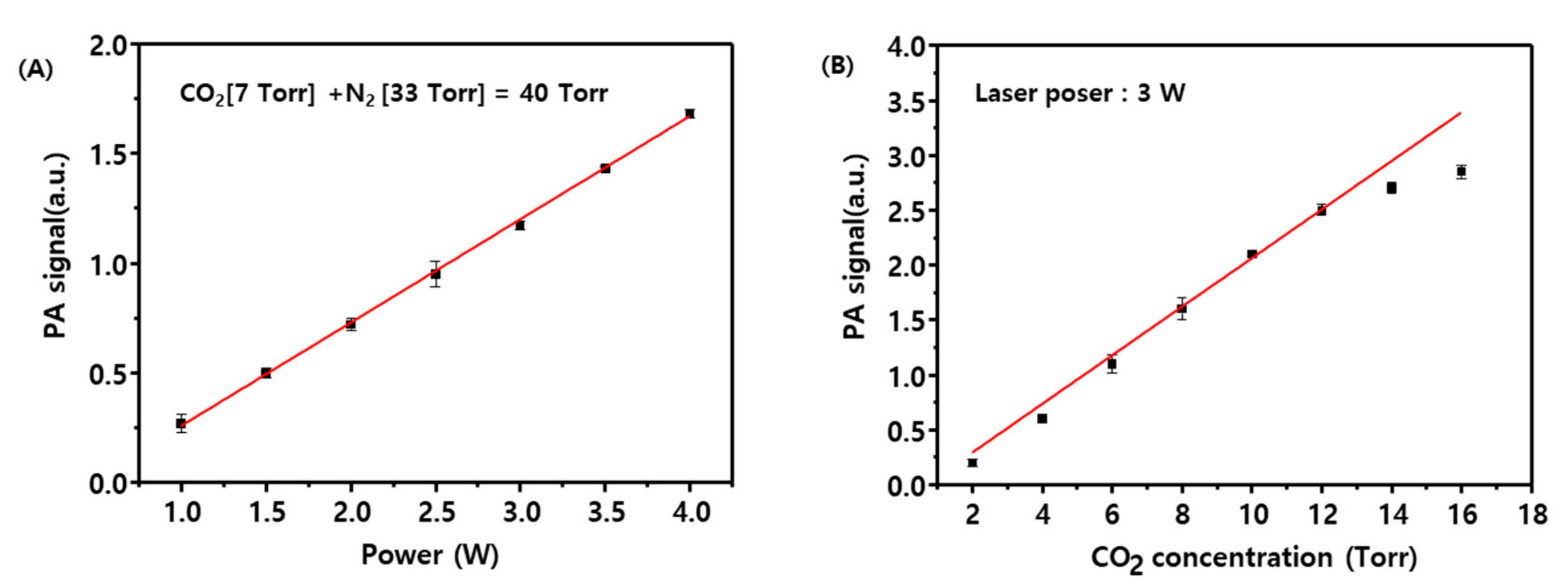
Figure 4A shows that the CO
2 photoacoustic signal as a function of laser power exhibits an optimal linearity in the range from 1–4 W.
Figure 4B displays the CO
2 photoacoustic signal measured as a function of CO
2 partial pressure and indicates that the signal is linear for CO
2 partial pressures below 12 Torr. Accordingly, subsequent measurements were performed with a CO
2 partial pressure below 12 Torr and a CO
2 laser power of 3 W. A blank test was performed by using a CO/O
2/N
2 (20/10/10 Torr) mixture in the temperature range of 25–400 °C, which revealed that variations in the CO
2 photoacoustic signal were absent. The effect of temperature on the rate of CO
2 formation for the catalytic reaction in the static reactor was investigated by using a CO/O
2/N
2 (20/10/10 Torr) mixture in the temperature range of 175–350 °C.
