1. Introduction
Water is considered to be an ideal reaction medium to proceed chemical reactions of organic molecules efficiently when organic molecules are dissolved in water. It is possible to proceed chemical reactions to produce molecules as planed in water because the temperature and the pressure of water are controlled. Understanding chemical reactions of organic molecules in water provides us with valuable information to extract the energy of biomass without drying because biomass contains large amount of water [
1]. CH
3OH is one of the simplest products by reformation of biomass and is the source of energy for a direct methanol fuel cell. It would be beneficial to study chemical reactions containing CH
3OH in water. We examine the oxidation of CH
3OH in water in this study.
The oxidation process of CH
3OH was extensively studied in the gas phase. The main channel for the oxidation of CH
3OH is identified using the detected OH absorption signals [
2]. An excited CH
3OH in a shock wave or flame is primarily decomposed into two radicals as
On the other hand, it is shown that CH3OH is decomposed into molecules in water.
Hack et al. [
3] examined the oxidation of CH
3OH in a flow reactor tube made of the alloy C-276 (Ni/Mo/Cr/Fe). They used a gas chromatography and a Fourier transform infrared spectrometer and found that the major product molecules for the oxidation of CH
3OH are H
2, CO
2, and CH
4. Hydrogen gas is the most abundant product in experiments. They measured the CH
3OH concentration by a Raman spectrometer as a function of time and determined the oxidation rate of CH
3OH in water as
/s at 653 K. The oxidation rate is dependent on the surface condition of the reactor, the heavy metals of which influence the oxidation rate of CH
3OH [
4]. Oxygen supply into a reactor is also shown to accelerate the oxidation of CH
3OH [
5].
It is interesting to investigate the effect of a reactor wall on the oxidation rate of CH
3OH. Hirsh and Franck [
6] determined the oxidation rate of CH
3OH in a Ni alloy reactor with the larger volume than that used by Hack et al. They discovered the slower oxidation rate of CH
3OH due to a smaller surface to volume ratio. DiLeo and Savage [
1] adopted a sealed quartz tube to perform experiments for the oxidation of CH
3OH in water without any catalyst as well as with catalyst of a Ni wire. This is the first experiment to measure the oxidation rate of CH
3OH without catalyst. They determined the oxidation rate of CH
3OH as
/s at 823 K, which is much slower than that given by Hack et al. (2005) in spite of the high temperature. They also found that the oxidation of CH
3OH is significantly accelerated by adding a Ni metal wire in the experiment. The catalytic effect of a Ni wire becomes weak after multiple uses in the experiments.
In our previous study, we revealed the catalytic role of H
2O molecules in chemical reactions in water by performing a number of quantum chemical simulations [
7,
8,
9,
10]. For example, H
2O molecules get involved in the the dehydration process of methanediol when a proton is carried from a hydroxyl of a methanediol to an oxygen atom of the other hydroxyl to form an H
2O molecule and a formaldehyde. An H
2O molecule assists the transfer of a proton with little change of the structure of a methanediol, reducing the energy barrier for the dehydration. The calculated dehydration rate of a methanediol in water was shown to agree with that determined by experimental studies. We apply the same method to the oxidation of CH
3OH.
In the present study, we examine the catalytic role of H
2O molecules in the oxidation of CH
3OH in water by performing quantum chemical simulations. We include H
2O molecules to catalyze the oxidation in a simulation and calculate the energy barrier for the oxidation of a CH
3OH as a function of the number of H
2O molecules. H
2O molecules have hydrogen bonds with neighbor H
2O molecules and form a cluster of H
2O molecules. We include a cluster of H
2O molecules in the present simulation because we showed that a cluster of H
2O molecules becomes an active catalyst in water [
7,
8,
10]. The effect of metal on the oxidation of CH
3OH is not considered at the present study even though it is essential when we consider the oxidation of CH
3OH in a metal reactor [
11]. We calculate the oxidation rate of CH
3OH and compare it with that determined by experiments.
2. Results and Discussion
First, we optimize a tentative structure of a molecule using the B3LYP [
12,
13,
14] fuctional with the 6-311+G(3df, 2p) basis set [
15,
16,
17,
18]. Then, we use the Gaussian 4 method to determine the optimized structure of the molecule. We examine the molecular oxidation process of a CH
3OH into an HCHO and an H
2. The optimized structures of a CH
3OH, an HCHO, and an H
2 are displayed in
Figure 1 using the MOLDEN software [
19]. The bond lengths and the angle of bonds calculated with the present simulations are compared with that determined by the experimental studies [
20,
21,
22]. One of the CH bond in a CH
3OH is shorter than the others because a hydrogen atom of the hydroxyl faces in the opposite direction with respect to the CH bond. We find the excellent agreement between the simulations and the experiments and confirm the Gaussian 4 method.
We locate a transition state by applying the synchronous transit guided quasi Newton method. The combinations of the H–O–C bend with the OH stretches brings a proton of the hydroxyl toward the carbon atom of a CH
3OH.
