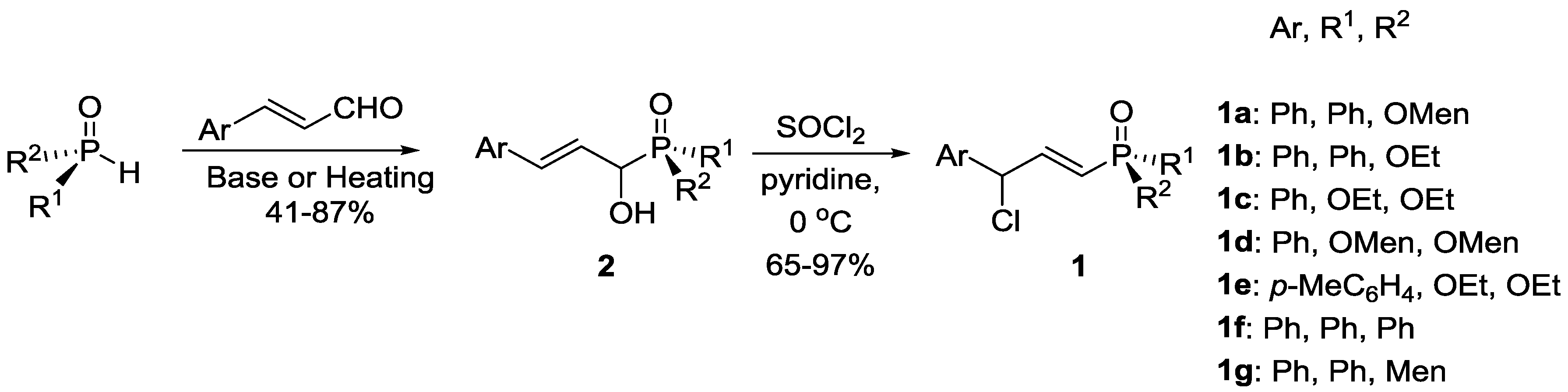
3.2. Synthesis
3.2.1. Addition of H-P Species to Cinnamaldehyde to Form α-Hydroxy Allyl Phosphorus Derivatives 2
(SP)-Menthyl-1-hydroxy-3-phenylallyl phenylphosphinate, 2a
A mixture of (RP)-menthyl phosphinate (10.0 g, 36 mmol) and cinnamaldehyde (5.5 mL, 43 mmol) was heated at 80 °C for 5 h under nitrogen to give a yellow solid, which was recrystallized with dichloromethane/ether to give pure 2a as a white solid. Yield 68%, (10.1 g), m.p. 186.0–188.4 °C. 31P-NMR (162 MHz, CDCl3) δ = 35.67 (s, 68%), 35.03 (s, 32%). 1H-NMR (400 MHz, CDCl3) δ = 7.87–7.73 (m, 2H), 7.55 (q, J = 7.7 Hz, 1H), 7.49–7.38 (m, 2H), 7.29 (dq, J = 16.9, 9.7 Hz, 5H), 6.66–6.49 (m, 1H), 6.27 (dtd, J = 55.0, 10.8, 10.1, 5.6 Hz, 1H), 4.81–4.60 (m, 1H), 4.42 (dq, J = 10.7, 6.1 Hz, 1H), 2.30 (d, J = 6.4 Hz, 1H), 1.80 (d, J = 11.4 Hz, 1H), 1.71–1.54 (m, 2H), 1.51–1.38 (m, 1H), 1.31 (s, 1H), 1.11–0.95 (m, 2H), 0.93 (d, J = 7.0 Hz, 2H), 0.88–0.82 (m, 3H), 0.79 (d, J = 6.9 Hz, 2H), 0.75 (dd, J = 6.4, 3.2 Hz, 4H).
Ethyl 1-hydroxy-3-phenylallyl phenylphosphinate, 2b
A solution of dichlorophenylphosphine (5 mL, 37 mmol) in acetonitrile (10 mL) was added dropwise to ethanol (4.7 mL, 81 mmol) with cooling in an ice bath. After the mixture was warmed to room temperature, potassium carbonate (6.4 g, 46 mmol) and cinnamaldehyde (5.1 mL, 40.7 mmol) were added. The resulting mixture was stirred at room temperature for 14 h. A saturated solution of ammonium chloride (10 mL) was added and the mixture was extracted with dichloromethane (3 × 30 mL) then washed with water (3 × 20 mL). The combined organic layer was dried over anhydrous magnesium sulfate and concentrated in vacuo to give the crude product, which was recrystallized with ether to give pure 2b as a white solid. Yield 55%, (6.1 g), m.p. 137.6–141.7 °C. 31P-NMR (162 MHz, CDCl3) δ = 33.62 (s, 32%), 32.73 (s, 68%). 1H-NMR (400 MHz, CDCl3) δ = 7.87–7.74 (m, 2H), 7.56 (dd, J = 14.6, 7.3 Hz, 1H), 7.51–7.39 (m, 2H), 7.39–7.19 (m, 5H), 6.65–6.54 (m, 1H), 6.35–6.17 (m, 1H), 4.78 (d, J = 25.4 Hz, 1H), 4.16 (dddd, J = 24.8, 21.1, 12.2, 6.7 Hz, 2H), 1.35 (td, J = 7.0, 2.9 Hz, 3H).
Diethyl 1-hydroxy-3-phenylallylphosphonate, 2c
To a solution of diethyl phosphite (3.6 mL, 28 mmol) in acetonitrile (10 mL), potassium carbonate (5.8 g, 42 mmol) and cinnamaldehyde (4.2 mL, 33.6 mmol) were added at room temperature. The mixture was stirred for 13 h at the same temperature. A saturated solution of ammonium chloride (10 mL) was added and the mixture was extracted with dichloromethane (3 × 30 mL) then washed with water (3 × 20 mL). The combined organic layer was dried over anhydrous magnesium sulfate and concentrated in vacuo. The crude product was recrystallized with ether to give pure 2c as a white solid. Yield 87%, 9.1 g, m.p. 98.7–100.0 °C. 31P-NMR (162 MHz, CDCl3) δ = 21.50 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.40 (d, J = 7.4 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.25 (d, J = 5.0 Hz, 1H), 6.78 (dd, J = 16.1, 4.1 Hz, 1H), 6.36–6.27 (m, 1H), 4.66 (dd, J = 13.0, 6.2 Hz, 1H), 4.23–4.15 (m, 4H), 1.33 (td, J = 7.0, 4.2 Hz, 6H).
Dimenthyl 1-hydroxy-3-phenylallylphosphonate, 2d
Compound 2d was prepared according a similar procedure to 2c, which was purified by chromatography with Rf = 0.22 (silica gel, petroleum ether). Pure 2d was obtained as yellow oil. Yield 60% (4.4 g). 31P-NMR (162 MHz, CDCl3) δ = 19.95 (s, 52%), 19.53 (s, 48%). 1H-NMR (400 MHz, CDCl3) δ = 7.41–7.35 (m, 2H), 7.31 (t, J = 6.8 Hz, 2H), 7.24 (d, J = 6.7 Hz, 1H), 6.82–6.73 (m, 1H), 6.33 (dt, J = 16.0, 5.1 Hz, 1H), 4.62 (d, J = 14.2 Hz, 1H), 4.36–4.21 (m, 2H), 2.41–2.10 (m, 4H), 1.66 (d, J = 7.5 Hz, 4H), 1.50–1.29 (m, 4H), 1.22–1.09 (m, 2H), 1.06–0.96 (m, 2H), 0.89 (dt, J = 10.8, 6.7 Hz, 12H), 0.85–0.77 (m, 9H).
Diethyl 1-hydroxy-3-p-tolyl allylphosphonate, 2e
Compound 2e was prepared according a similar procedure to 2c, which was recrystallized with ether. The pure 2e was obtained as a white solid. Yield 80% (6.4 g), m.p. 108.1–113.0 °C. 31P-NMR (162 MHz, CDCl3) δ = 21.45 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.30 (d, J = 7.7 Hz, 2H), 7.13 (d, J = 7.7 Hz, 2H), 6.75 (d, J = 16.1 Hz, 1H), 6.26 (dt, J = 15.9, 5.9 Hz, 1H), 4.71–4.60 (m, 1H), 4.19 (m, 4H), 2.34 (s, 3H), 1.34 (t, J = 7.1 Hz, 6H).
Diphenyl 1-hydroxy-3-phenylallylphosphine oxide, 2f
To a solution of chlorodiphenyl phosphine (6 mL, 29 mmol) in acetonitrile (20 mL), water (0.5 mL) was added dropwise with cooling in an ice bath, then potassium carbonate (6.0 g, 43.5 mmol) and cinnamaldehyde (4.4 mL, 34.8 mmol) were added. The resulting mixture was stirred at room temperature for 18 h. A saturated solution of ammonium chloride (10 mL) was added and the resulting pink solid was collected by filtration and recrystallized with ethanol/ether to give pure 2f as a white solid. Yield 41% (3.9 g), m.p. 168.9–170.3 °C. 31P-NMR (162 MHz, CDCl3) δ = 26.31 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.91–7.80 (m, 4H), 7.55 (dd, J = 15.1, 7.7 Hz, 2H), 7.50–7.43 (m, 4H), 7.27–7.20 (m, 5H), 6.58 (dd, J = 16.0, 4.4 Hz, 1H), 6.31–6.23 (m, 1H), 5.16 (t, J = 6.4 Hz, 1H).
(SP)-Menthyl 1-hydroxy-3-phenylallylphenylphosphine oxide, 2g
Compound 2g was prepared according a similar procedure to 2a, which was recrystallized with ether, and was obtained as a white solid. Yield 54% (8.1 g), m.p. 151.5–156.4 °C. 31P-NMR (162 MHz, CDCl3) δ = 40.12 (s, 58%), 36.47 (s, 42%). 1H-NMR (400 MHz, CDCl3) δ = 7.75 (dt, J = 42.3, 8.9 Hz, 2H), 7.56–7.36 (m, 3H), 7.23–7.12 (m, 5H), 6.44 (dd, J = 59.9, 16.5 Hz, 1H), 6.17–6.03 (m, 1H), 4.94 (d, J = 23.5 Hz, 1H), 2.52–2.20 (m, 2H), 1.95–1.63 (m, 4H), 1.46 (d, J = 30.6 Hz, 2H), 1.18–1.00 (m, 2H), 0.97 (d, J = 5.9 Hz, 1H), 0.91 (t, J = 5.9 Hz, 3H), 0.76 (d, J = 6.8 Hz, 1H), 0.70 (d, J = 6.8 Hz, 2H), 0.29 (d, J = 6.5 Hz, 1H).
3.2.2. Chlorination of α-Hydroxy Allyl Phosphorus Derivatives 2 to Form 1a–1g
(SP)-Menthyl 3-chloro-3-phenylprop-1-en-1-yl phenylphosphinate, 1a
To an ice-cooled solution of 2a (2.0 g, 4.8 mmol) and pyridine (0.6 mL, 7.2 mmol) in dichloromethane (10 mL), thionyl chloride (0.5 mL, 7.2 mmol) was added dropwise. The mixture was allowed to warm to room temperature slowly over 5 h. Water (5 mL) was added and the aqueous layer was extracted with dichloromethane (3 × 10 mL). The combined organic layer was dried over anhydrous magnesium sulfate and concentrated in vacuo. The crude product was recrystallized with petroleum ether (30–60 °C) to give pure 1a as a white solid. Yield 97% (2.1 g), m.p. 148.7–154.1 °C. 31P-NMR (162 MHz, CDCl3) δ = 27.45 (s, 23%), 27.25 (s, 77%). 1H-NMR (400 MHz, CDCl3) δ = 7.83–7.73 (m, 2H), 7.57–7.51 (m, 1H), 7.47 (dt, J = 7.2, 3.6 Hz, 2H), 7.38–7.29 (m, 5H), 6.98 (dddd, J = 19.1, 16.6, 5.8, 2.8 Hz, 1H), 6.19 (dd, J = 20.5, 16.6 Hz, 1H), 5.53 (d, J = 5.6 Hz, 1H), 4.31–4.16 (m, 1H), 2.10 (d, J = 13.6 Hz, 2H), 2.03 (s, 1H), 1.65 (d, J = 6.9 Hz, 2H), 1.41 (d, J = 9.4 Hz, 2H), 1.20–1.09 (m, 1H), 0.93 (dd, J = 9.1, 7.1 Hz, 3H), 0.86–0.81 (m, 4H), 0.80 (s, 1H), 0.75 (d, J = 6.9 Hz, 2H).