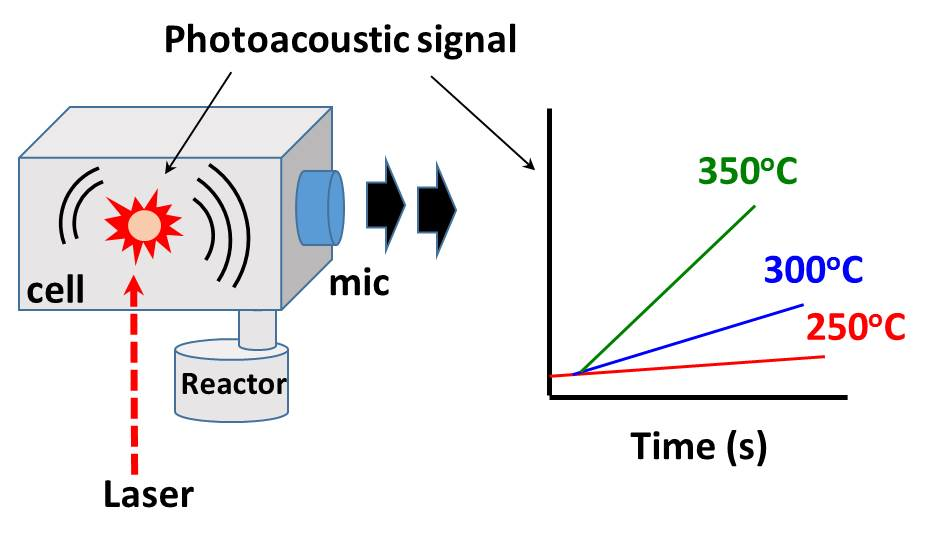
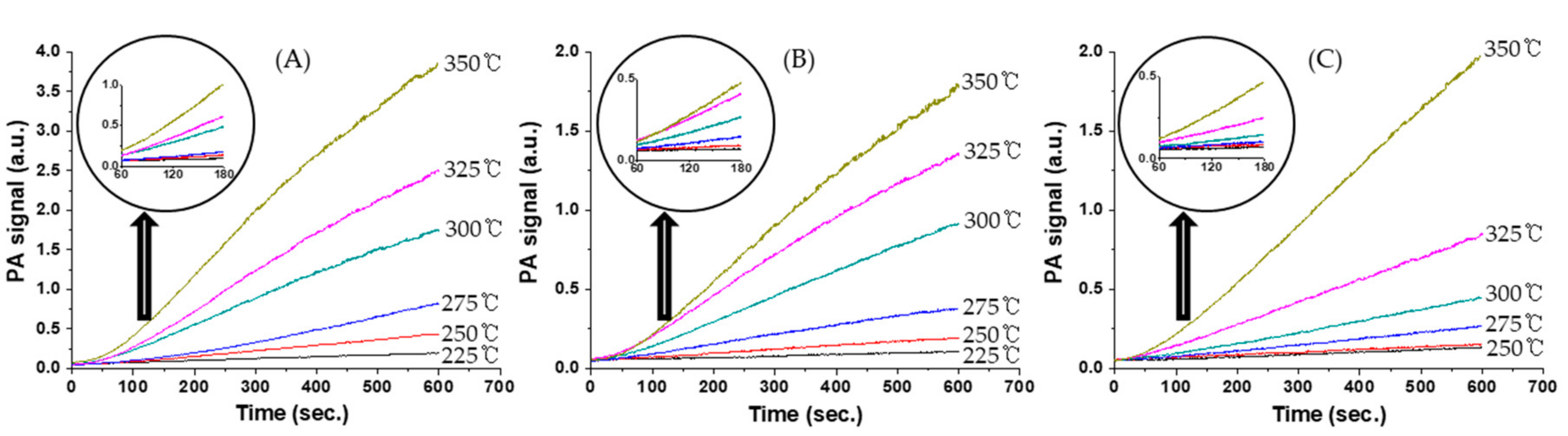
Figure 5 shows variations in the CO
2 photoacoustic signal relative to time for CO oxidation over catalysts at various temperatures. The rates of CO
2 formation were calculated from the CO
2 photoacoustic signals obtained in the reaction period range of 60–180 s after the injection of the reaction mixture into the static reactor.