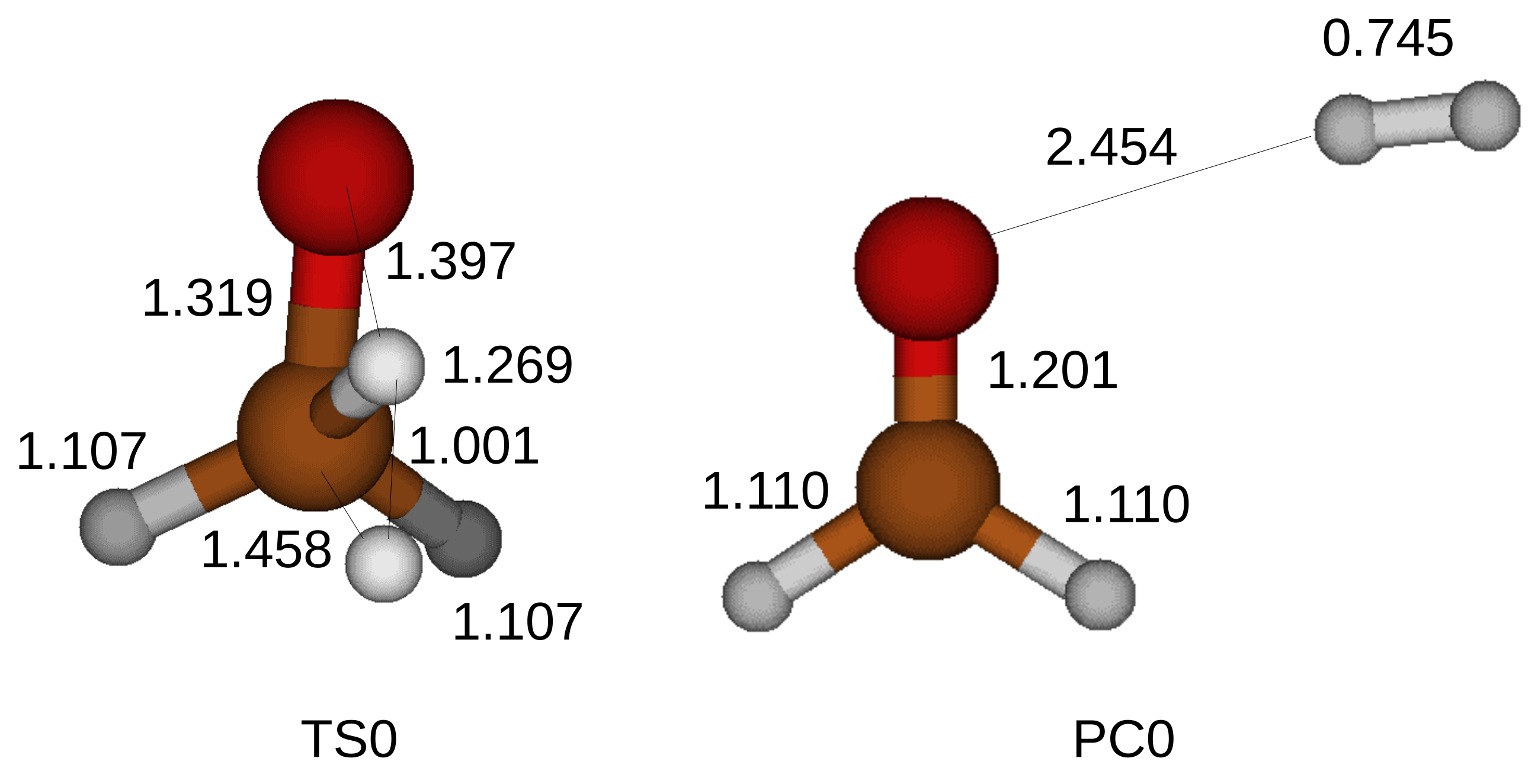
Figure 2 shows a transition state for the oxidation of the CH
3OH. A bond is formed between the proton and the carbon atom, while another proton of the methyl is liberated. We find a single imaginary frequency of vibration in a transition state and confirm the discovered transition state. We execute the IRC calculation to trail the minimum energy pathway and discover local lowest energy points, which correspond to a CH
3OH and a product compound. The product compound consists of an HCHO and an H
2.
It was shown that the decomposition of formic acid and methanediol is efficiently accelerated by H
2O molecules [
7,
8,
9]. H
2O molecules need to be incorporated in the simulations for the oxidation of CH
3OH in water. We use the Amber 16 [
12] and find the number density distribution of H
2O molecules as a function of the distance from a CH
3OH. We carry out a molecular dynamical simulation of a CH
3OH and 2689 H
2O molecules distributed in a box (46.9 Å, 47.7 Å, and 48.3 Å) for 100 pico seconds. The pressure and the temperature are controlled to keep constant values of 1 atm and 300 K.
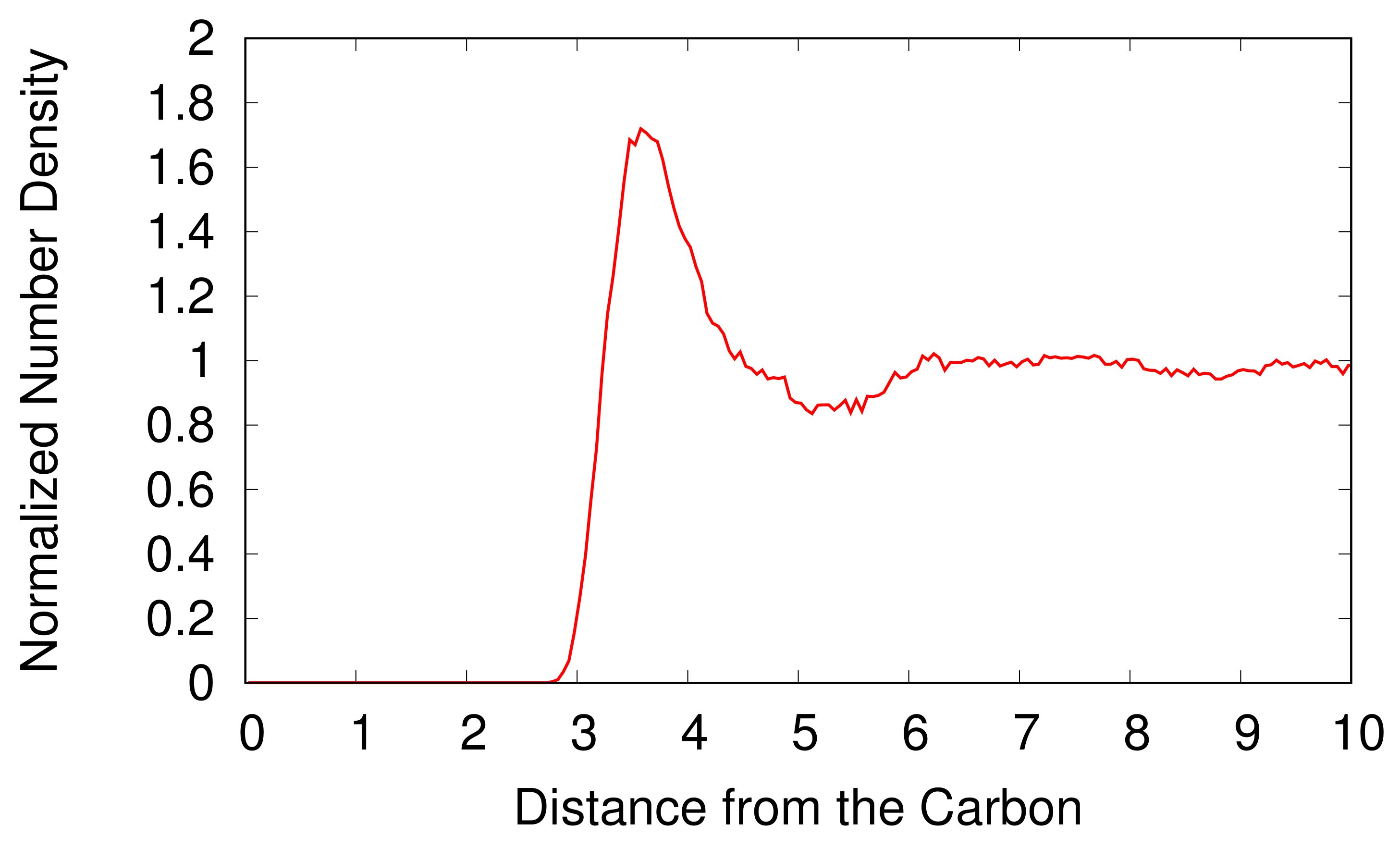
The radial number density distribution of H
2O molecules is shown in
Figure 3 as a function of the distance from the carbon atom of the CH
3OH. We normalize the number density of the H
2O molecules by the background number density of H
2O molecules. A CH
3OH expels adjacent H
2O molecules and forms an empty space with the radius of 2.8 Å around the CH
3OH. The normalized number density of H
2O molecules increases rapidly with the increasing distance from the CH
3OH and has a maximum at 3.6 Å, which is a first hydration shell of the CH
3OH. Beyond the first hydration shell, the normalized number density of the H
2O molecules decreases and converges to one as the distance from the CH
3OH increases because H
2O molecules distant from a CH
3OH are not influenced by the CH
3OH.