Ethyl 3-chloro-3-phenylprop-1-en-1-ylphenylphosphinate, 1b
The compound 1b was prepared similar to 1a, which was purified with flash column chromatography with Rf = 0.46 (silica gel, petroleum ether/ethyl acetate = 2:1 as eluent), and was obtained as a yellow oil. Yield 70% (1.5 g). 31P-NMR (162 MHz, CDCl3) δ = 33.18 (s, 50%), 33.10 (s, 50%). 1H-NMR (400 MHz, CDCl3) δ = 7.83–7.63 (m, 2H), 7.55–7.43 (m, 1H), 7.43–7.33 (m, 2H), 7.35–7.11 (m, 5H), 6.87 (dddd, J = 19.1, 16.7, 5.8, 2.3 Hz, 1H), 6.22–6.05 (m, 1H), 5.50–5.40 (m, 1H), 4.07–3.83 (m, 2H), 1.32–1.18 (m, 3H).
Diethyl 3-chloro-3-phenylprop-1-en-1-ylphosphonate, 1c
The compound 1c was prepared similar to 1a, which was purified with flash column chromatography with Rf = 0.52 (silica gel, petroleum ether/ethyl acetate = 3:1 as eluent), and was obtained as a yellow oil. Yield 65% (1.4 g). 31P-NMR (162 MHz, CDCl3) δ = 16.82 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.47–7.31 (m, 5H), 6.96 (ddd, J = 22.5, 16.7, 5.9 Hz, 1H), 6.08–5.91 (m, 1H), 5.52 (d, J = 4.6 Hz, 1H), 4.21–3.98 (m, 4H), 1.40–1.23 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ = 149.0 (d, J = 7.0 Hz), 138.1 (s), 128.9 (d, J=1.4 Hz), 127.5 (s), 120.1 (s), 118.2 (s), 62.0 (dd, J = 5.6, 2.9 Hz), 61.8 (d, J = 25.2 Hz), 16.3 (d, J = 6.3 Hz). HRMS (ESI+) Calcd. for C13H18ClO3P [M + Na+]: 311.0580, Found: 311.0587.
Dimenthyl 3-chloro-3-phenylprop-1-en-1-ylphosphonate, 1d
The compound 1d was prepared similar to 1a, which was purified with flash column chromatography with Rf = 0.48 (silica gel, petroleum ether/ethyl acetate = 5:1 as eluent), and was obtained as a yellow oil. Yield 92% (1.9 g). 31P-NMR (162 MHz, CDCl3) δ = 14.80 (s, 51%), 14.65 (s, 49%). 1H-NMR (400 MHz, CDCl3) δ = 7.36 (d, J = 2.3 Hz, 4H), 7.25 (dd, J = 13.4, 5.9 Hz, 1H), 7.00–6.86 (m, 1H), 6.05–5.88 (m, 1H), 5.50 (d, J = 5.0 Hz, 1H), 4.34–4.04 (m, 2H), 2.19 (dd, J = 40.1, 9.0 Hz, 3H), 1.98 (d, J = 5.8 Hz, 1H), 1.64 (d, J = 8.6 Hz, 4H), 1.52–1.24 (m, 4H), 1.22–1.07 (m, 2H), 1.07–0.76 (m, 20H), 0.73 (d, J = 6.9 Hz, 1H), 0.66 (d, J = 6.9 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ = 147.8 (dd, J = 14.9, 7.5 Hz), 138.4 (d, J = 3.8 Hz), 128.8 (d, J = 4.4 Hz), 127.5 (d, J = 5.0 Hz), 121.2 (d, J = 188.5 Hz), 77.7 (dd, J = 6.7, 3.8 Hz), 62.0 (dd, J = 25.5, 3.4 Hz), 48.4 (dd, J = 6.7, 3.6 Hz), 43.49 (s), 43.1 (d, J = 4.4 Hz), 34.0 (s), 31.7–31.3 (m), 25.5 (d, J = 6.7 Hz), 22.8 (s), 21.9 (d, J = 3.1 Hz), 20.9 (d, J = 2.5 Hz), 15.8 (d, J = 1.4 Hz), 15.6 (d, J = 13.1 Hz). HRMS (ESI+) Calcd. for C29H46ClO3P [M + Na+]: 531.2771, Found: 531.2766.
Diethyl 3-chloro-3-p-tolylprop-1-en-1-ylphosphonate, 1e
The compound 1e was prepared similar to 1a, which was purified with flash column chromatography (silica gel, petroleum ether/ethyl acetate = 1:1 as eluent), and was obtained as a yellow oil. Yield 65% (1.4 g). 31P-NMR (162 MHz, CDCl3) δ = 16.93 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.27–7.22 (m, 2H), 7.18 (d, J = 7.8 Hz, 2H), 6.95 (ddd, J = 21.6, 16.7, 5.9 Hz, 1H), 6.03–5.90 (m, 1H), 5.50 (d, J = 5.6 Hz, 1H), 4.09 (dq, J = 12.1, 6.5, 6.0 Hz, 4H), 2.35 (s, 3H), 1.34 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ = 150.5 (dd, J = 250.0, 6.2 Hz), 138.4 (d, J = 89.2 Hz), 135.3 (d, J = 24.5 Hz), 129.5 (s), 129.3 (s), 129.1 (s), 127.5 (s), 127.1 (s), 126.8 (s), 126.5 (s), 117.4 (dd, J = 300.6, 187.1 Hz), 82.9 (d, J = 21.3 Hz), 62.0 (dd, J = 6.5, 3.5 Hz), 61.7 (d, J = 6.1 Hz), 56.5 (s), 21.1 (d, J = 1.2 Hz), 16.3 (d, J = 6.3 Hz). HRMS (ESI+) Calcd. for C14H20ClO3P [M + Na+]: 325.0736, Found: 325.0746.
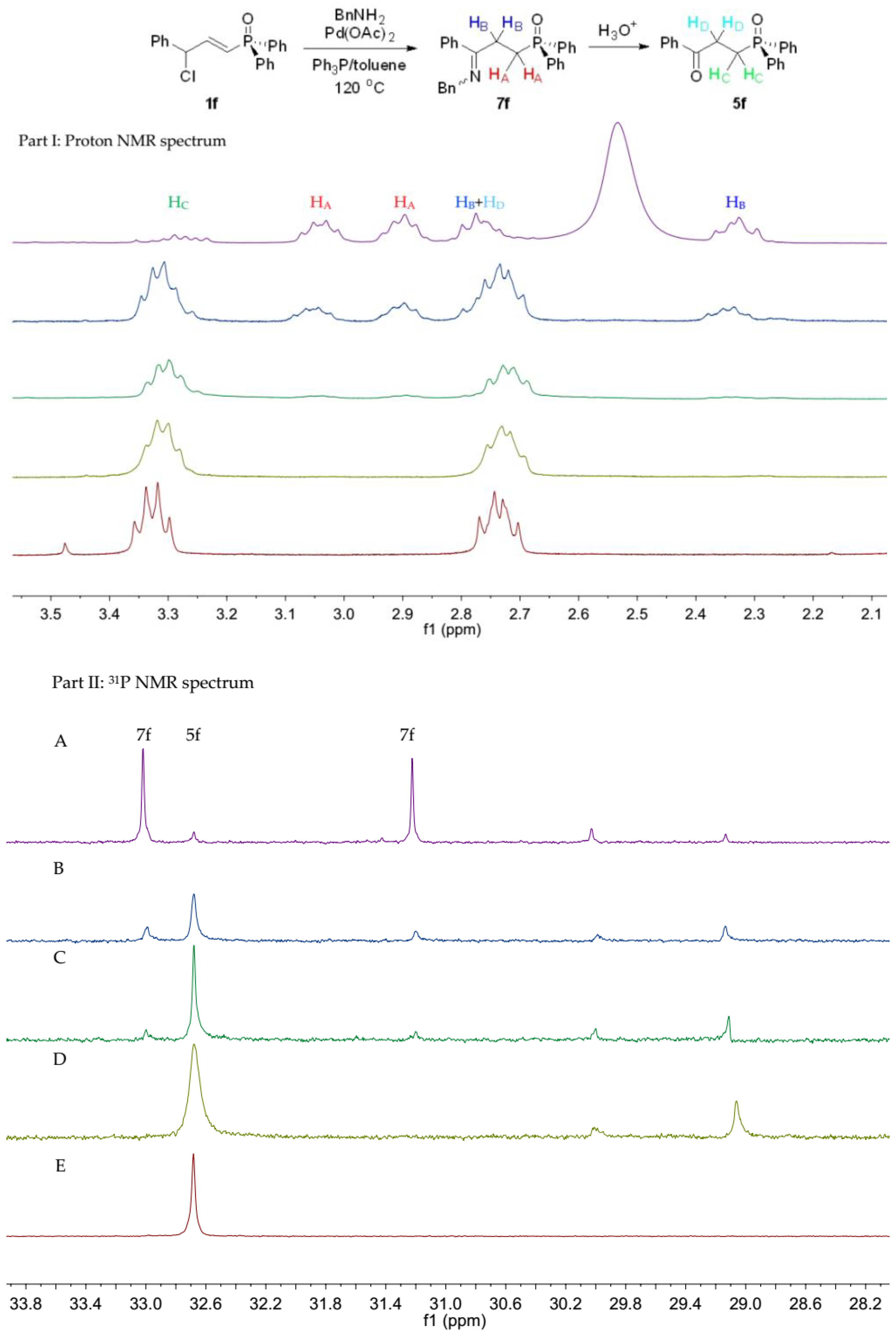
Diphenyl 3-chloro-3-phenylprop-1-en-1-ylphosphine oxide, 1f
The compound 1f was prepared similar to 1a, which was recrystallized with dichloromethane/ether, and was obtained as a yellow solid. Yield 90% (1.9 g), m.p. 109.2–112.4 °C. 31P-NMR (162 MHz, CDCl3) δ = 18.16 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.73–7.64 (m, 4H), 7.59–7.52 (m, 2H), 7.48 (dddd, J = 8.4, 7.0, 4.5, 2.3 Hz, 4H), 7.39–7.31 (m, 5H), 6.99 (ddd, J = 18.2, 16.6, 5.6 Hz, 1H), 6.62 (ddd, J = 22.5, 16.6, 1.5 Hz, 1H), 5.62 (dt, J = 5.6, 2.0 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ = 148.2 (d, J = 4.1 Hz), 138.3 (s), 132.1 (d, J = 2.5 Hz), 131.3 (dd, J = 10.0, 1.5 Hz), 129.0 (s), 128.8 (d, J = 1.4 Hz), 128.7 (d, J = 1.4 Hz), 127.6 (s), 124.3 (d, J = 99.0 Hz), 65.8 (s), 62.3 (d, J = 18.7 Hz), 15.3 (s). HRMS (ESI+) Calcd. for C21H18ClOP [M + Na+]: 375.0681, Found: 375.0692.