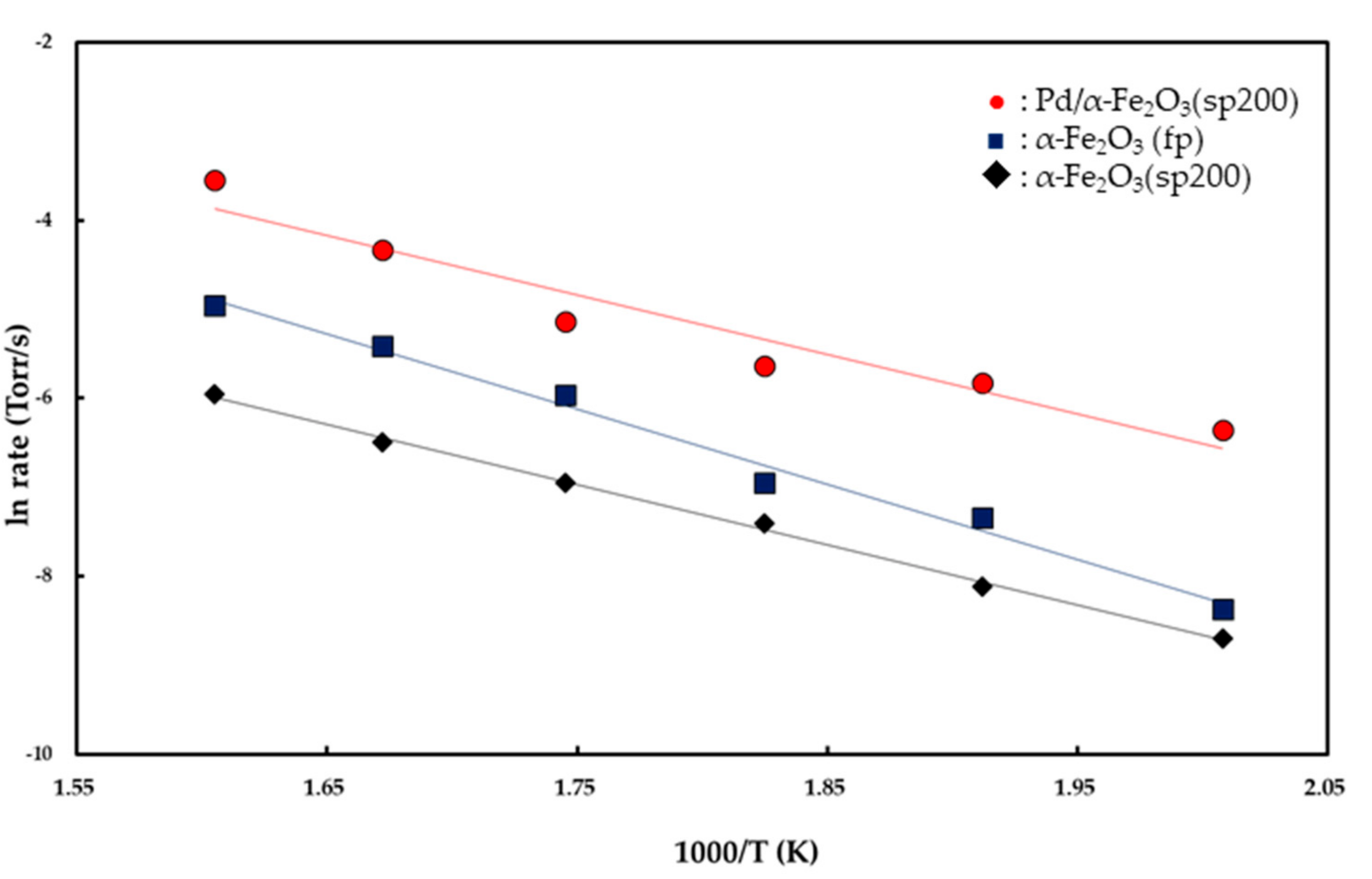
The rates were plotted as a function of reciprocal temperature according to the Arrhenius-type equation given by rate ∞ exp(
−Ea/
RT) as shown in
Figure 6. From the slopes of the linear curves in the temperature range of 225–350 °C, apparent activation energies were calculated as 16.8 (±0.3) kcal/mol for the α-Fe
2O
3 (fp), 13.4 (±0.1) kcal/mol for the α-Fe
2O
3 (sp200), and 13.2 (±0.5) kcal/mol for the Pd/α-Fe
2O
3 (sp200). Reaction orders with respect to
PCO and
Po
2 were determined from the rates measured at various partial pressures of CO and O
2 at 350 °C. The rates were plotted as a function of partial pressures of CO and O
2 which based on the power rate law should follow rate =
k(
Pco)
m(
Po
2)
n.