The oxidation of CH
3OH proceeds efficiently in water with the help of H
2O molecules located near a CH
3OH. The enhanced number density of H
2O molecules at the first hydration shell of a CH
3OH suggests the active catalytic role of H
2O molecules in the oxidation of a CH
3OH. We showed that H
2O molecules assist the decomposition processes of formic acid and methanediol as catalyst [
7,
8]. H
2O molecules are required to be included in a quantum chemical simulation to describe the oxidation process of a CH
3OH in water. We consider two structures of a supermolecule in the present study: (1) a CH
3OH and H
2O molecules form a ring structure and (2) a CH
3OH and a cluster of H
2O molecules are bonded with hydrogen bonds.
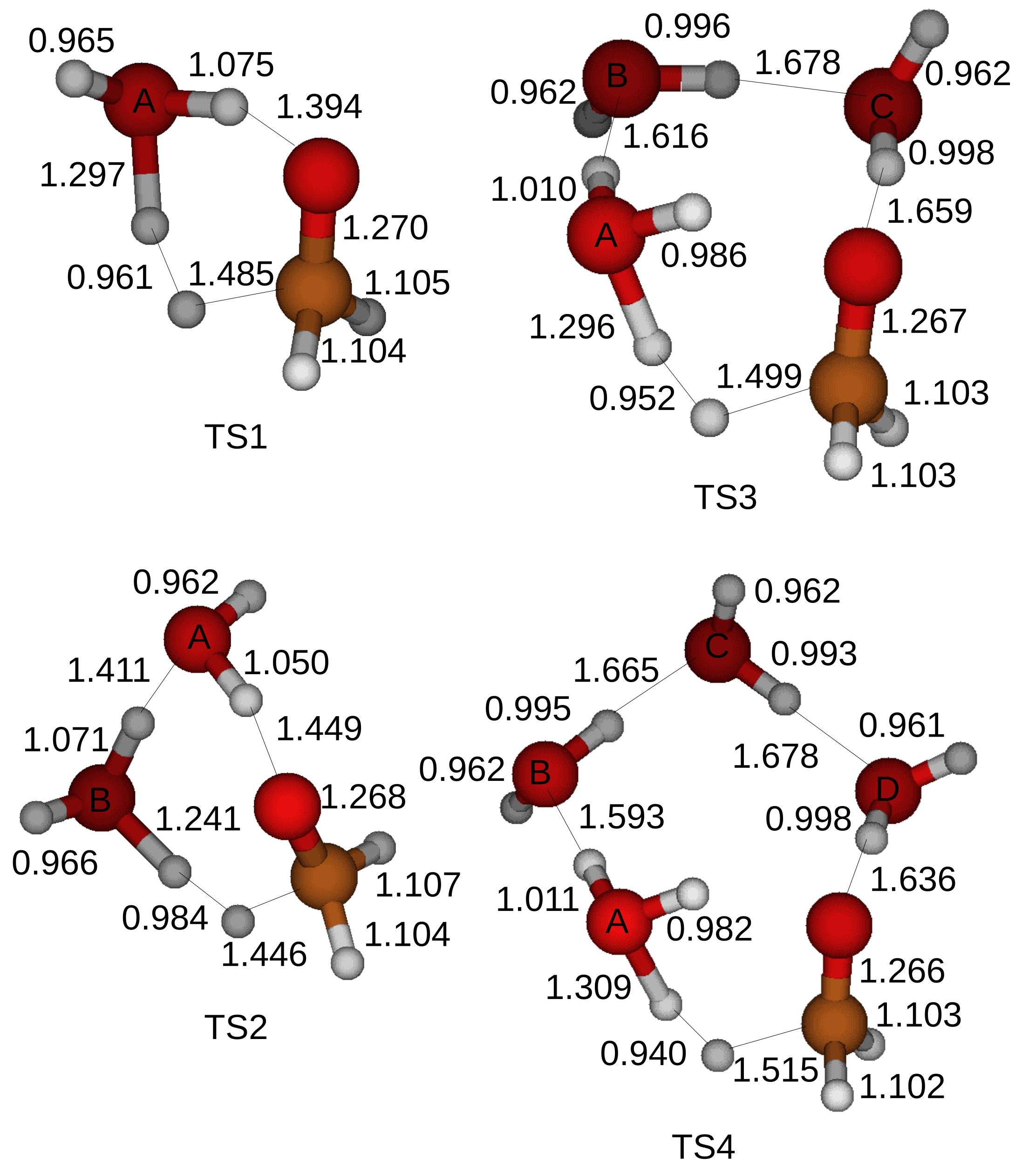
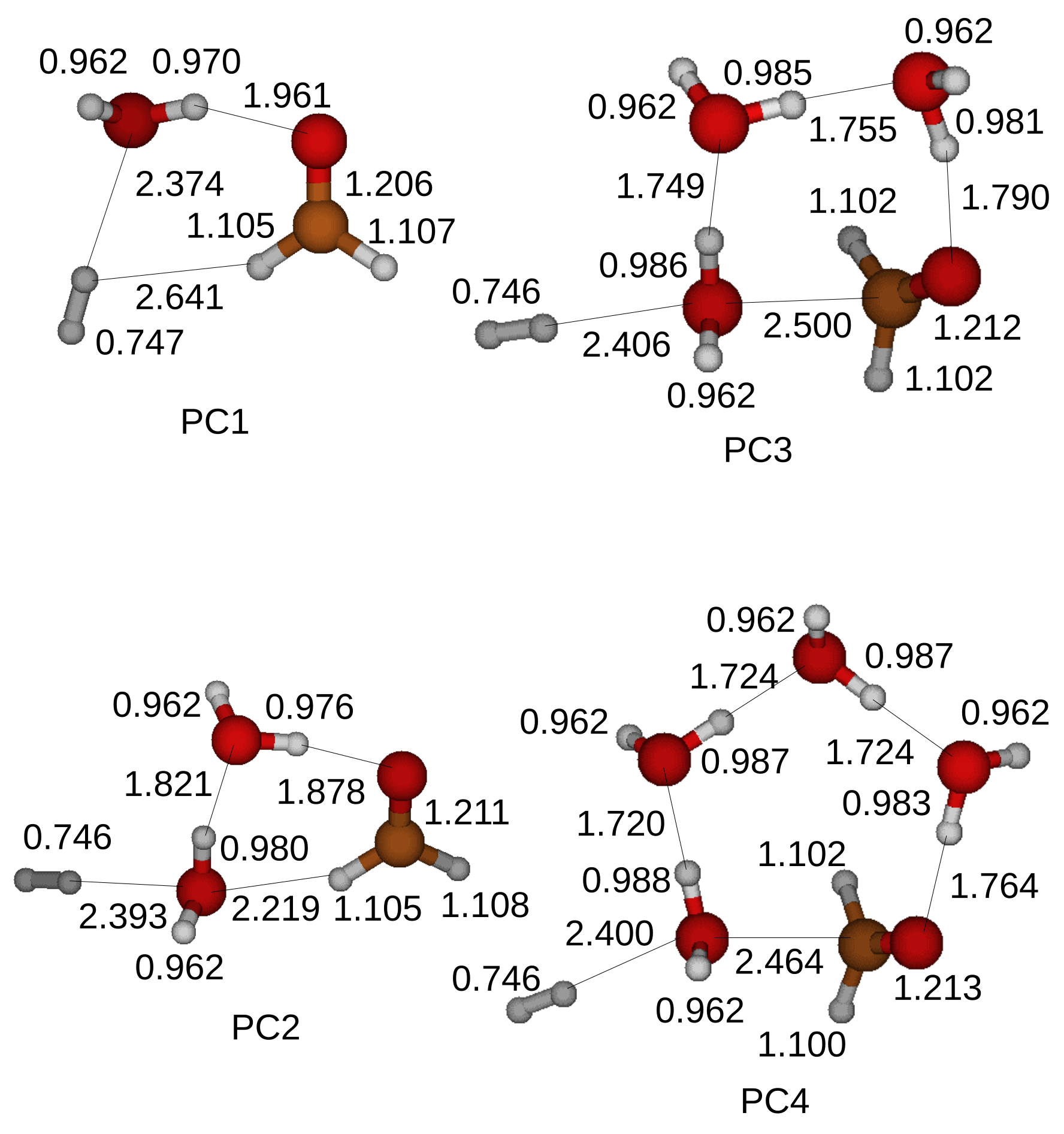
In the first structure a CH
3OH and H
2O molecules are attracted by hydrogen bonds and form a ring structure in a transition state. The optimized structures of the transition states for the oxidation of a CH
3OH are shown in
Figure 4 when up to four H
2O molecules are included in the simulations. A transition state cannot be located when five H
2O molecules are introduced in a simulation.
The oxidation process of a CH
3OH starts when a proton of a hydroxyl of a CH
3OH is shared with a first neighbor H