(SP)-Menthyl-3-chloro-3-phenylprop-1-en-1-yl phenylphosphine oxide, 1g
The compound 1g was prepared similar to 1a, which was recrystallized with ether, and was obtained as a white solid. Yield 80% (1.6 g), m.p. 138.9–143.6 °C. 31P-NMR (162 MHz, CDCl3) δ = 32.80 (s, 32%), 32.67 (s, 68%). 1H-NMR δ = 7.77–7.65 (m, 2H), 7.48 (s, 3H), 7.34 (d, J = 8.2 Hz, 5H), 6.96 (td, J = 16.6, 5.5 Hz, 1H), 6.56–6.28 (m, 1H), 5.60 (d, J = 5.4 Hz, 1H), 2.15 (s, 1H), 2.07–1.94 (m, 1H), 1.80–1.48 (m, 4H), 1.02 (dq, J = 26.1, 14.2, 13.3 Hz, 2H), 0.89 (d, J = 6.5 Hz, 2H), 0.84 (d, J = 6.5 Hz, 3H), 0.78 (d, J = 6.6 Hz, 2H), 0.48 (t, J = 6.2 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 146.6 (dd, J = 140.6, 2.5 Hz), 138.7 (d, J = 7.0 Hz), 131.3 (d, J = 2.6 Hz), 130.3 (dd, J = 8.6, 4.7 Hz), 129.0 (s), 128.8 (d, J = 5.1 Hz), 128.6 (dd, J = 11.4, 6.1 Hz), 127.6 (d, J = 1.5 Hz), 62.8–62.2 (m), 43.7–43.0 (m), 41.0 (dd, J = 71.6, 30.3 Hz), 35.6 (d, J = 95.2 Hz), 34.3 (s), 33.2 (dd, J = 13.7, 11.4 Hz), 28.3 (dd, J = 7.6, 2.9 Hz), 24.6 (d, J = 12.8 Hz), 22.5 (s), 21.5 (d, J = 6.3 Hz), 15.4 (d, J = 42.7 Hz).HRMS (ESI+) Calcd. for C25H32ClOP [M + Na+]: 437.1777, Found: 437.1773.
3.2.3. The Preparation of (SP)-Menthyl 3-Chloro-3-Phenylallyl Phenyl Phosphine Oxide, 4g
The compound 1g (50 mg, 0.131 mmol) was dissolved in toluene (0.3 mL) and then heated at 120 °C for 14 h. After removing the solvent in vacuo, 4g was obtained as a yellow oil. Yield 98% (49 mg). 31P-NMR (162 MHz, CDCl3) δ = 41.60 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.78–7.70 (m, 2H), 7.45 (s, 3H), 7.40–7.33 (m, 2H), 7.27 (dd, J = 3.9, 1.6 Hz, 3H), 6.16 (dd, J=14.4, 7.3, 1H), 3.27–3.03 (m, 2H), 2.21–1.99 (m, 2H), 1.90 (d, J = 6.2 Hz, 1H), 1.73 (d, J = 7.8 Hz, 4H), 1.43–1.24 (m, 2H), 1.03 (dd, J = 25.0, 12.6 Hz, 1H), 0.94 (dd, J = 8.3, 5.2 Hz, 4H), 0.85–0.78 (m, 3H), 0.42–0.35 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ = 137.67 (d, J = 2.3 Hz), 135.81 (d, J = 12.8 Hz), 134.54 (s), 133.65 (s), 131.21 (d, J = 2.7 Hz), 130.32 (d, J = 8.7 Hz), 128.65 (s), 128.33 (s), 128.21 (d, J = 1.5 Hz), 126.46 (d, J = 1.2 Hz), 117.91 (d, J = 7.2 Hz), 43.32 (d, J = 3.5 Hz), 41.06 (d, J = 66.1 Hz), 35.26 (d, J = 2.4 Hz), 34.20 (d, J = 1.2 Hz), 33.23 (d, J = 13.2 Hz), 31.19 (d, J = 62.9 Hz), 28.31 (d, J = 2.8 Hz), 24.61 (d, J = 12.2 Hz), 22.59 (s), 21.53 (s), 15.20 (s). HRMS (ESI+) Calcd. for C25H32ClOP [M + Na+]: 437.1777, Found: 437.1773.
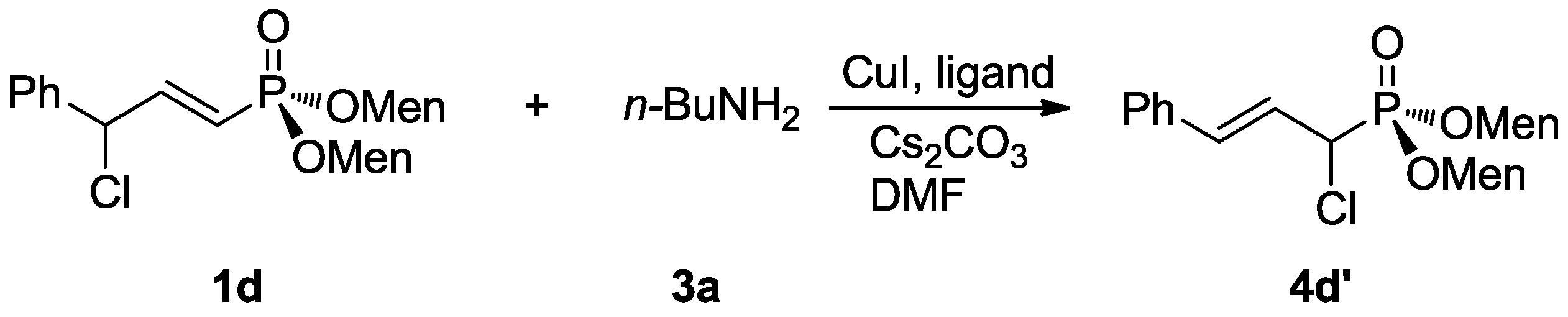
3.2.4. The Preparation of α-Chlorovinylphosphinate 4d’
Dimenthyl 1-chloro-3-phenylprop-1-en-1-ylphosphonate, 4d’
To a suspension of 1d (38 mg, 0.075 mmol), cuprous iodide (0.8 mg, 0.004 mmol, 5 mol %), 1,10-phenanthroline hydrate (14 mg, 0.008 mmol, 10 mol %), and cesium carbonate (29 mg, 0.09 mmol) in DMF (0.2 mL), 3a (7.1 µL, 0.075 mmol) was added. The mixture was heated at 80 °C for 16 h. A saturated aqueous solution of ammonium chloride (3 mL) was added and the mixture was extracted with dichloromethane (3 × 5 mL). The combined organic layer was washed with water (3 × 3 mL), dried over anhydrous magnesium sulfate, and concentrated in vacuo. The crude product was purified with preparative TLC with Rf = 0.50 (silica gel, petroleum ether/ethyl acetate = 3:1). Compound 4d’ was obtained as a yellow oil. Yield 65% (25 mg), 31P-NMR (162 MHz, CDCl3) δ = 16.31 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.41 (t, J = 7.7 Hz, 4H), 7.17 (d, J = 7.7 Hz, 2H), 7.09 (dd, J = 21.1 Hz, 17.1, 1H), 5.46–5.31 (m, 1H), 4.12–3.94 (m, 2H), 2.11 (d, J = 11.8 Hz, 1H), 1.98 (dd, J = 17.3, 10.0 Hz, 2H), 1.85–1.76 (m, 1H), 1.37 (s, 3H), 1.17 (dd, J = 22.9, 12.1 Hz, 3H), 1.03 (dd, J = 23.3, 12.0 Hz, 2H), 0.93 (s, 2H), 0.91–0.85 (m, 10H), 0.83 (dd, J = 6.9, 2.6 Hz, 7H), 0.78 (d, J = 12.5 Hz, 1H), 0.70 (d, J = 6.9 Hz, 3H), 0.59 (d, J = 6.9 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 130.2 (s), 128.5 (s), 77.2 (d, J = 1.9 Hz), 48.3 (dd, J = 6.8, 2.6 Hz), 43.3 (d, J = 59.4 Hz), 34.0 (s), 31.5 (d, J = 3.8 Hz), 25.4 (d, J = 11.9 Hz), 22.8 (d, J = 7.0 Hz), 21.9 (d, J = 7.8 Hz), 21.0 (s), 15.7 (d, J = 12.8 Hz). HRMS (ESI+) Calcd. for C29H49ClO3P [M + H+]: 509.2951, Found: 509.2954.
3.2.5. Preparation of γ-Keto Phosphorus Derivatives 5
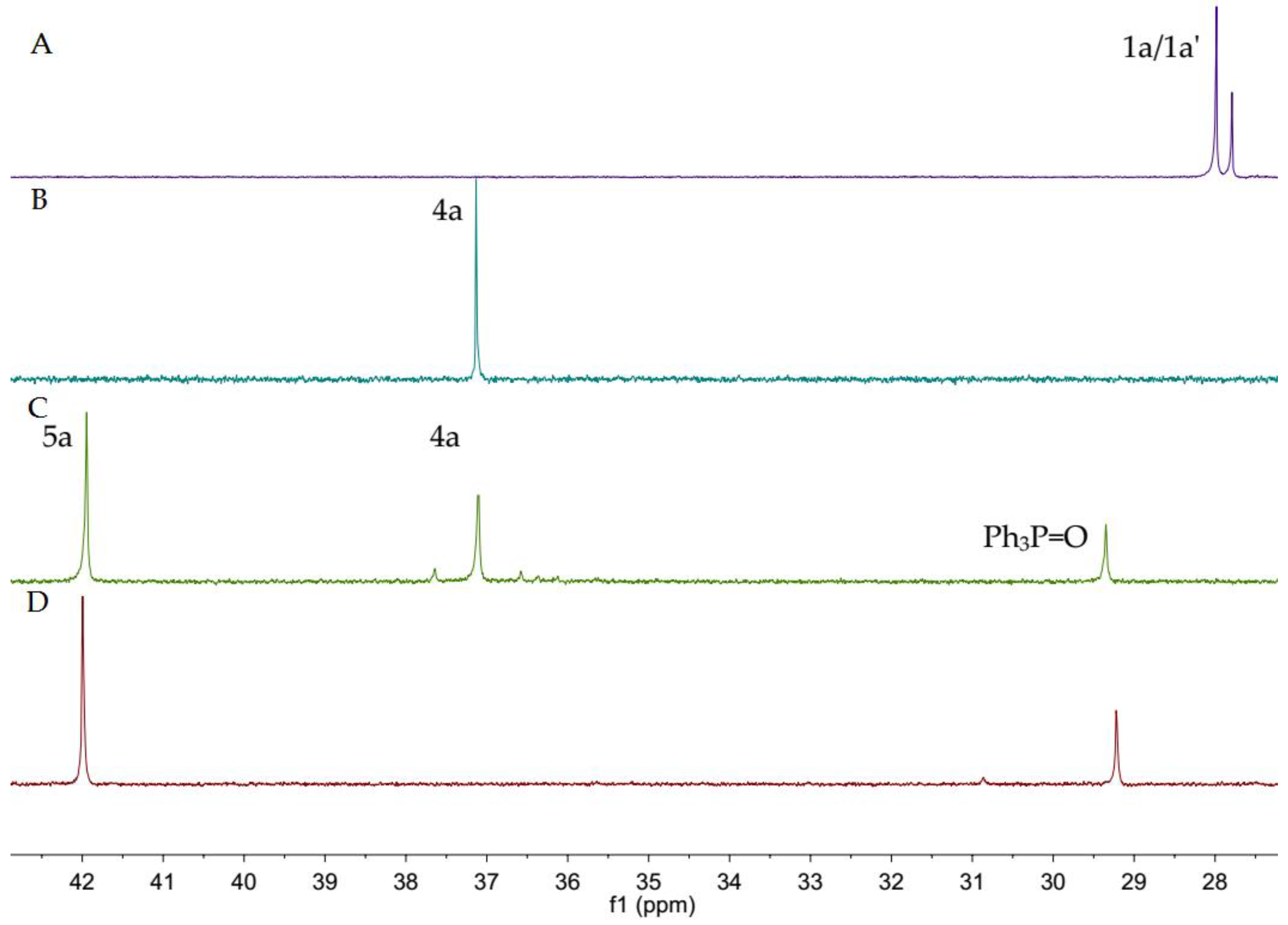
(SP)-Menthyl 3-oxo-3-phenylpropyl phenylphosphinate, 5a
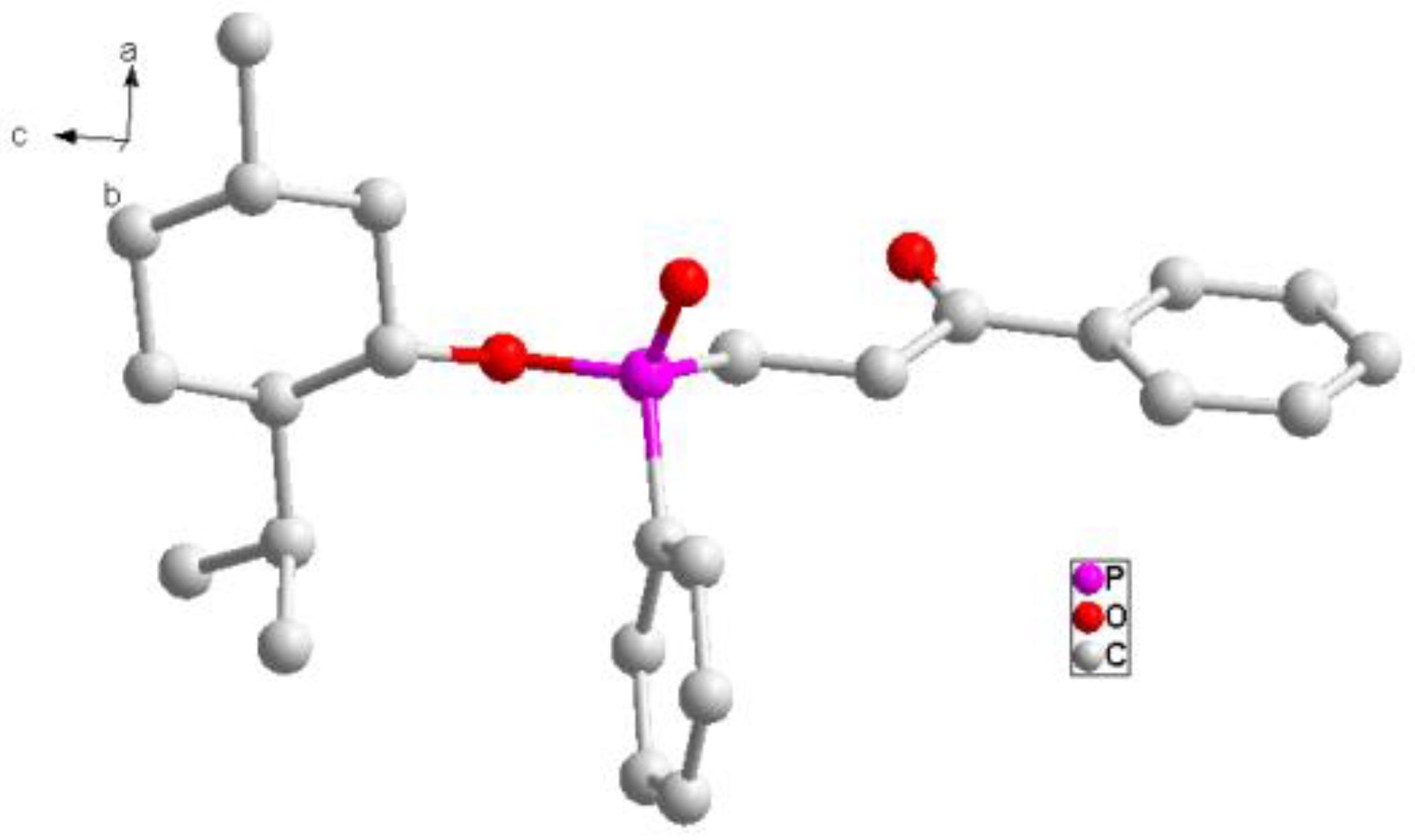