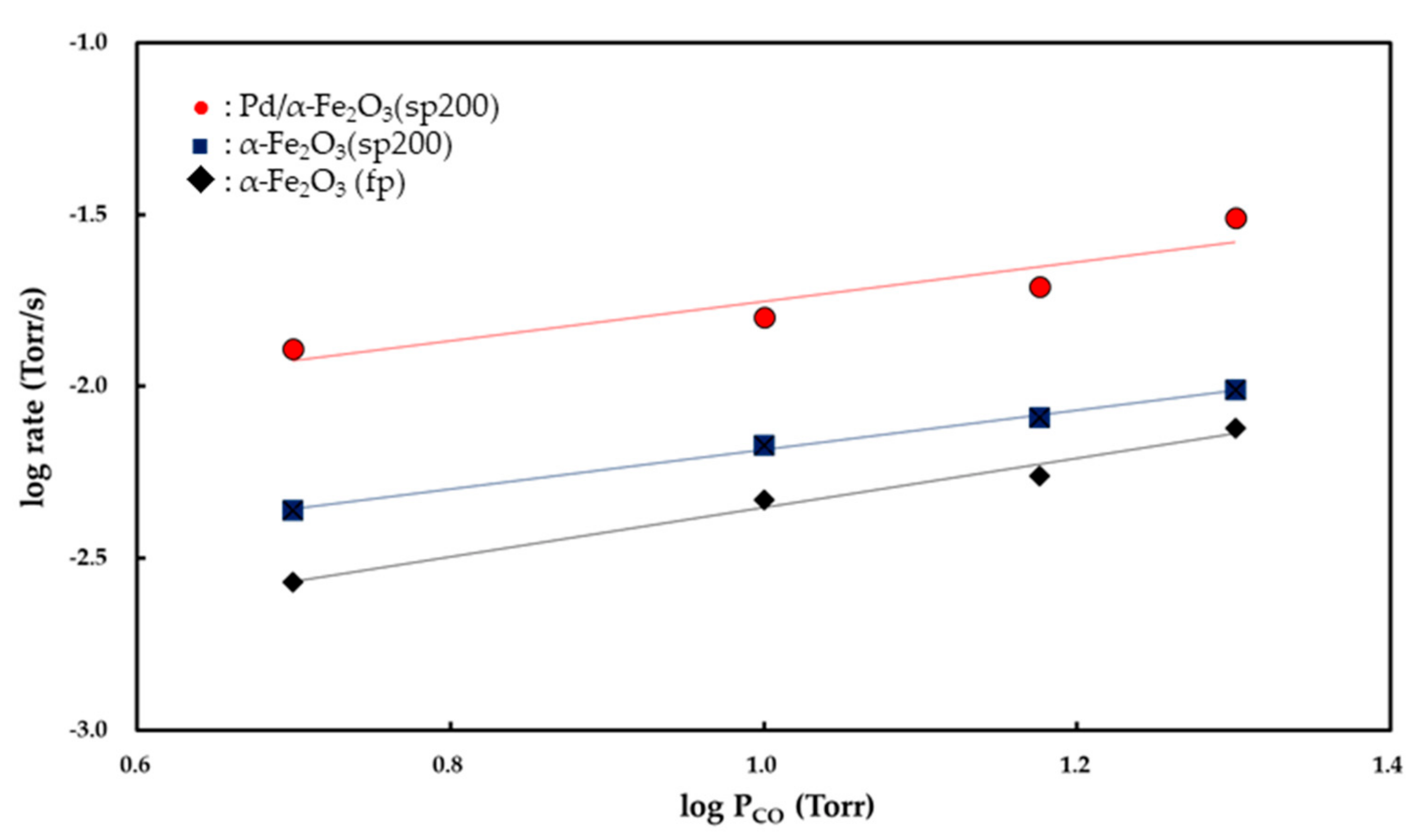
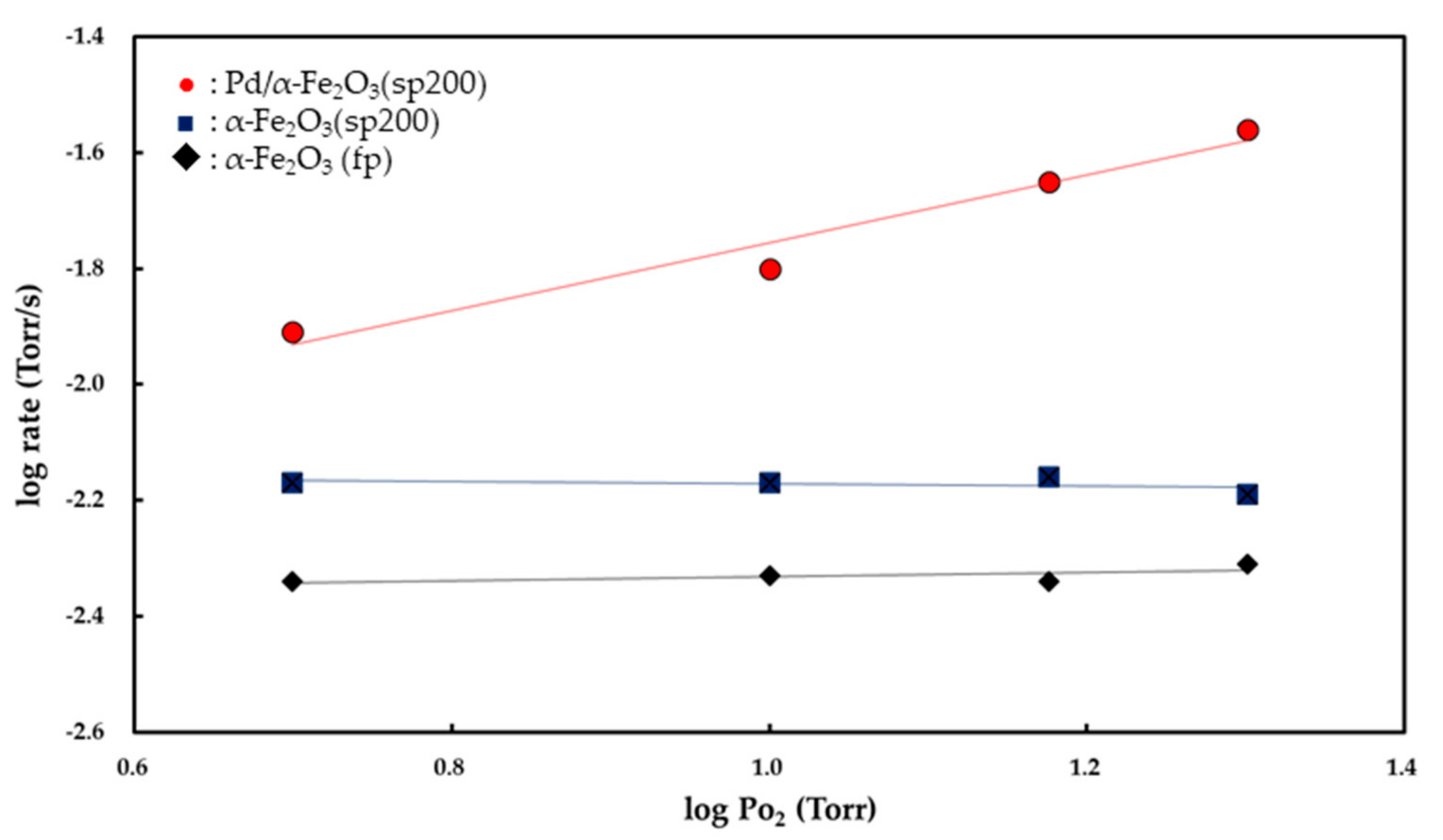
Figure 7 and
Figure 8 show the
Pco and
Po
2 dependencies of the rate of CO
2 formation, respectively. Partial orders with respect to
Pco and
Po
2 were determined from the slopes of the linear curves. Both the α-Fe
2O
3 (fp) and α-Fe
2O
3 (sp200) catalysts exhibited zero-order kinetics for
Po
2 while the Pd/α-Fe
2O
3 (sp200) catalyst showed 0.48 (±0.03)-order with respect to
Po
2. The partial order with respect to
PCO was determined to be 0.85 (±0.03)-order for the α-Fe
2O
3 (fp) catalyst, 0.58 (±0.1)-order for the α-Fe
2O
3 (sp200) catalyst, and 0.54 (±0.06)-order for the Pd/α-Fe
2O
3 (sp200) catalyst.