2O molecule with the label A. The shared proton is completely transferred to the first H
2O molecule with the label A after another proton of the first H
2O molecule with the label A is shared with a second H
2O molecule with the label B when a CH
3OH and two H
2O molecules form a ring structure. On the other hand, a proton cannot be transferred to a second H
2O molecule with the label B when more than three H
2O molecules are included in the simulations. A transition state is formed when a proton is detached from the CH bond of the CH
3OH as shown in
Figure 4.
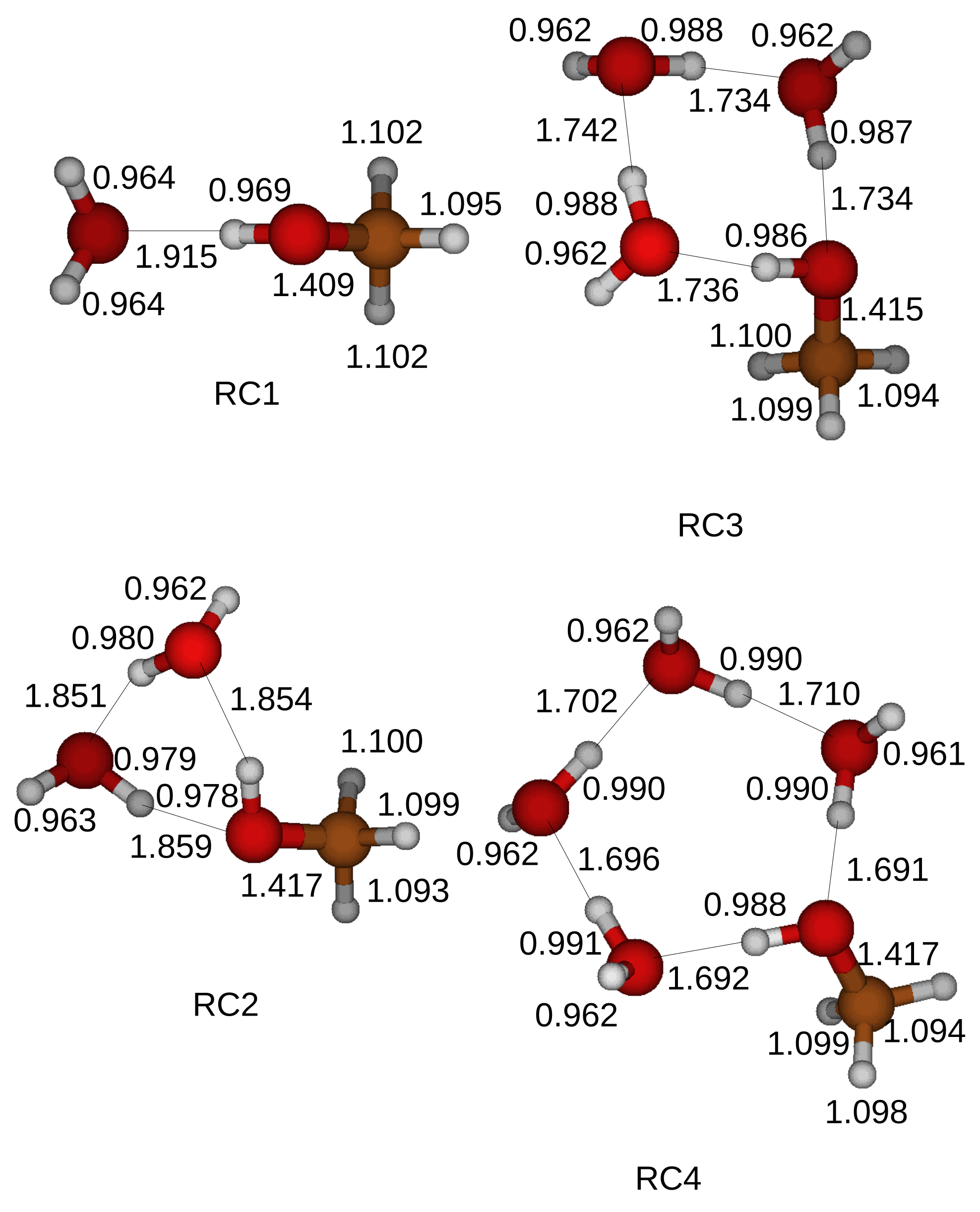
We adopt the transition states and carry out the IRC calculations with the 6-311+G(3df, 2p) basis set to follow the minimum energy reaction pathway. Reactant and product compounds are located at local minimums. We further apply the Gaussian 4 method to obtain the optimized structures of the reactant and product compounds. The optimized structures of the reactant compounds and the product compounds are shown in
Figure 5 and
Figure 6. A hydrogen bond is formed when an oxygen atom faces a neighbor hydrogen atom. A hydrogen bond network contains a hydroxyl of a CH
3OH as shown in
Figure 5. The total energy of a reactant compound is reduced by the formed hydrogen bonds inside the reactant compound.