To a solution of 1a (50 mg, 0.116 mmol), palladium acetate (1.3 mg, 0.058 mmol, 5 mol %), and triphenyl phosphine (6.1 mg, 0.232 mmol, 20 mol %) in toluene (0.5 mL), 3a (25 µL, 0.255 mmol) was added. The mixture was heated at 120 °C for 24 h and monitored with TLC. After removing the solvent in vacuo, the residue was dissolved in dichloromethane, filtered over silica gel, and washed with dichloromethane. After removing the solvent in vacuo, the residue was purified with preparative TLC with Rf = 0.55 (silica gel, petroleum ether/ethyl acetate = 1/1 as eluent). The pure 5a was obtained as a yellow solid. Yield 89% (43 mg), m.p. 95.6–100.7 °C. 31P-NMR (162 MHz, CDCl3) δ = 41.90 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.91 (d, J = 7.8 Hz, 2H), 7.84 (dd, J = 11.6, 7.4 Hz, 2H), 7.55 (d, J = 7.6 Hz, 2H), 7.51–7.42 (m, 4H), 4.36–4.28 (m, 1H), 3.33 (s, 1H), 3.11 (s, 1H), 2.45–2.30 (m, 1H), 2.30–2.20 (m, 2H), 1.74 (d, J = 11.5 Hz, 1H), 1.63 (d, J = 27.1 Hz, 4H), 1.44–1.34 (m, 1H), 1.26 (s, 1H), 1.04–0.97 (m, 1H), 0.91 (dd, J = 14.0, 7.0 Hz, 7H), 0.80 (d, J = 12.3 Hz, 1H), 0.74 (d, J = 6.6 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 197.7 (d, J = 15.7 Hz), 136.2 (s), 133.3 (s), 133.1 (s), 132.1 (s), 131.9 (s), 131.3 (d, J = 9.9 Hz), 128.6 (s), 128.6 (s), 128.4 (s), 128.0 (s), 76.6 (s), 48.7 (d, J = 6.0 Hz), 43.1 (s), 34.0 (s), 31.4 (s), 30.9 (s), 25.7 (s), 24.4 (d, J = 103.3 Hz), 22.8 (s), 21.9 (s), 21.1 (s), 15.7 (s). HRMS (ESI+) Calcd. for C25H33O3P [M + H+]: 413.2246, Found: 413.2244.
Ethyl 3-oxo-3-phenylpropyl phenylphosphinate, 5b
The compound 5b was obtained as a yellow solid. Yield 81% (38 mg, containing 79% of 5b and ca. 21% of triphenyl phosphine oxide). Rf = 0.55 (petroleum ether/ethyl acetate = 1:1). 31P-NMR (162 MHz, CDCl3) δ = 44.36 (s, 81%), 29.14(s, 19%). 1H-NMR (400 MHz, CDCl3) δ = 7.93–7.88 (m, 2H), 7.83–7.76 (m, 2H), 7.68–7.61 (m, 2H), 7.56–7.44 (m, 6H), 7.44–7.39 (m, 3H), 4.08 (dd, J = 10.1, 7.1 Hz, 1H), 3.85 (dd, J = 10.1, 7.1 Hz, 1H), 3.34 (dddd, J = 18.1, 11.0, 9.1, 5.1 Hz, 1H), 3.16 (dddd, J = 18.1, 10.8, 9.6, 4.9 Hz, 1H), 2.44–2.20 (m, 3H), 1.26 (t, J = 7.1 Hz, 3H).
Diethyl 3-oxo-3-phenyl propylphosphonate, 5c
The pure 5c was obtained as a yellow solid. Yield 83% (39 mg) Rf = 0.63 (petroleum ether/ethyl acetate = 1:1), m.p. 78.5–83.6 °C. 31P-NMR (162 MHz, CDCl3) δ = 31.77 (s). 1H-NMR (400 MHz, CDCl3) δ = 8.01–7.91 (m, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 4.19–4.02 (m, 4H), 3.30 (ddd, J = 10.6, 7.9, 4.8 Hz, 2H), 2.19 (dddd, J = 39.0, 37.6, 20.4, 17.3 Hz, 2H), 1.37–1.24 (m, 6H). 13C-NMR (101 MHz, CDCl3) δ = 197.4 (d, J = 15.7 Hz), 136.3 (s), 133.3 (s), 128.7 (s), 128.0 (s), 61.7 (d, J = 6.5 Hz), 39.4 (s), 31.7 (d, J = 3.0 Hz), 23.3 (s), 20.6–18.9 (m), 16.4 (d, J = 6.0 Hz), 13.7 (s). HRMS (ESI+) Calcd. for C13H19O4P [M]: 271.1099, Found: 271.1103.
Dimenthyl 3-oxo-3-phenylpropyl phenylphosphonate, 5d
The pure 5d was obtained as a yellow oil. Yield 89% (43 mg). Rf = 0.54 (petroleum ether/ethyl acetate = 5:1). 31P-NMR (400 MHz, CDCl3) δ = 29.24 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.98 (d, J = 7.9 Hz, 2H), 7.58 (t, J = 7.3 Hz, 1H), 7.48 (t, J = 7.3 Hz, 2H), 4.27–4.18 (m, 2H), 3.29 (dd, J = 16.0, 8.7 Hz, 2H), 2.26 (s, 2H), 2.22–2.05 (m, 4H), 1.66 (d, J = 11.6 Hz, 4H), 1.45 (s, 2H), 1.31 (s, 2H), 1.20–1.09 (m, 2H), 1.01 (dd, J = 24.9, 13.2 Hz, 3H), 0.91 (d, J = 4.0 Hz, 12H), 0.84 (dd, J = 14.0, 7.7 Hz, 7H). 13C-NMR (101 MHz, CDCl3) δ = 197.8 (d, J = 16.7 Hz,), 136.4 (s), 133.2 (d, J = 2.8 Hz), 128.6 (s), 128.0 (s), 77.2 (d, J = 7.4 Hz), 48.6 (s), 43.7 (s), 43.1 (s), 34.1 (s), 32.2 (d, J = 3.2 Hz), 31.5 (d, J = 6.5 Hz), 25.7 (s), 25.4 (s), 22.8 (s), 22.5 (s), 21.9 (s), 21.0 (s), 15.8 (s), 15.6 (s). HRMS (ESI+) Calcd. for C29H47O4P [M + H+]: 491.3290, Found: 491.3297.
Diethyl 3-oxo-3-p-tolyl propylphosphonate, 5e
The compound 5e was obtained as a yellow solid. Yield 88% (41 mg, containing 90% of 5e and ca. 10% of triphenyl phosphine oxide, 5e was obtained in a yield of 88%). Rf= 0.58 (petroleum ether/ethyl acetate = 1:1). 31P-NMR (162 MHz, CDCl3) δ = 31.90 (s, 93%), 29.13 (s, 7%). 1H-NMR (400 MHz, CDCl3) δ = 7.86 (d, J = 8.0 Hz, 2H), 7.66 (dd, J = 12.0, 7.1 Hz, 0.58H), 7.58–7.50 (m, 0.28H), 7.46 (d, J = 5.0 Hz, 0.59H), 7.25 (d, J = 7.9 Hz, 2H), 4.19–4.02 (m, 4H), 3.29–3.18 (m, 2H), 2.40 (s, 3H), 2.22–2.10 (m, 2H), 1.32 (t, J = 7.1 Hz, 6H).
Diphenyl 3-oxo-3-phenyl propylphosphine oxide, 5f
The pure 5f was obtained as a yellow solid. Yield 87% (41 mg). Rf = 0.48 (petroleum ether/ethyl acetate = 1:1), m.p. 94.3–99.1 °C. 31P-NMR (162 MHz, CDCl3) δ = 28.03 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.92 (d, J = 8.3 Hz, 2H), 7.79 (dd, J = 11.4, 8.1 Hz, 4H), 7.71–7.34 (m, 9H), 3.33 (dd, J = 15.8, 7.9 Hz, 2H), 2.74 (dd, J = 16.1, 10.4 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ = 197.8 (d, J = 14.1 Hz), 136.2 (s), 133.4 (s), 133.0 (s), 131.9 (d, J = 2.7 Hz), 130.8 (d, J = 9.3 Hz), 128.8 (s), 128.7 (s), 128.6 (s), 128.1 (s), 30.7 (s), 23.7 (d, J = 73.9 Hz). HRMS (ESI+) Calcd. for C21H19O2P [M]: 335.1201, Found: 335.1201.