3. Discussion
With respect to CO oxidation over Fe
2O
3, it was reported that Fe
2O
3 acts as a catalyst in the presence of O
2 (g) and as a direct oxidant for CO (g) in the absence of O
2 (g) [
6,
7]. In the presence of O
2, the Fe
2O
3 surface is primarily reduced by CO (g), and the reduced Fe
2O
3 surface is successively re-oxidized by O
2 (g). In the catalytic oxidation of CO, the CO disproportion reaction (the Boudourd reaction corresponding to 2CO (g) ⇄ (s) + CO
2 (g)) is considered as a side reaction because it is thermodynamically feasible below 630 °C based on the assumption of the standard free energy change Δ
G0 = 0. According to Li et al. [
6], with respect to CO oxidation on Fe
2O
3 nanopowder, the CO disproportion reaction occurs above 300 °C in the absence of O
2 (g). The photoacoustic measurements in our work were performed by using a CO/O
2 mixture at a total pressure of 40 Torr, and accordingly, the occurrence of CO disproportion is considered as infeasible under the reaction conditions.
As shown in
Figure 6, the apparent activation energies for CO oxidation were determined as 16.8 (±0.3) kcal/mol for the Fe
2O
3 (fp) catalyst and 13.4 (±0.1) kcal/mol for the Fe
2O
3 (sp200) catalyst in the temperature range of 225–350 °C. It is expected from the magnitude of the apparent activation energies that the reactants are chemisorbed on the catalyst surface. Specifically, Fe
2O
3 is oxygen deficient and oxygen vacancies are considered as the predominant defect for the oxide. Oxygen vacancies are readily generated in the oxide when Fe
2O
3 is heated at a low pressure [
8,
9]. Typically, oxygen vacancies in metal oxides act as adsorption sites for O
2 (g) because they serve as electron donors. When O
2 (g) is adsorbed (ads) on the oxygen vacancy, it is dissociated into two oxygen atoms as follows:
where e
− represents an electron trapped at the oxygen vacancy site. Given that an O
− (ads) ion is active, CO (g) reacts with O
− (ads) to form CO
2− (ads): CO (g) + O
− (ads) ⇄ CO
2− (ads). The resultant CO
2− (ads) ion is desorbed from the surface to produce CO
2 (g) and the oxygen vacancy is simultaneously regenerated in the oxide.
If the O
− (ads) ions act as adsorption sites for CO (g) to form CO
2 (g), then the partial order with respect to O
2 is derived to be 0.5 [
4]. In this study, as shown in
Figure 8, the rates of CO
2 formation for both the Fe
2O
3 (fp) and Fe
2O
3 (sp200) powders were obtained as independent of
Po
2, and this indicates that the surface is continuously saturated by O
2 (g). The zero-order kinetics for
Po
2 suggests that CO (g) preferably reacts with O
2− (latt) as opposed to the O
− (ads) ion. The interaction between CO (g) and the lattice (latt) oxygen of Fe
2O
3 is described by the following equilibrium:
When the CO
2− (ads) ion reacts further with O
2− (latt), a carbonate ion is formed as follows:
The carbonate ion is decomposed into CO
2 (g) according to the following reaction: CO
32− (ads) → CO
2 (g) + O
2− (latt). If Equations (3) and (4) are included in the reaction mechanism, then the rate law is derived as first-order kinetics for
Pco based on the assumption of constant [O
2− (latt)] [
4]. In this study, the partial order to
Pco is determined as 0.85 (±0.03)-order for the α-Fe
2O
3 (fp) catalyst and 0.58 (±0.1)-order for the α-Fe
2O
3 (sp200) catalyst, thereby suggesting that an inhibition process by CO
2 is included in the reaction mechanism under the reaction conditions.