Figure 6 shows the structures of the product compounds, an HCHO, an H
2, and H
2O molecules, when a CH
3OH and H
2O molecules form a ring structure in a transition state. An HCHO in a product compound has approximately the same structure with that shown in
Figure 1. The hydrogen bonds stretch the CO bond weakly by a factor of 1.01. We confirm that the final products yielded from the oxidation of a CH
3OH are an HCHO and an H
2. The total energy of a product compound is also reduced due to the formed hydrogen bonds inside the product compound. A ring structure of a supermolecule is reconstructed after the oxidation.
We apply the transition state theory to calculate the oxidation rate of a CH
3OH with Equation (
3). The oxidation rate of a CH
3OH is strongly dependent on the energy barrier for the oxidation. We calculate the energy barrier from the difference of the electronic energies corrected by the zero point energy between a transition state and a reactant compound. We find the electronic energy and the zero point energy with the Gaussian 4 method.
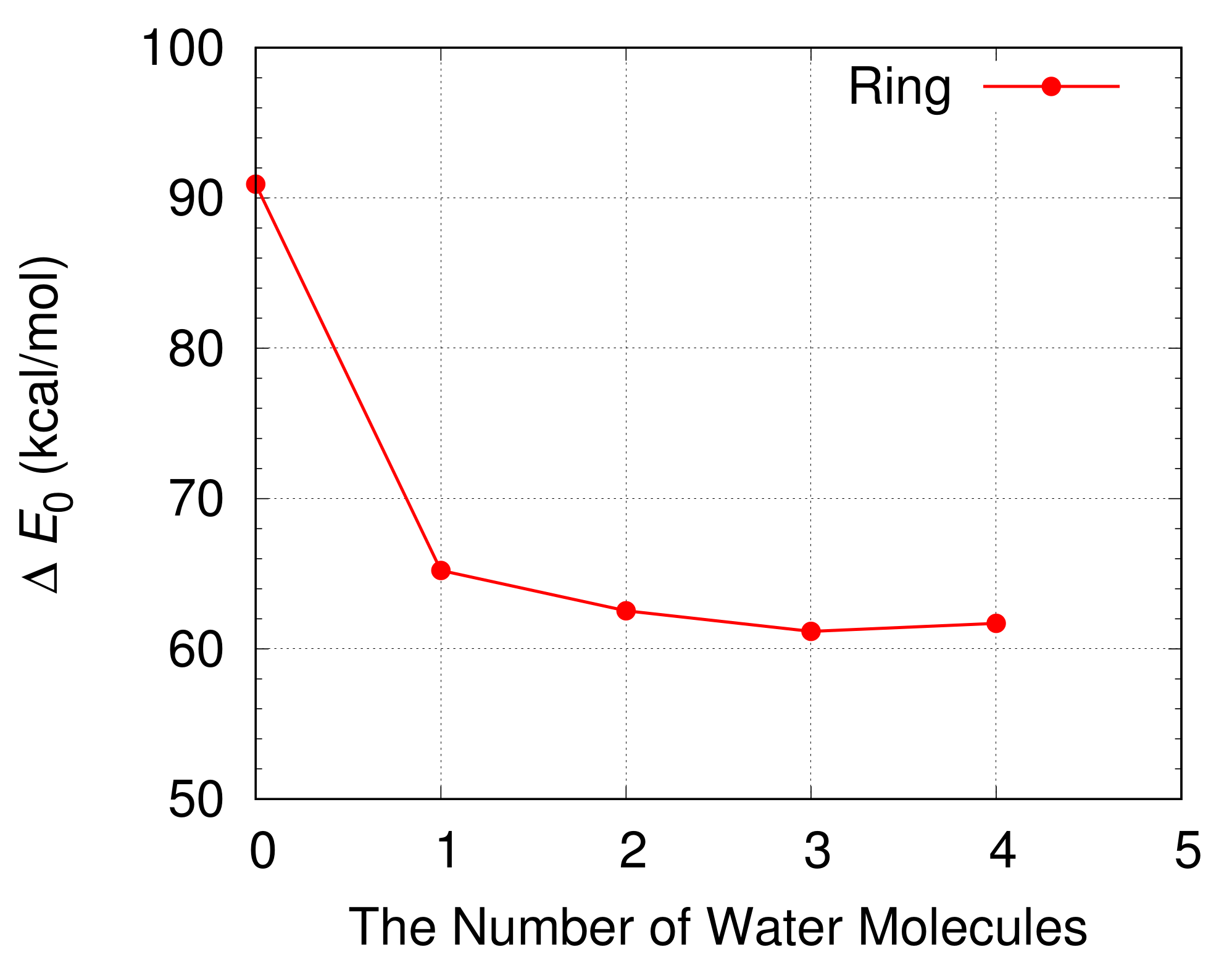
The energy barrier for the oxidation of a CH
3OH is shown in
Figure 7 and
Table 1. The free energy differences between a reactant compound and a transition state at 298.15 K are also displayed in
Table 1. The energy barrier for the oxidation of a CH
3OH decreases from 90.9 kcal/mol to 65.2 kcal/mol when a single H
2O molecule is included in a simulation. This indicates the catalytic role of a H
2O molecule during the oxidation of a CH
3OH. The energy barrier decreases further when more H
2O molecules are introduced in the simulation and has a minimum value of 61.2 kcal/mol when three H
2O molecules are included in the simulation.
A cluster of H
2O molecules was shown to play an active role as catalyst in the decomposition of formic acid and methanediol [
7,
8]. A hydrogen bond in liquid water forms when a proton of an H
2O molecule faces an oxygen atom of a neighbor H
2O molecule. It is natural to consider that H
2O molecules in liquid water develop a network of hydrogen bonds and frequently organize a cluster of H
2O molecules. Experimental studies with the far-infrared vibrational-rotational-tunneling spectroscopy confirmed small clusters consisted of 2, 3, 4, and 5 H
2O molecules [
23,
24,
25,
26]. A cluster of H
2O molecules has a cyclic network of hydrogen bonds. The intermolecular distance between H
2O molecules decreases by the cooperative many body interactions in a cluster of H
2O molecules. A proton is efficiently transferred between H
2O molecules within a cluster of H
2O molecules.