(SP)-Menthyl 3-oxo-3-phenylpropyl phenylphosphine oxide, 5g/5g’
Compound 5g/5g’ was obtained as a yellow solid. Yield 85% (40 mg). Rf = 0.43 (petroleum ether/ethyl acetate = 1:1), m.p. 96.5–100.8 °C. 31P-NMR (162 MHz, CDCl3) δ = 45.87 (s, 20%), 43.78 (s, 80%). 1H-NMR (400 MHz, CDCl3) δ = 7.88 (dd, J = 17.0, 7.7 Hz, 2H), 7.79–7.68 (m, 2H), 7.62–7.44 (m, 4H), 7.40 (t, J = 7.6 Hz, 2H), 3.42 (ddd, J = 18.2, 11.6, 6.2 Hz, 1H), 3.00–2.43 (m, 1H), 2.33–2.14 (m, 2H), 2.11–1.88 (m, 3H), 1.73 (d, J = 8.8 Hz, 3H), 1.46–1.19 (m, 3H), 1.03 (dd, J = 29.4, 16.0 Hz, 1H), 0.92 (d, J = 6.3 Hz, 3H), 0.86 (t, J = 6.3 Hz, 4H), 0.79 (d, J = 6.9 Hz, 1H), 0.39 (d, J = 6.7 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ = 198.4 (d, J = 13.0 Hz), 133.7 (d, J = 93.4 Hz), 131.6–130.9 (m), 130.6 (d, J = 8.6 Hz), 128.5 (t, J = 5.4 Hz), 128.0 (d, J = 4.4 Hz), 43.4 (d, J = 3.3 Hz), 41.5 (dd, J = 67.3, 51.8 Hz), 35.8 (d, J = 78.0 Hz), 34.2 (s), 33.2 (dd, J = 13.2, 7.4 Hz), 30.6 (s), 28.4 (d, J = 19.9 Hz), 24.6 (d, J = 12.3 Hz), 22.2 (d, J = 57.4 Hz), 21.5–21.2 (m), 15.4 (d, J = 55.2 Hz). HRMS (ESI+) Calcd. for C25H33O2P [M]: 397.2296, Found: 397.2303.
3.2.6. Preparation of γ-Amino Phosphorous Derivatives 11
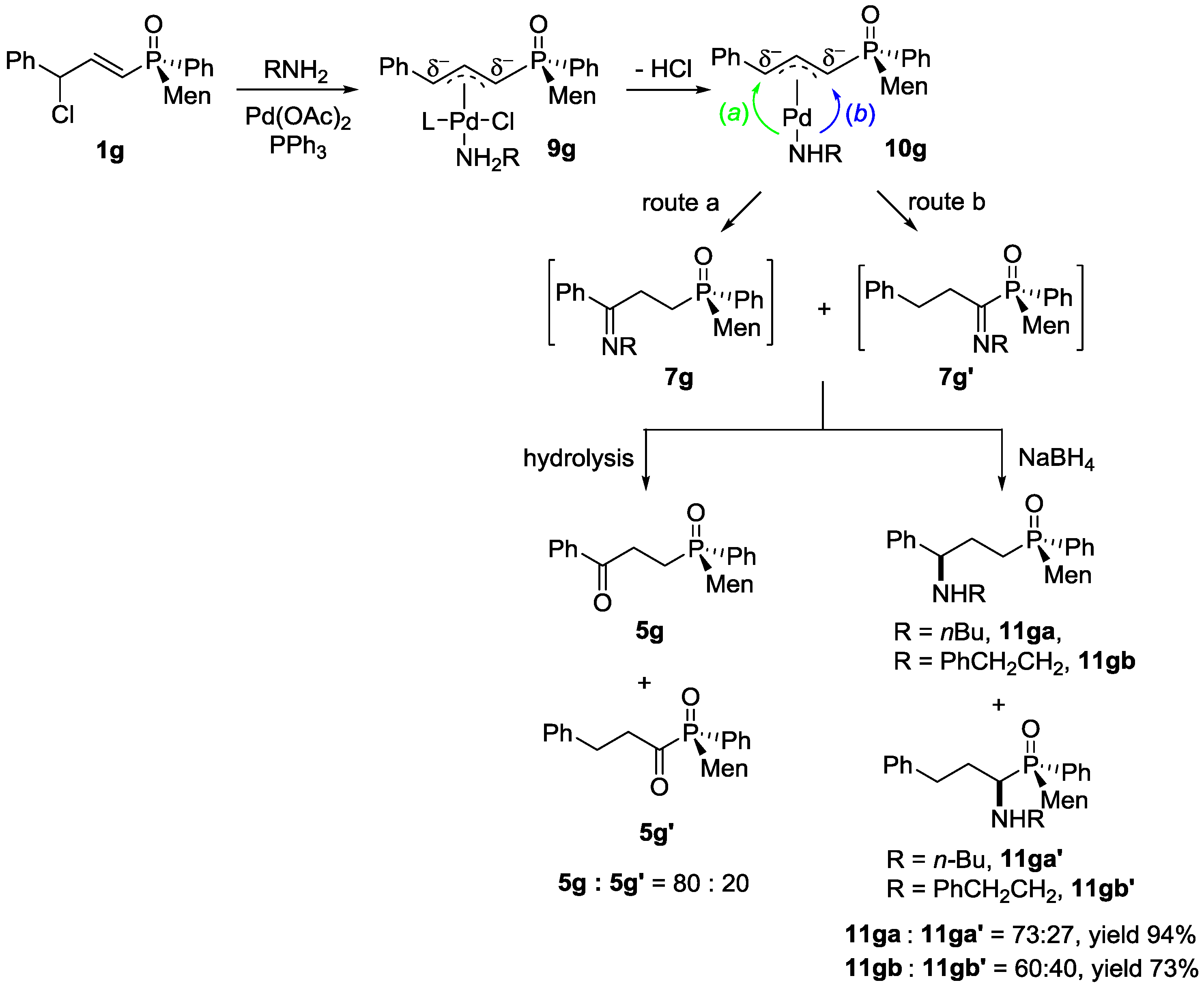
(SP)-Menthyl-3-butylamino-3-phenylpropyl phenylphosphinate, 11aa
To a solution of 1a (50 mg, 0.116 mmol), palladium acetate (1.3 mg, 0.058 mmol, 5 mol %), and triphenyl phosphine (6.1 mg, 0.232 mmol, 20 mol %) in toluene (0.5 mL), 3a (22 µL, 0.225 mmol) was added. The mixture was heated at 120 °C for 24 h, monitored with TLC, and then cooled to room temperature. Ethanol (2 mL) and sodium borohydride (8.7 mg, 0.232 mmol) were added. The mixture was stirred at room temperature for 10 h. After a saturated solution of ammonium chloride (3 mL) was added, the mixture was extracted with dichloromethane (3 × 5 mL) and washed with water (3 × 3 mL). The residue was purified with preparative TLC with Rf = 0.40 (silica gel, methanol/dichloromethane = 1/20 as eluent). The pure 11aa was obtained as a yellow oil. Yield 87% (47 mg). 31P-NMR (162 MHz, CDCl3) δ = 37.39 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.77–7.65 (m, 2H), 7.52 (t, J = 7.3 Hz, 1H), 7.49–7.39 (m, 2H), 7.29 (dd, J = 12.3, 5.9 Hz, 5H), 4.27–4.09 (m, 1H), 3.77 (d, J = 33.2 Hz, 1H), 2.46 (d, J = 7.1 Hz, 2H), 2.23–1.93 (m, 3H), 1.84 (dt, J = 36.0, 14.3 Hz, 2H), 1.74–1.55 (m, 4H), 1.55–1.44 (m, 2H), 1.38–1.18 (m, 4H), 1.03–0.88 (m, 5H), 0.82 (ddd, J = 12.1, 7.0, 3.6 Hz, 6H), 0.77–0.68 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ = 143.1 (s), 133.5 (d, J = 22.0 Hz), 132.3 (d, J = 21.5 Hz), 131.8 (d, J = 2.6 Hz), 131.4 (d, J = 2.2 Hz), 131.3 (d, J = 2.2 Hz), 128.5–128.3 (m), 128.2 (s), 127.2 (t, J = 7.1 Hz), 76.4–76.1 (m), 64.0–63.2 (m), 48.8 (d, J = 4.4 Hz), 47.3 (d, J = 4.3 Hz), 43.2 (s), 34.1 (s), 32.3 (s), 31.4 (s), 29.7 (s), 27.0 (dd, J = 100.8, 10.1 Hz), 25.6 (s), 22.8 (s), 21.9 (s), 21.1 (s), 20.4 (s), 15.7 (d, J = 2.6 Hz), 13.9 (s). HRMS (ESI+) Calcd. for C29H44NO2P [M]: 470.3188, Found: 470.3188.
(SP)-Menthyl-3-phenethylamino-3-phenylpropyl phenylphosphinate, 11ab
The pure 11ab was obtained as a yellow oil. Yield 83% (35 mg). Rf = 0.46 (methanol/dichloromethane = 1:20). 31P-NMR (162 MHz, CDCl3) δ = 43.35 (s, 48%), 43.20 (s, 52%). 1H-NMR (400 MHz, CDCl3) δ = 7.69 (dd, J = 19.5, 10.4 Hz, 2H), 7.57–7.38 (m, 4H), 7.29 (s, 2H), 7.24 (d, J = 7.4 Hz, 2H), 7.15 (dd, J = 22.6, 10.8 Hz, 5H), 4.01–3.86 (m, 1H), 3.59 (s, 1H), 2.80–2.59 (m, 4H), 2.30 (s, 1H), 2.00–1.78 (m, 5H), 1.60 (s, 4H), 1.31–1.10 (m, 3H), 0.84 (dd, J = 37.1, 6.5 Hz, 8H), 0.36–0.21 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ = 142.8 (s), 139.9 (s), 131.9 (s), 131.4 (dd, J = 9.7, 5.2 Hz), 130.7 (d, J = 11.8 Hz), 128.6 (s), 128.4 (d, J = 1.6 Hz), 128.4 (d, J = 3.6 Hz), 128.3 (s), 127.3 (d, J = 3.0 Hz), 127.2 (s), 126.1 (s), 76.6 (d, J = 2.4 Hz), 63.4 (dd, J = 16.4, 9.0 Hz), 48.7 (d, J = 6.7 Hz), 48.6 (d, J = 3.6 Hz), 43.9 (s), 36.3 (s), 34.1 (s), 31.5 (s), 29.6 (s), 26.5 (d, J = 101.8 Hz), 25.3 (s), 22.6 (s), 22.0 (s), 21.0 (s), 15.1 (d, J = 4.6 Hz). HRMS (ESI+) Calcd. for C33H44NO2P [M]: 518.3188, Found: 518.3185.
(SP)-Menthyl 3-phenyl-3-pyrrolidin-1-yl propylphosphinate, 11ac
The pure 11ac was obtained as a yellow oil. Yield 83% (35 mg). Rf = 0.40 (methanol/dichloromethane = 1:20). 31P-NMR (162 MHz, CDCl3) δ = 42.71 (s, 51%), 42.50 (s, 49%). 1H-NMR (400 MHz, CDCl3) δ = 7.65 (dd, J = 18.9, 10.6 Hz, 2H), 7.48 (s, 1H), 7.40 (s, 2H), 7.29–7.19 (m, 3H), 7.16 (t, J = 7.1 Hz, 2H), 4.28–4.13 (m, 1H), 3.04 (d, J = 5.5 Hz, 1H), 2.45 (s, 2H), 2.27 (s, 2H), 2.20–2.04 (m, 2H), 1.97 (s, 2H), 1.77–1.43 (m, 9H), 1.30 (dd, J = 26.6, 13.7 Hz, 2H), 1.03–0.84 (m, 5H), 0.80 (t, J = 7.7 Hz, 3H), 0.70 (t, J = 5.4 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 141.9 (d, J = 19.1 Hz), 132.9 (dd, J = 124.2, 57.6 Hz), 131.9–131.7 (m), 131.3 (dd, J = 12.2, 9.8 Hz), 128.3 (s), 128.2 (s), 128.2 (s), 128.1 (s), 127.1 (d, J = 7.5 Hz), 76.2 (dd, J = 18.1, 7.4 Hz), 70.9 (dd, J = 37.9, 17.2 Hz), 52.7 (s), 52.4 (s), 48.9–48.6 (m), 43.1 (d, J = 7.7 Hz), 34.0 (s), 31.4 (d, J = 1.7 Hz), 27.6 (d, J = 4.7 Hz), 26.5 (dd, J = 101.2, 19.3 Hz), 25.6 (s), 23.2 (d, J = 2.5 Hz), 22.8 (s), 21.9 (s), 21.1 (s), 15.6 (d, J = 3.8 Hz). HRMS (ESI+) Calcd. for C29H42NO2P [M + H+]: 468.3031, Found: 468.3027.