Previous studies of CO
2 adsorption on Fe
2O
3 [
10,
11] indicate that CO
2 is chemisorbed on the surface of iron oxide to form bidentate carbonate. Several studies suggested that oxygen vacancies in metal oxide act as adsorption sites for CO
2 as well as O
2, wherein CO
2 (g) is chemisorbed on oxygen vacancies and the resultant CO
2 (ads) further reacts with O
2− (latt) to form carbonate ions [
12,
13,
14,
15,
16,
17]. Oxygen vacancies are considered as predominant defects in Fe
2O
3, and thus it is feasible to form a carbonate species on the surface of Fe
2O
3. If CO
2 is adsorbed on the lattice oxygen of Fe
2O
3, then CO
2 (g) is in equilibrium with carbonate ions according to the following equilibrium: CO
2 (g) + O
2− (latt) CO
32− (ads). When both CO and CO
2 competitively interact with the lattice oxygen, the partial order with respect to
Pco is observed as a value less than 1. As shown in
Figure 7, the partial order to
Pco for CO oxidation over Fe
2O
3 (sp200) was observed as 0.54 (±0.06), and this value is lower than 0.85 (±0.03) for the reaction relative to Fe
2O
3 (fp). These results suggest that the inhibition process by CO
2 is more feasible on Fe
2O
3 (sp200) as opposed to the Fe
2O
3 (fp).
Conversely, the apparent activation energy for CO oxidation on the Pd/Fe
2O
3 (sp200) catalyst is 13.2 kcal/mol, as shown in
Figure 6. This value is in good agreement with 13.3 kcal/mol for CO oxidation on 1 wt % Pd/FeO
x in the temperature range of 120–160 °C [
2] and 12.5 kcal/mol on 5 wt % Pd/Al
2O
3 in the temperature range of 120–160 °C [
18], although it slightly exceeds 8.2 kcal/mol on 1.9 wt % Pd/FeO
x in the temperature range of 5–80 °C [
19]. As shown in
Figure 8, the partial order with respect to
Po
2 on the Pd/Fe
2O
3 (sp200) catalyst is 0.48 (±0.03)-order, and this is entirely different from the zero-order kinetics observed for both the Fe
2O
3 (sp200) and Fe
2O
3 (fp) catalysts. The 0.48 (±0.03)-order is close to 0.5-order, and thus it is possible to consider that O
2 (g) is dissociatively adsorbed on the Pd surface to form two oxygen atoms (O
− (ads)) based on equilibrium (1). Therefore, CO (g) is easily adsorbed on the active O
− (ads) ion to form CO
2− (ads) as follows:
When CO (g) reacts with the O
− (ads) ion to produce CO
2 (g) based on Reactions (5) and (6), the partial order to
Po
2 is derived to be 0.5-order [
8]. The 0.48 (±0.03)-order obtained in this study supports that O
2 (g) is dissociatively adsorbed on the surface of Pd. The partial order to
Pco for the Pd/Fe
2O
3 (sp200) catalyst, determined to be 0.58 (±0.1), implies that the inhibition process by CO
2 is involved in the reaction mechanism. The O
− (ads) ions formed on the Pd surface act as an adsorption site for CO
2 (g). If CO
2 (g) is adsorbed on O
− (ads), then carbonate ions are formed according to the following equilibrium:
This implies that CO2 (g) inhibits CO2 formation via the reaction between CO (g) and O− (ads). Although it is not possible to exclude oxygen spillover from Fe2O3 to the Pd surface in the reaction mechanism, the kinetic results for CO oxidation over Pd/Fe2O3 (sp200) suggest that CO (g) favorably reacts with O− (ads) to form CO2− (ads). The CO2− (ads) further reacts with O− (ads) to produce CO32− (ads), and the resultant CO32− (ads) decomposes into CO2 (g). When both CO (g) and CO2 (g) are competitively adsorbed on the O− (ads) on the Pd surface, the partial order to Pco is observed as a value below 1 as obtained for the Pd/Fe2O3 (sp200) catalyst (0.58 (±0.1)-order).