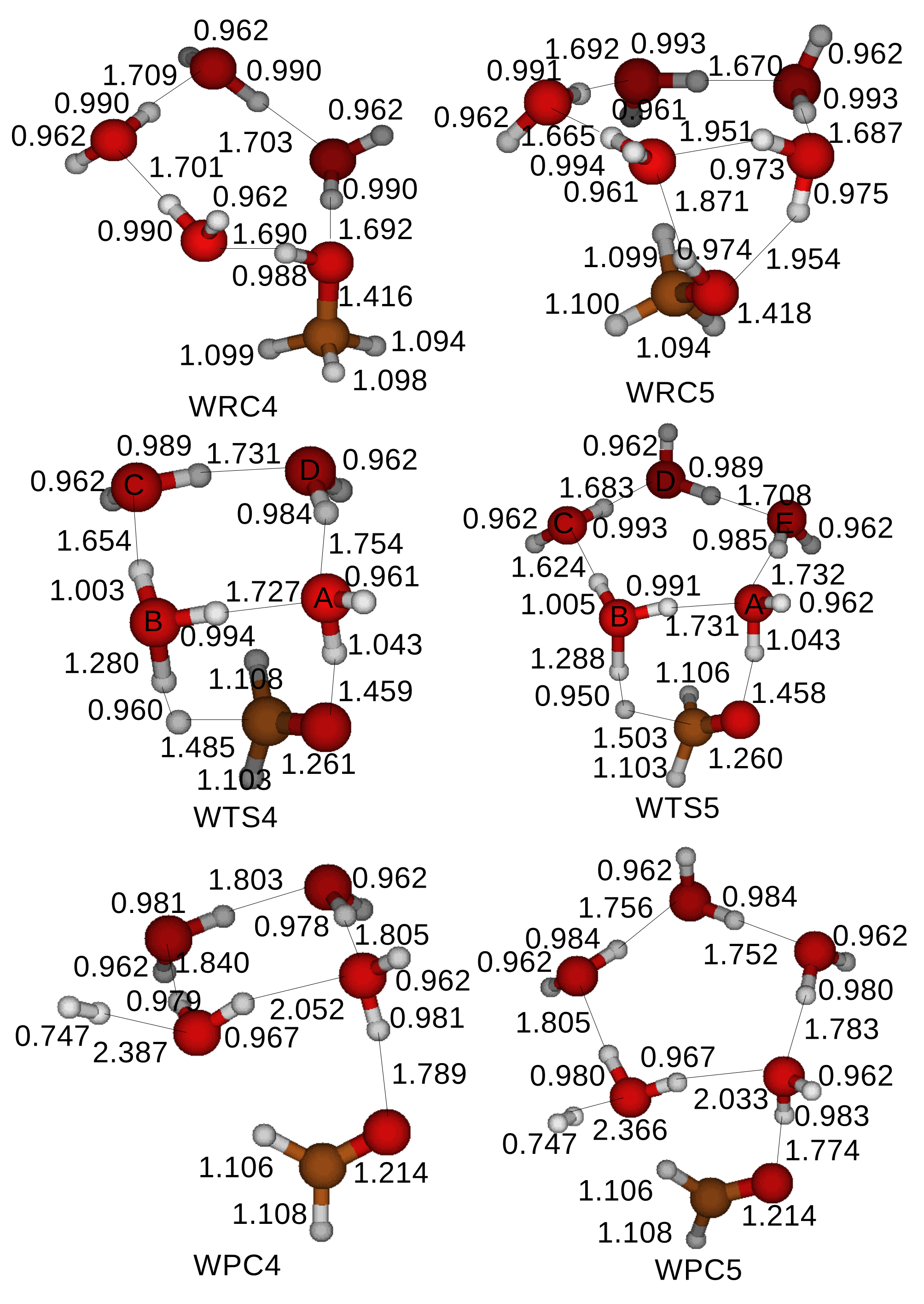
A cluster of H
2O molecules is expected to reduce the energy barrier in the oxidation of CH
3OH as well. We include clusters with 4 and 5 H
2O molecules in the simulations and examine the catalytic role of a cluster of H
2O molecules in the oxidation of CH
3OH.
Figure 8 shows the optimized structures of the reactant compounds, the transition states, and the product compounds for the oxidation of a CH
3OH catalyzed by a cluster of H
2O molecules. A CH
3OH uses two H
2O molecules with the labels A and B within a cluster of H
2O molecules to transfer a proton of the hydroxyl of a CH
3OH, while the rest of H
2O molecules in the cluster supports the two H
2O molecules with hydrogen bonds. The oxidation process starts when a proton of an H
2O molecule with the label A is transferred to an H
2O molecule with the label B within a cluster of H
2O molecules. An H
2O molecule with the label A supplements the lost proton with a proton from a hydroxyl of a CH
3OH. A transition state is formed when a proton is detached from a methyl of a CH
3OH. An H
2O molecule with the label B loses an extra proton, which is combined with a free proton detached from the methyl of a CH
3OH, yielding an H
2. Clusters of four and five H
2O molecules are recovered in product compounds and might catalyze another oxidation of a CH
3OH.
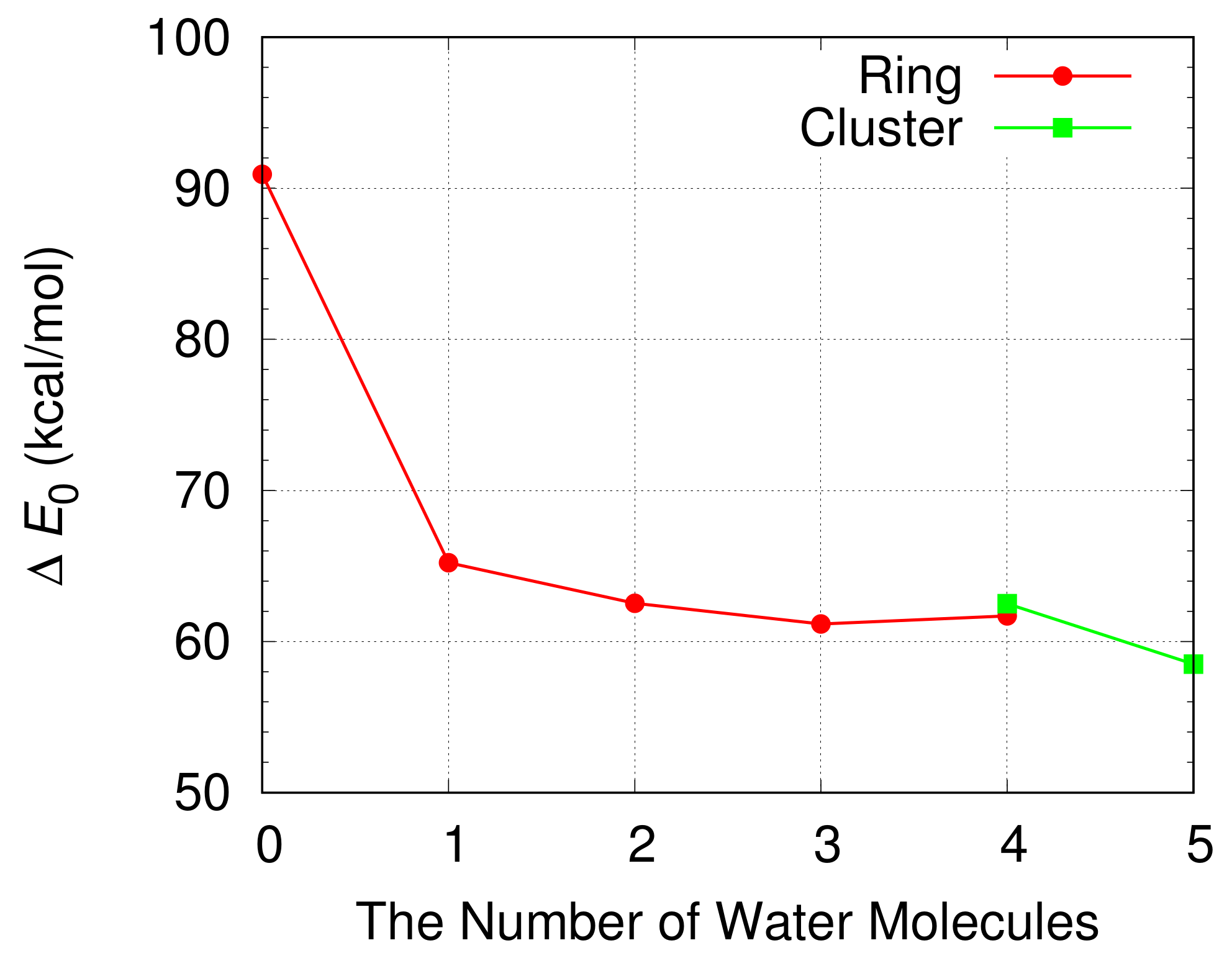
The energy barrier for the oxidation of a CH
3OH is shown in
Figure 9 when a cluster of H
2O molecules catalyzes the oxidation. For comparison the energy barrier is also shown when a CH
3OH and H
2O molecules form a ring structure. The energy barrier for the oxidation of a CH
3OH catalyzed by a cluster of four H
2O molecules is nearly the same with that for the oxidation of a CH
3OH by four H
2O molecules in a ring structure. The energy barrier is reduced when a cluster of five H
2O molecules catalyzes the oxidation. We obtain the lowest energy barrier of 58.5 kcal/mol when a cluster with five H