Ethyl 3-butylamino-3-phenylpropyl phenylphosphinate, 11ba
The pure 11ba was obtained as a yellow oil. Yield 90% (50 mg). Rf = 0.41 (ethyl acetate). 31P-NMR (162 MHz, CDCl3) δ = 44.85 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.70 (ddd, J = 11.6, 9.2, 6.8 Hz, 2H), 7.57–7.49 (m, 1H), 7.49–7.37 (m, 2H), 7.30 (d, J = 7.0 Hz, 1H), 7.25–7.21 (m, 1H), 7.18 (d, J = 7.2 Hz, 2H), 4.03 (dt, J = 20.1, 6.4 Hz, 1H), 3.80 (dt, J = 12.4, 7.3 Hz, 1H), 3.55 (dd, J = 12.8, 6.5 Hz, 1H), 2.37 (dt, J = 6.9, 5.1 Hz, 2H), 2.05–1.63 (m, 5H), 1.52 (s, 1H), 1.36 (d, J = 4.2 Hz, 2H), 1.25 (td, J = 7.0, 3.8 Hz, 5H), 0.84 (t, J = 7.3 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 143.1 (s), 132.1 (d, J = 2.7 Hz), 131.6 (s), 131.5 (s), 130.2 (d, J = 15.7 Hz), 128.6 (s), 128.5 (s), 128.4 (s), 127.5–127.0 (m), 63.6 (d, J = 18.8 Hz), 60.5 (d, J = 6.2 Hz), 47.3 (d, J = 3.8 Hz), 32.3 (s), 29.6 (d, J = 2.8 Hz), 26.3 (dd, J = 101.3, 21.0 Hz), 20.4 (s), 16.4 (d, J = 6.5 Hz), 13.9 (s). HRMS (ESI+) Calcd. for C21H30NO2P [M]: 360.2092, Found: 360.2094.
Ethyl 3-phenethylamino-3-phenylpropyl phenylphosphinate, 11bb
The pure 11bb was obtained as a yellow oil. Yield 85% (54 mg). Rf = 0.32 (ethyl acetate). 31P-NMR (162 MHz, CDCl3) δ = 44.76 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.68 (ddd, J = 11.3, 9.9, 4.3 Hz, 2H), 7.53 (t, J = 6.8 Hz, 1H), 7.44 (dd, J = 8.9, 5.8 Hz, 2H), 7.26 (dt, J = 10.8, 7.3 Hz, 5H), 7.18 (d, J = 7.3 Hz, 1H), 7.12 (t, J = 8.3 Hz, 4H), 4.07–3.95 (m, 1H), 3.85–3.72 (m, 1H), 3.56 (dd, J = 12.5, 6.0 Hz, 1H), 2.78–2.59 (m, 4H), 2.01–1.60 (m, 5H), 1.24 (td, J = 7.0, 2.7 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 142.8 (s), 139.9 (s), 132.1 (d, J = 2.7 Hz), 131.6 (s), 131.5 (s), 131.4–130.0 (m), 128.6 (s), 128.5 (d, J = 1.5 Hz), 128.4 (s), 127.2 (d, J = 4.4 Hz), 127.1 (s), 126.1 (s), 63.4 (d, J = 17.4 Hz), 60.5 (d, J = 4.3 Hz), 48.6 (d, J = 3.4 Hz), 36.4 (s), 29.6 (d, J = 2.8 Hz), 26.3 (dd, J = 101.4, 21.2 Hz), 16.4 (d, J = 6.3 Hz). HRMS (ESI+) Calcd. for C25H30NO2P [M]: 408.2092, Found: 408.2107.
Diethyl 3-butylamino-3-phenyl propylphosphonate, 11ca
The pure 11ca was obtained as a yellow oil. Yield 86% (49 mg). Rf = 0.36 (ethyl acetate). 31P-NMR (162 MHz, CDCl3) δ = 32.49 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.35–7.28 (m, 2H), 7.25 (d, J = 7.3 Hz, 3H), 4.10–3.90 (m, 4H), 3.59 (t, J = 6.7 Hz, 1H), 2.50–2.31 (m, 2H), 1.93 (tdd, J = 23.7, 21.5, 16.4 Hz, 3H), 1.79–1.50 (m, 3H), 1.40 (dt, J = 11.9, 7.0 Hz, 2H), 1.32–1.16 (m, 8H), 0.86 (t, J = 7.3 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 143.1 (s), 128.5 (s), 127.2 (s), 127.1 (s), 63.5 (d, J = 17.9 Hz), 61.5 (d, J = 1.4 Hz), 61.4 (d, J = 1.2 Hz), 47.3 (s), 32.3 (s), 30.4 (d, J = 4.4 Hz), 23.0 (s), 21.6 (s), 20.4 (s), 16.4 (d, J = 1.3 Hz), 16.3 (d, J = 2.0 Hz), 13.9 (s). HRMS (ESI+) Calcd. for C17H30NO3P [M]: 328.2042, Found: 328.2083.
Diethyl 3-phenylethylamino-3-phenyl propylphosphonate, 11cb
The pure 11cb was obtained as a yellow oil. Yield 79% (51 mg). Rf = 0.85 (petroleum ether:ethyl acetate = 1:3). 31P-NMR (162 MHz, CDCl3) δ = 32.38 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.33–7.28 (m, 2H), 7.25 (dd, J = 6.5, 3.2 Hz, 3H), 7.22–7.17 (m, 3H), 7.14 (d, J = 6.9 Hz, 2H), 4.11–3.94 (m, 4H), 3.67–3.56 (m, 1H), 2.78–2.67 (m, 4H), 1.70–1.47 (m, 4H), 1.27 (td, J = 7.0, 3.5 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ = 142.8 (s), 139.9 (s), 128.7 (s), 128.5 (s), 128.4 (s), 127.3 (s), 127.1 (s), 126.1 (s), 110.0 (s), 63.3 (d, J = 17.8 Hz), 61.4 (d, J = 6.4 Hz), 48.6 (s), 36.4 (s), 30.4 (d, J = 4.3 Hz), 23.0 (s), 21.6 (s), 16.4 (dd, J = 6.0, 1.6 Hz). HRMS (ESI+) Calcd. for C21H30NO3P [M]: 376.2042, Found: 376.2041.
Dimenthyl 3-butylamino-3-phenyl propylphosphonate, 11da
The pure 11da was obtained as a yellow oil. Yield 77% (49 mg). Rf = 0.78 (petroleum ether:ethyl acetate = 1:3). 31P-NMR (162 MHz, CDCl3) δ = 30.07 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.32 (d, J = 7.0 Hz, 2H), 7.30–7.24 (m, 3H), 4.21–4.04 (m, 2H), 3.62 (t, J = 6.7 Hz, 1H), 2.46 (d, J = 7.6 Hz, 2H), 2.21 (d, J = 11.4 Hz, 2H), 2.16–2.07 (m, 1H), 1.97 (ddd, J = 26.2, 16.9, 8.2 Hz, 3H), 1.77–1.51 (m, 10H), 1.50–1.37 (m, 5H), 1.37–1.22 (m, 5H), 1.15–0.96 (m, 2H), 0.91 (dd, J = 5.2, 2.0 Hz, 11H), 0.88 (dd, J = 6.9, 3.7 Hz, 3H), 0.81–0.78 (m, 3H), 0.74 (d, J = 6.9, 1H), 0.69 (d, J = 6.9, 1H). 13C-NMR (101 MHz, CDCl3) δ = 143.3 (s), 128.4 (s), 127.2 (dd, J = 8.8, 5.8 Hz), 77.3 (s), 77.1–76.5 (m), 63.7 (d, J = 18.0 Hz), 48.6 (d, J = 6.5 Hz), 47.4 (s), 43.7 (d, J = 7.9 Hz), 43.0 (d, J = 15.4 Hz), 34.1 (s), 32.4 (s), 31.6–31.2 (m), 30.9 (s), 25.6–25.1 (m), 23.7 (s), 22.7 (t, J = 5.6 Hz), 21.9 (s), 21.03 (s), 20.0 (s), 15.7 (dd, J = 26.2, 7.1 Hz), 13.9 (s). HRMS (ESI+) Calcd. for C33H58NO3P [M + H+]: 548.4233, Found: 548.4224.
Dimenthyl 3-phenylethylamino-3-phenyl propylphosphonate, 11db
The pure 11db was obtained as a yellow oil. Yield 77% (49 mg). Rf = 0.66 (petroleum ether:ethyl acetate = 1:3). 31P-NMR (162 MHz, CDCl3) δ = 29.96 (s, 50%), 29.94 (s, 50%). 1H-NMR (400 MHz, CDCl3) δ = 7.29 (d, J = 7.3 Hz, 3H), 7.23 (d, J = 5.6 Hz, 2H), 7.20 (d, J = 8.0 Hz, 3H), 7.14 (d, J = 8.0 Hz, 2H), 4.17–4.03 (m, 2H), 3.61 (dd, J = 13.1, 5.4 Hz, 1H), 2.73 (t, J = 6.3 Hz, 4H), 2.12 (d, J = 34.5 Hz, 3H), 1.94 (dd, J = 13.8, 7.0 Hz, 2H), 1.69–1.35 (m, 10H), 1.25 (s, 2H), 1.14–1.02 (m, 2H), 0.95 (dd, J = 21.1, 9.0 Hz, 3H), 0.91–0.84 (m, 12H), 0.78 (t, J = 8.4 Hz, 4H), 0.70 (d, J = 6.9 Hz, 1H), 0.65 (d, J = 6.9 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ = 142.9 (s), 140.0 (s), 128.7–128.3 (m), 127.2 (d, J = 6.7 Hz), 126.1 (s), 77.3 (s), 77.0 (s), 76.7 (s), 63.5 (d, J = 17.8 Hz), 49.1–48.1 (m), 43.7 (d, J = 8.2 Hz), 43.0 (d, J = 15.8 Hz), 36.4 (s), 34.1 (s), 32.0–31.3 (m), 30.9 (s), 29.7 (s), 25.3 (t, J = 18.1 Hz), 23.6 (d, J = 11.0 Hz), 22.7 (t, J = 5.5 Hz), 22.0 (d, J = 3.5 Hz), 21.1 (s), 15.8 (d, J = 5.7 Hz), 15.5 (d, J = 7.7 Hz). HRMS (ESI+) Calcd. for C37H58NO3P [M + H+]: 596.4233, Found: 596.4224.
Dimenthyl 3-phenyl-3-pyrrolidin-1-yl propylphosphonate, 11dc
The pure 11dc was obtained as a yellow oil. Yield 77% (49 mg). Rf = 0.80 (petroleum ether:ethyl acetate = 1:3). 31P-NMR (162 MHz, CDCl3) δ = 30.23 (s, 61%), 30.19 (s, 39%). 1H-NMR (400 MHz, CDCl3) δ = 7.26 (t, J = 13.1 Hz, 5H), 4.15–4.02 (m, 2H), 3.08 (d, J = 5.7 Hz, 1H), 2.53 (s, 1H), 2.36 (s, 2H), 2.15 (s, 3H), 1.99 (s, 2H), 1.67 (dd, J = 32.6, 15.7 Hz, 11H), 1.41 (s, 4H), 1.25 (s, 4H), 1.07 (dd, J = 22.6, 10.9 Hz, 2H), 0.87 (dd, J = 11.9, 6.6 Hz, 12H), 0.79–0.75 (m, 3H), 0.71 (d, J = 7.0 Hz, 2H), 0.63 (d, J = 6.9 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ = 142.1 (s), 128.3–127.9 (m), 127.1 (s), 77.3 (s), 77.0 (s), 76.7 (s), 71.1 (s), 52.7 (s), 48.5 (s), 43.8 (s), 43.0 (d, J = 18.4 Hz), 34.1 (s), 31.5 (d, J = 4.6 Hz), 29.7 (s), 29.0 (s), 25.7–24.8 (m), 23.3 (s), 22.7 (s), 21.9 (s), 21.1 (s), 15.6 (dd, J = 25.7, 12.3 Hz). HRMS (ESI+) Calcd. for C33H56NO3P [M + H+]: 546.4076, Found: 546.4069.