4. Experimental
The α-Fe
2O
3 spherical submicron particles were synthesized from FeCl
3·6H
2O (97%, Aldrich- Sigma, St. Louis, MO, USA), sodium acetate trihydrate (NaAc) (≥99%, Sigma-Aldrich, St. Louis, MO, USA), ethylene glycol (EG) (≥99%, Sigma-Aldrich, St. Louis, MO, USA), and diethylene glycol (DEG) (≥99%, Sigma-Aldrich, St. Louis, MO, USA) by the solvothermal method [
20,
21]. Specifically, NaAc (1.5 g) was dissolved in a mixture of EG (20 mL) and DEG (20 mL) at 60 °C with constant stirring for 30 min. Subsequently, FeCl
3·6H
2O (1.08 g) was dissolved in the NaAc solution with vigorous stirring for 30 min. The formation of viscous slurry was transferred into a teflon-line stainless-steel autoclave with 50 mL capacity. The autoclave was maintained at 200 °C for 10 h and cooled to room temperature to obtain precipitates. The resulting precipitates were rinsed with distilled water and absolute alcohol several times, dried at 60 °C for 6 h, and a black powder was obtained. The black powder (Fe
3O
4) was calcined at 500 °C in air, cooled to room temperature, and a red powder (α-Fe
2O
3) was obtained as a final product. Furthermore, α-Fe
2O
3-supported Pd was prepared according to the following procedures: the Fe
2O
3 submicron particles were dispersed in ethanol, followed by sonication for 3 h at room temperature by using an ultrasonic cleaner, and PdCl
2 was added into the mixture. After sonication for 3 h, a hydrazine solution was dropped into the suspension while stirring, in which Pd
2+ was reduced to Pd
0 by hydrazine [
22,
23]. The precipitates were separated by centrifugation, washed with ethanol five times, and the Pd/Fe
2O
3 sample was obtained. In this study, α-Fe
2O
3 fine powder, used as a reference material, was prepared by the wet method by using FeCl
3 and NH
4OH solution [
24].
The phase identification of the sample was performed by using XRD and using a D/max-RB diffractometer (Rigaku, The Woodlands, TX, USA). The size, morphology, and chemical composition of the spherical particles were determined by using FE-SEM (JEOL-6701F, JEOL, Tokyo, Japan) equipped with an energy dispersive X-ray spectroscopy (EDS). In order to measure the surface area and porosity of samples, N2 adsorption measurements were conducted with a Micromeritics Autosorb-iQ 2ST/MP instrument (Quantachrome Instruments, Boynton Beach, FL, USA).
Kinetic measurements of CO oxidation were performed in a static reactor by using a CO
2 laser-based photoacoustic technique. As indicated in a previous study [
4], the photoacoustic technique with a differential photoacoustic cell was proven as a suitable technique for in situ monitoring of the change in the CO
2 concentration during the catalytic oxidation of CO. The differential photoacoustic cell was composed of a reference cell and a sample cell that were separated from each other by a ZnSe window. The reference cell was filled with a gaseous mixture of CO
2 (0.2 Torr) and N
2 (39.8 Torr), and the sample cell was directly connected to the reactor that was composed of quartz tubing with a volume of 21 cm
3. The photoacoustic cell was a Helmholtz resonator with a diameter of 19 mm and a length of 33 mm, with an adjoining tube with a diameter of 1 mm and a length of 28 mm. Additionally, CO
2 photoacoustic signals from the microphones attached to the sample (signal A) and reference (signal B) cells were detected by a lock-in amplifier (EG & G Princeton Applied Research Model 5210). The signal ratio (A/B) was recorded as a function of time by using a personal computer. The total pressure (CO/O
2/N
2) in the reactor was maintained at 40 Torr and filled with N
2 as a buffer gas. The purity of gases exceeded 99.999%, and the gases were dehydrated with suitable filters. The output beam of a continuous wave (cw) CO
2 laser (Synrad Series 48-1-28) operating in multilines of 10.6 μm was modulated at the nonresonance condition of 20 Hz. The rates of CO
2 formation for the catalytic reaction were estimated from the CO
2 photoacoustic data in the early reaction stage.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