2O molecules is included in the simulation. The energy barrier is about 3 kcal/mol lower than the lowest energy barrier obtained when a ring structure is formed by a CH
3OH and H
2O molecules.
In order to describe the entire decomposition processes of CH
3OH, we need to consider the sequence of decompositions after HCHO: (1) from HCHO to CH
2(OH)
2, (2) from CH
2(OH)
2 to HCOOH, and (3) from HCOOH to CO or CO
2. We performed the ONIOM(QM/MM) simulations [
27,
28,
29,
30] and revealed that HCHO is hydrated to become CH
2(OH)
2 with the low energy barrier of about 20 kcal/mol [
9]. It is possible to consider the decomposition path from HCHO to CO, but it was shown that the energy barrier for this path becomes 61 kcal/mol and is higher than the energy barrier of 49 kcal/mol for the decomposition of CH
2(OH)
2 into HCOOH [
10]. We further examined the decomposition of HCOOH and calculated the energy barriers for the decomposition of HCOOH into CO or CO
2. Both energy barriers are shown to be lower than 40 kcal/mol [
7]. The energy barriers for the decomposition of HCHO into CO or CO
2 are much lower than the energy barrier of 58.5 kcal/mol to decompose CH
3OH into HCHO. The rate constant for the decomposition of CH
3OH into CO or CO
2 is mainly determined by the rate constant to oxidize CH
3OH into HCHO. We calculate the rate constant with Equation (
3) and compare it with the decomposition rate of CH
3OH determined by laboratory experiments.
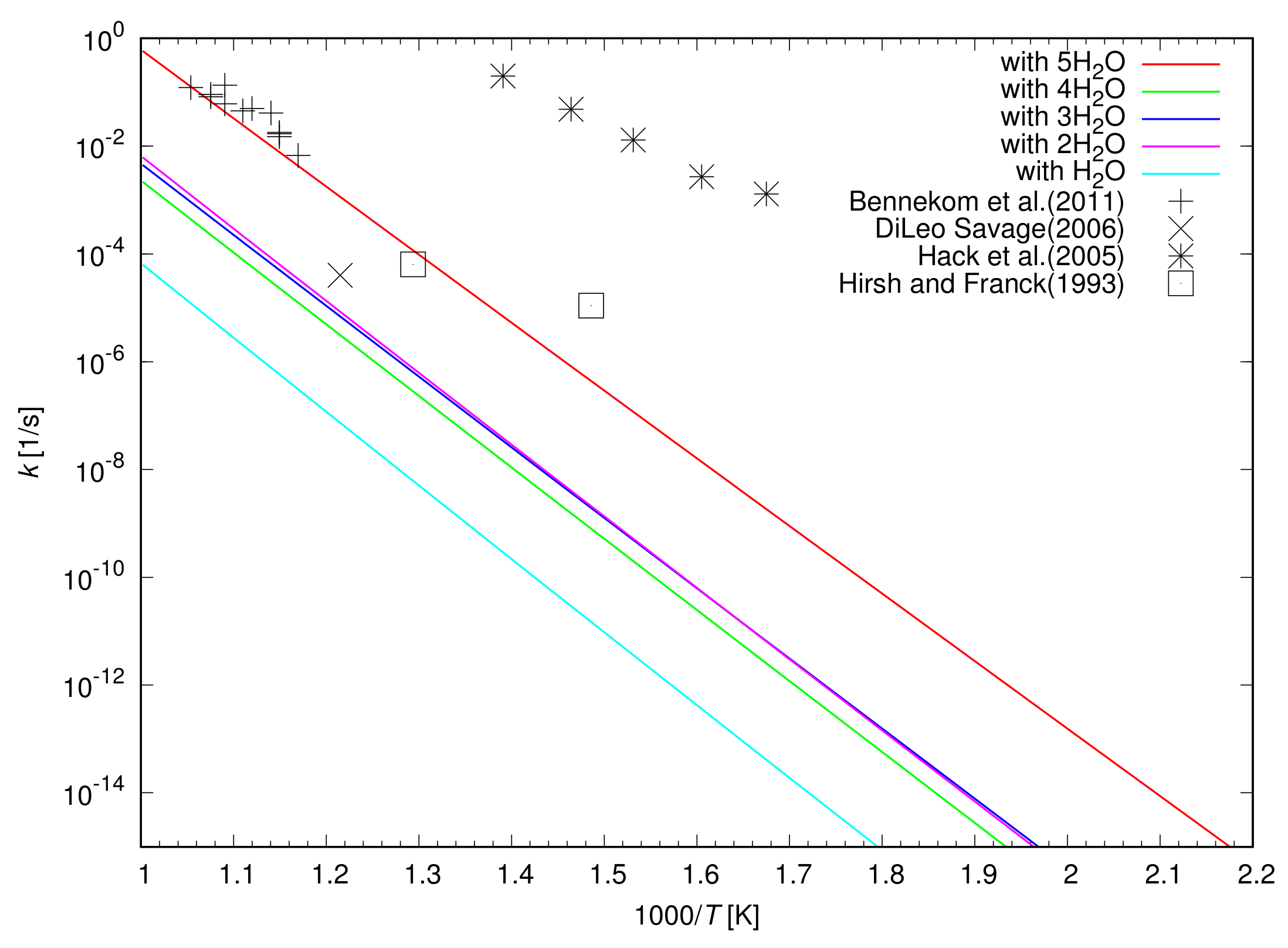
Figure 10 shows the calculated rate constants for the oxidation of a CH
3OH catalyzed by up to five H
2O molecules as a function of the temperature. The rate constant for the oxidation of a CH
3OH increases with an increase in the temperature regardless of the number of H
2O molecules included in the simulations. When H
2O molecules and a CH
3OH form a ring structure in a simulation, the highest rate constant is obtained when two H
2O molecules catalyzes the oxidation of a CH
3OH. The rate constant is further enhanced by the catalyst of a cluster of five H
2O molecules by a factor of 94.