Dimenthyl-3-methyl phenylamino-3-phenyl propylphosphonate, 11dd
The pure 11dd was obtained as a yellow oil. Yield 62% (35 mg). Rf = 0.74 (petroleum ether:ethyl acetate = 1:3). 31P-NMR (162 MHz, CDCl3) δ = 30.28 (s, 50%), 30.26 (s, 50%). 1H-NMR (400 MHz, CDCl3) δ = 7.36–7.28 (m, 3H), 7.23 (dd, J = 18.7, 7.4 Hz, 6H), 7.13 (d, J = 7.4 Hz, 1H), 4.10 (s, 2H), 3.68 (dd, J = 29.0, 6.3 Hz, 1H), 3.48–3.24 (m, 1H), 2.13 (d, J = 41.4 Hz, 2H), 2.00 (s, 1H), 1.89–1.79 (m, 1H), 1.63 (d, J = 10.8 Hz, 8H), 1.40 (s, 2H), 1.32 (d, J = 6.3 Hz, 1H), 1.24 (d, J = 7.0 Hz, 4H), 1.10–0.97 (m, 2H), 0.87 (d, J = 6.3 Hz, 12H), 0.82 (d, J = 11.6 Hz, 2H), 0.77 (t, J = 7.1 Hz, 3H), 0.70 (d, J = 6.5 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 128.4 (d, J = 9.3 Hz), 127.4–126.6 (m), 126.5 (s), 77.3 (s), 77.1–76.5 (m), 60.2 (d, J = 18.3 Hz), 54.6 (s), 48.5 (s), 43.7 (s), 43.0 (d, J = 9.3 Hz), 34.1 (s), 31.5 (d, J = 10.2 Hz), 30.2 (s), 29.7 (s), 25.4 (dd, J = 28.7, 10.2 Hz), 23.0–22.6 (m), 22.3 (d, J = 12.6 Hz), 22.0 (d, J = 2.9 Hz), 21.1 (s), 15.8 (d, J = 7.0 Hz), 15.6 (s), 14.0 (s). HRMS (ESI+) Calcd. for C36H56NO3P [M + H+]: 582.4076, Found: 582.4080.
Diethyl 3-butylamino-3-p-tolyl propylphosphonate, 11ea
The pure 11ea was obtained as a yellow oil. Yield 83% (47 mg). Rf = 0.70 (dichloromethane/methanol = 20:1). 31P-NMR (162 MHz, CDCl3) δ = 32.57 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.12 (d, J = 8.6 Hz, 4H), 4.09–3.97 (m, 4H), 3.56 (t, J = 6.7 Hz, 1H), 2.51–2.38 (m, 2H), 2.33 (s, 3H), 2.04–1.79 (m, 2H), 1.64 (dddd, J = 26.4, 19.3, 15.4, 4.8 Hz, 3H), 1.48–1.36 (m, 2H), 1.34–1.21 (m, 8H), 0.86 (t, J = 7.2 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 140.1 (s), 136.7 (s), 129.1 (s), 127.0 (s), 63.2 (d, J = 17.9 Hz), 61.4 (d, J = 2.2 Hz), 61.3 (d, J=2.1 Hz), 53.4 (s), 47.3 (s), 32.4 (s), 30.4 (d, J = 4.3 Hz), 29.6 (s), 23.1 (s), 21.7 (s), 21.0 (s), 20.4 (s), 16.4 (d, J = 6.0 Hz), 13.9 (s). HRMS (ESI+) Calcd. for C18H32NO3P [M]: 342.2198, Found: 342.2234.
Diethyl 3-phenethylamino-3-p-tolyl propylphosphonate, 11eb
The pure 11eb was obtained as a yellow oil. Yield 77% (49 mg). Rf = 0.65 (dichloromethane/methanol = 20:1). 31P-NMR (162 MHz, CDCl3) δ = 32.46 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.25 (dd, J = 8.4, 5.8 Hz, 2H), 7.22–7.05 (m, 7H), 4.26–3.89 (m, 4H), 3.57 (t, J = 6.7 Hz, 1H), 2.95–2.55 (m, 4H), 2.32 (s, 3H), 2.29–1.81 (m, 2H), 1.74–1.47 (m, 3H), 1.26 (td, J = 7.0, 3.1 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ = 139.9 (d, J = 21.9 Hz), 136.8 (s), 129.1 (s), 128.6 (s), 128.3 (s), 127.0 (s), 126.0 (s), 63.0 (d, J = 17.9 Hz), 61.4 (d, J = 1.1 Hz), 61.3 (d, J = 1.1 Hz), 53.4 (s), 48.6 (s), 36.4 (s), 30.4 (d, J = 4.3 Hz), 23.0 (s), 21.6 (s), 21.0 (s), 16.4 (d, J = 0.7 Hz). HRMS (ESI+) Calcd. for C22H32NO3P [M]: 391.2276, Found: 391.2232.
Diphenyl 3-butylamino-3-phenylpropyl phosphine oxide, 11fa
The pure 11fa was obtained as a yellow oil. Yield 80% (44 mg). Rf = 0.50 (dichloromethane/methanol = 20:1). 31P-NMR (162 MHz, CDCl3) δ = 33.05 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.62 (dd, J = 10.8, 7.9 Hz, 4H), 7.52–7.45 (m, 2H), 7.41 (qd, J = 7.1, 3.5 Hz, 4H), 7.31 (t, J = 7.2 Hz, 2H), 7.28–7.24 (m, 1H), 7.21 (t, J = 6.4 Hz, 2H), 3.66–3.58 (m, 1H), 2.45–2.32 (m, 2H), 2.30–2.18 (m, 1H), 2.18–1.96 (m, 3H), 1.91 (ddd, J = 20.6, 12.6, 6.4 Hz, 1H), 1.44–1.32 (m, 2H), 1.31–1.18 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 143.0 (s), 132.9 (dd, J = 98.4, 46.3 Hz), 131.6 (s), 130.8 (s), 130.7 (d, J = 2.2 Hz), 130.6 (s), 128.6 (s), 128.5 (d, J = 4.3 Hz), 127.2 (s), 63.6 (d, J = 14.6 Hz), 47.3 (s), 32.3 (s), 29.3 (d, J = 3.0 Hz), 26.0 (d, J = 72.6 Hz), 20.4 (s), 14.0 (s). HRMS (ESI+) Calcd. for C25H30NOP [M]: 392.2143, Found: 392.2147.
To a solution of 1a (1.02 g, 2.89 mmol), palladium acetate (32.5 mg, 0.145 mmol, 5 mol %), and triphenyl phosphine (0.152 g, 0.580 mmol, 20 mol %) in toluene (2 mL), 3a (0.63 mL, 6.36 mmol) was added. The mixture was heated at 120 °C for 24 h and monitored with TLC. After the reaction was completed, the mixture was cooled to room temperature. Ethanol (5 mL) and sodium borohydride (0.44 g, 11.6 mmol) were added. The mixture was stirred at room temperature for 14 h. After a saturated solution of ammonium chloride (10 mL) was added, the mixture was extracted with dichloromethane (3 × 15 mL) and washed with water (3 × 10 mL). The crude product was obtained as a red oil in a 66% yield (estimated by a 31P-NMR spectrum). The residue was dissolved in ethanol (5 mL), acidified to pH = 2 with diluted hydrochloric acid (ca. 7%, 6 mL), and then the ethanol was removed in vacuo. The residue was washed with ether (5 × 5 mL), and the aqueous phase was neutralized with saturated sodium bicarbonate solution (10 mL) until pH = 9. The mixture was extracted with dichloromethane (3 × 15 mL). After drying and removal of the solvent, 11fa was obtained as a yellow oil. Yield 59% (0.67 g). 11fa gave similar spectrum data to those above.
Diphenyl 3-phenethylamino-3-phenyl propylphosphine oxide, 11fb
The pure 11fb was obtained as a yellow oil. Yield 82% (51 mg). Rf = 0.53 (dichloromethane/methanol = 15:1). 31P-NMR δ = 33.02 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.60 (dd, J = 10.0, 8.5 Hz, 4H), 7.54–7.45 (m, 2H), 7.41 (t, J = 7.2 Hz, 4H), 7.33–7.21 (m, 5H), 7.20–7.08 (m, 5H), 3.63 (t, J = 6.4 Hz, 1H), 2.81–2.60 (m, 4H), 2.28–2.15 (m, 1H), 2.14–2.04 (m, 1H), 2.04–1.94 (m, 1H), 1.94–1.81 (m, 1H). 13C-NMR (101 MHz, CDCl3) δ = 142.7 (s), 139.9 (s), 133.3 (d, J = 43.1 Hz), 132.4 (d, J = 43.0 Hz), 131.6 (s), 130.8 (s), 130.7 (s), 130.6 (s), 128.7 (d, J = 2.7 Hz), 128.5 (d, J = 2.1 Hz), 128.4 (s), 127.3 (d, J = 9.0 Hz), 126.1 (s), 63.4 (d, J = 14.6 Hz), 48.5 (s), 36.3 (s), 29.3 (d, J = 3.3 Hz), 26.0 (d, J = 72.6 Hz). HRMS (ESI+) Calcd. for C29H30NOP [M]: 440.2143, Found: 440.2142.
(SP)-Menthyl-3-butylamino-3-phenylpropyl phenylphosphine oxide, 11gaA/11gaB
Compound 11gaA/11gaB was obtained as a yellow oil. Yield 85% (56 mg). Rf = 0.46 (dichloromethane/methanol = 15:1). 31P-NMR (162 MHz, CDCl3) δ = 43.39 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.58–7.51 (m, 2H), 7.48–7.39 (m, 3H), 7.29 (dd, J = 13.9, 6.5 Hz, 3H), 7.16 (d, J = 6.7 Hz, 2H), 3.59–3.51 (m, 1H), 2.43–2.27 (m, 2H), 2.13–1.89 (m, 5H), 1.88–1.77 (m, 1H), 1.67 (d, J = 10.8 Hz, 3H), 1.54–1.42 (m, 2H), 1.39–1.19 (m, 5H), 1.17–1.03 (m, 2H), 0.95 (dd, J = 23.2, 12.7 Hz, 2H), 0.86 (d, J = 6.6 Hz, 4H), 0.82 (d, J = 7.2 Hz, 2H), 0.78 (d, J = 6.8 Hz, 2H), 0.29 (d, J = 6.7 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ = 143.2 (s), 134.2 (d, J = 87.4 Hz), 130.8 (d, J = 2.6 Hz), 130.5 (d, J = 8.4 Hz), 128.4–128.2 (m), 127.4 (s), 127.1 (s), 63.9 (d, J = 13.6 Hz), 53.4 (s), 47.3 (s), 43.1 (s), 41.1 (s), 40.4 (s), 35.1 (s), 34.2 (s), 33.1 (d, J = 14.7 Hz), 32.2 (s), 29.1 (s), 28.1 (s), 24.6 (s), 24.3 (d, J = 2.3 Hz), 23.9 (s), 22.5 (s), 21.5 (s), 20.3 (s), 15.1 (s), 13.9 (s). HRMS (ESI+) Calcd. for C29H44NOP [M]: 454.3239, Found: 454.3248.