Figure 10 also shows that the rate constants for the oxidation of CH
3OH determined by laboratory experiments. The oxidation of CH
3OH was examined using a Ni alloy reactor in experiments [
3,
6,
31]. The rate constants determined by Hack et al. [
3] is much larger than that calculated in the present study, indicating the catalytic role of the Ni alloy surface of a reactor. It was confirmed that the reactor surface acts as catalyst for the oxidation of CH
3OH and increases the rate constant considerably. The oxidation rate is dependent on the surface condition of a reactor and is reduced by a factor of 1000 when the reactor surface is treated with HNO
3/H
2O
2 before an experiment [
3]. Hirsh and Franck [
6] and Bennekom et al. [
31] also used Ni alloy reactors to study the oxidation of CH
3OH, however, their reactors have a surface to volume ratio smaller than that used by Hack et al. (2005), suggesting the weaker catalytic role of the surface of the reactor. It is shown in
Figure 10 that the rate constants given by Hirsh and Franck (1993) and Bennekom et al. (2011) are much slower than that given by Hack et al. (2005). On the other hand, DiLeo and Savage [
1] performed experiments for the oxidation of CH
3OH in a sealed quartz tube to determine the oxidation rate of CH
3OH without any catalytic effect of a reactor surface. The slowest oxidation of CH
3OH was discovered due to the lack of catalytic effect of a reactor wall as shown in
Figure 10, even though only a single data point was given.
The calculated rate constant is smaller than that given by DiLeo and Savage (2006) when we only consider the catalyst of H2O molecules in a ring structure with a CH3OH. The oxidation of CH3OH catalyzed by a cluster of five H2O molecules is required to be included to explain the measured oxidation rate of DiLeo and Savage (2006). The oxidation of CH3OH determined by the experiment is well explained if we consider three percent of CH3OH molecules are decomposed with the help of the catalyst of a cluster of five H2O molecules. The catalytic effect of Ni metal surface needs to be considered when we explain the oxidation rate of CH3OH determined by Hack et al. (2005) in a Ni alloy reactor.
3. Computational Method
A simple model for the oxidation process of a CH
3OH in water is used at the present study. We consider the molecular oxidation of a CH
3OH, the product of which are a formaldehyde, HCHO, and a hydrogen molecule, H
2, given by
even though a CH
3OH is expected to decompose into radicals in the gas phase at the high temperature. The interaction of a CH
3OH with another CH
3OH is not considered in this study. Single carbon compounds such as methane might be formed in an experiment, however we do not consider any reactions involving single carbon compounds other than a CH
3OH. A CH
3OH decomposes into an HCHO and an H
2 and interacts with nothing but some H
2O molecules when H
2O molecules act as catalyst for the oxidation of a CH
3OH.
The previous studies showed a crucial role of H
2O molecules to lower the energy barrier for the oxidation of formic acid and methanediol in water [
7,
8]. We perform molecular dynamical simulations with the Amber 16 [
32] and find the distribution of H
2O molecules around a CH
3OH even though a molecular dynamical simulation is not useful to study the formation and breaking of molecular bonds. A CH
3OH and H
2O molecules are located in a box and interact with each other. The temperature and the pressure of a box are regulated to be constant values,
K and 1 atm. A three point model of TIP3P is used to describe a H
2O molecule [
33]. We adopt the periodic boundary conditions with the particle mesh Ewald method to take into account the electric forces of H
2O molecules outside of the box. The positions of the CH
3OH and H
2O molecules are recorded every 0.1 ps. We obtain the number density distribution of the H
2O molecules around the CH
3OH by averaging 1000 coordinate data.
A proton is transferred from a reactant to a neighbor H
2O molecule to promote the oxidation process efficiently. We need to include some H
2O molecules in quantum chemical simulations to follow the formation and breaking processes of bonds between a proton and an H
2O molecule. We adopt the Gaussian 16 software [
34] to perform quantum simulations for the oxidation of a CH
3OH in water. We deal with a supermolecule consisting of a CH
3OH and H
2O molecules when we examine the catalytic role of H
2O molecules in the oxidation of CH
3OH. We use the Gaussian 4 method and optimize the structure of a supermolecule because the Gaussian 4 method is reliable to calculate the enthalpy of formation [
35]. The electronic energies as well as the vibrational frequencies at 0 K of a reactant compound, a transition state, and a product compound are calculated using the optimized structures of a supermolecule. We add thermal correction as a result of translation, rotation, and vibration of atomic nuclei to the electronic energy and obtain the total energy of a supermolecule. The energy barrier,
, for the oxidation of a CH
3OH is given by the difference of the electronic energies corrected by the zero point energy between a transition state and a reactant compound.
Transition state theory expresses the oxidation rate of a CH
3OH as
where
is an enhanced factor due to the tunneling of a proton through an energy barrier,
and
are the partition functions of a transition state and a reactant compound, and
h,
, and
T, are the Planck constant, the Boltzmann constant, and the temperature, respectively. We calculate the enhanced factor due to the tunneling following our previous study [
7] by approximating the energy barrier with the unsymmetrical Eckart potential. We carry out the IRC calculation and follow the minimum energy pathway to find a reactant compound and a product compound. The discovered reactant compound and product compound are further optimized with the Gaussian 4 method.
The partition function is calculated from the product of the partition functions due to translation, rotation, and vibration. Furthermore, the oxidation rate relies only on the partition functions of the rotation and the vibration because both a reactant compound and a transition state have nearly the same partition function for the translation. The rotational partition function is calculated from
where
and
are the rotational temperatures. The vibrational partition function of a supermolecule is given by
where
is the frequency of the
i-th vibrational mode and
N is the total number of atoms contained in the supermolecule. The rotational temperatures and the vibrational frequencies are shown in the output file of the Gaussian 4 and used to obtain the partition functions of a transition state and a reactant compound with Equations (
4) and (
5).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}