(SP)-Menthyl 3-phenethylamino-3-phenylpropyl phenylphosphine oxide, 11gbA/11gbB, 11gbA’/11gbB’
Compound 11gbA/11gbB, 11gbA’/11gbB’ was obtained as a yellow oil. Yield 87% (52 mg). Rf = 0.41 (dichloromethane/methanol = 20:1). 31P-NMR (162 MHz, CDCl3) δ = 45.80 (s, 17%), 45.34 (s, 23%), 43.44 (s, 31%), 43.28 (s, 29%). 1H-NMR (400 MHz, CDCl3) δ = 7.62–7.48 (m, 3H), 7.43 (dd, J = 7.4, 5.8 Hz, 2H), 7.35–7.22 (m, 5H), 7.21–7.07 (m, 5H), 3.65–3.53 (m, 1H), 2.79–2.46 (m, 4H), 2.10–1.81 (m, 4H), 1.78–1.41 (m, 7H), 1.26 (s, 2H), 1.16–0.94 (m, 2H), 0.87 (dd, J = 8.6, 6.5 Hz, 2H), 0.78 (dd, J = 9.3, 4.7 Hz, 4H), 0.75–0.70 (m, 1H), 0.31 (dd, J = 10.9, 6.8 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ = 143.0 (dd, J = 8.5, 6.1 Hz), 140.2–139.9 (m), 134.8–134.4 (m), 133.8 (d, J = 20.0 Hz), 131.2–130.9 (m), 130.8 (d, J = 3.3 Hz), 130.5 (dd, J = 8.5, 2.4 Hz), 128.6 (d, J = 4.4 Hz), 128.4 (d, J = 1.3 Hz), 128.3 (s), 128.2 (s), 128.0 (d, J = 4.2 Hz), 127.4 (s), 127.2–127.1 (m), 127.0 (d, J = 3.8 Hz), 126.0 (d, J = 1.5 Hz), 63.7–63.1 (m), 48.5 (dd, J = 13.3, 7.8 Hz), 43.5–43.1 (m), 41.6–40.4 (m), 37.0–36.8 (m), 36.2 (t, J = 11.7 Hz), 35.2 (dd, J = 15.5, 2.3 Hz), 34.2 (s), 33.2 (ddd, J = 13.3, 7.4, 4.1 Hz), 29.7 (s), 29.6–29.0 (m), 28.3 (dd, J = 22.1, 5.1 Hz), 25.3 (d, J = 4.4 Hz), 24.8–24.5 (m), 24.1 (d, J = 66.2 Hz), 22.5 (d, J = 3.3 Hz), 21.5–21.2 (m), 15.7–15.0 (m). HRMS (ESI+) Calcd. for C33H44NOP [M]: 502.3239, Found: 502.3238.
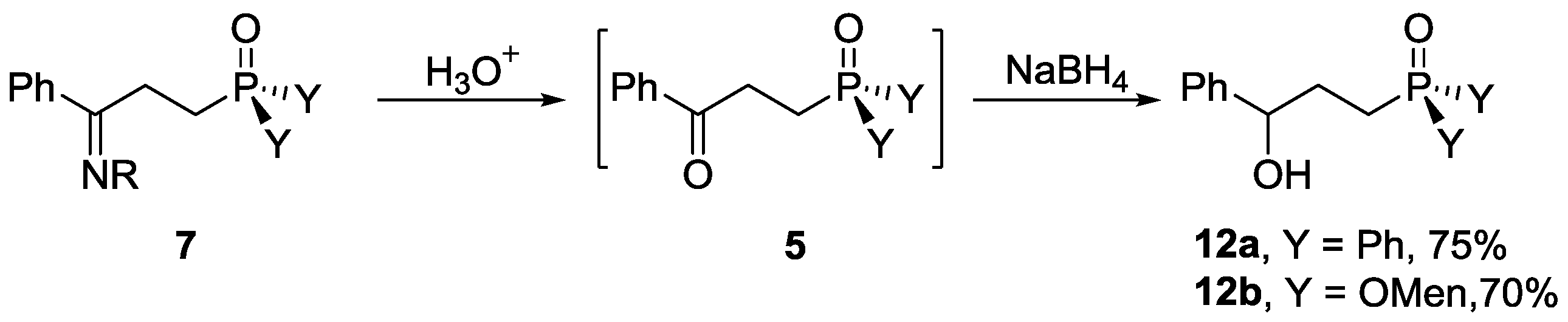
3.2.7. Preparation of γ-Hydroxy Phosphorous Derivatives 12
Diphenyl 3-hydroxy-3-phenylprop-1-en-1-ylphosphine oxide, 12a
To a solution of 1f (50 mg, 0.142 mmol), palladium acetate (1.6 mg, 0.007 mmol), and triphenyl phosphine (7.4 mg, 0.284 mmol) in toluene (0.5 mL), 3a (31 µL, 0.312 mmol) was added. The mixture was heated at 120 °C for 24 h and monitored with TLC, and then cooled to room temperature. Water (3 µL, 0.142 mmol) and sodium borohydride (10.8 mg, 0.284 mmol) were added. The mixture was stirred at room temperature for 13 h. After a saturated solution of ammonium chloride (3 mL) was added, the mixture was extracted with dichloromethane (3 × 5 mL) and washed with water (3 × 3 mL). The combined organic layer was dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was purified with preparative TLC. Rf = 0.63 (silica gel, petroleum ether/ethyl acetate = 1/5 as eluent). The pure 12a was obtained as a white solid. Yield 70% (33 mg), m.p. 142.2–144.6 °C. 31P-NMR (162 MHz, CDCl3) δ = 34.53 (s). 1H-NMR (400 MHz, CDCl3) δ = 7.73–7.62 (m, 4H), 7.54–7.47 (m, 2H), 7.44 (dd, J = 6.9, 3.3 Hz, 4H), 7.29 (dd, J = 8.1, 4.8 Hz, 4H), 7.25–7.19 (m, 1H), 4.81 (s, 1H), 4.19 (s, 1 H), 2.48–2.27 (m, 2H), 2.16–1.93 (m, 2H). 13C-NMR (101 MHz, CDCl3) δ = 139.3 (s), 128.1 (d, J = 18.6 Hz), 127.1 (s), 126.1 (dd, J = 9.4, 4.4 Hz), 123.9 (dd, J = 11.7, 1.6 Hz), 123.6 (s), 122.6 (s), 121.0 (s), 72.6 (s), 72.3 (s), 71.9 (s), 68.8 (d, J = 10.0 Hz), 26.6 (s), 21.5 (s), 20.8 (s). HRMS (ESI+) Calcd. for C21H21O2P [M + H+]: 337.1357, Found: 337.1362.
Dimenthyl 3-hydroxy-3-phenylprop-1-en-1-ylphosphonate, 12b
Compound 12b was prepared according to a similar procedure to that for the preparation of 12a, which was purified with preparative TLC with Rf = 0.68 (silica gel, petroleum ether/ethyl acetate = 1/3 as eluent), and 12b was obtained as a yellow oil. Yield 55% (26 mg). 31P-NMR (162 MHz, CDCl3) δ = 30.88 (s, 50%), 30.86 (s, 50%). 1H-NMR (400 MHz, CDCl3) δ = 7.76–7.48 (m, 1H), 7.35 (d, J = 4.2 Hz, 3H), 7.27 (s, 1H), 4.80 (s, 1H), 4.24–4.09 (m, 2H), 3.04 (d, J = 12.4 Hz, 1H), 2.22 (s, 2H), 2.17–2.00 (m, 4H), 1.83–1.70 (m, 3H), 1.68–1.58 (m, 7H), 1.44 (d, J = 7.6 Hz, 3H), 1.25 (s, 6H), 1.17–1.06 (m, 2H), 0.90 (d, J = 6.2 Hz, 9H), 0.81–0.77 (m, 3H), 0.76–0.73 (m, 1H). 13C-NMR (101 MHz, CDCl3) δ = 144.0 (s), 128.4 (s), 127.5 (d, J = 2.2 Hz), 125.8 (s), 125.7 (s), 77.2 (d, J = 4.4 Hz), 73.9 (dd, J = 13.2, 6.1 Hz), 48.7–48.6 (m), 48.6–48.5 (m), 43.7 (d, J = 1.8 Hz), 43.1 (d, J = 6.0 Hz), 41.3 (s), 34.6 (d, J = 15.5 Hz), 34.1 (s), 32.2 (dd, J = 5.0, 2.4 Hz), 31.6 (d, J = 1.7 Hz), 31.5 (s), 29.0 (s), 26.9 (s), 25.5 (d, J = 4.9 Hz), 25.3 (s), 22.8 (d, J = 3.3 Hz), 22.6 (d, J = 3.8 Hz), 21.9 (d, J = 2.9 Hz), 21.0 (d, J = 1.7 Hz), 15.8 (d, J = 2.3 Hz), 15.6 (d, J = 2.8 Hz), 14.1 (s), 11.4 (s). HRMS (ESI+) Calcd. for C29H49O4P [M + H+]: 493.3447, Found: 493.3447.
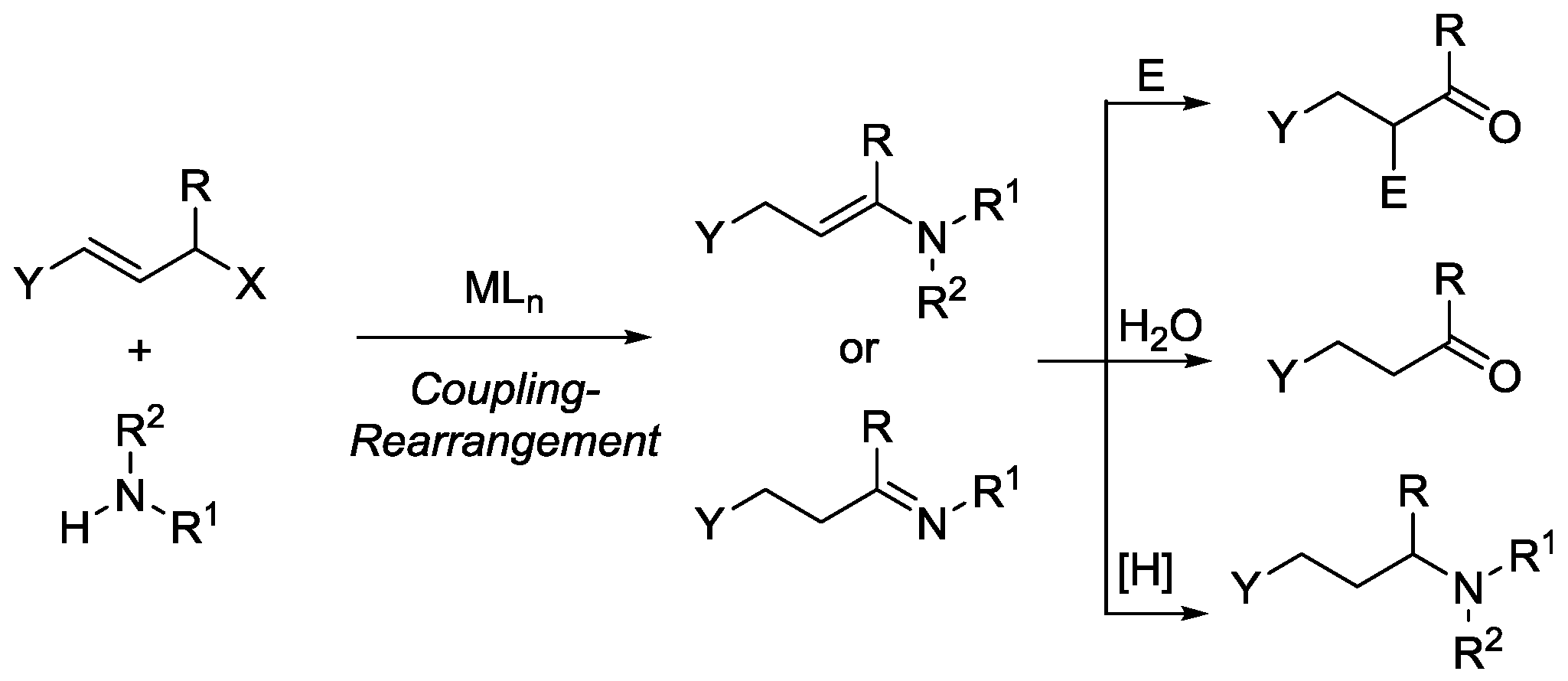
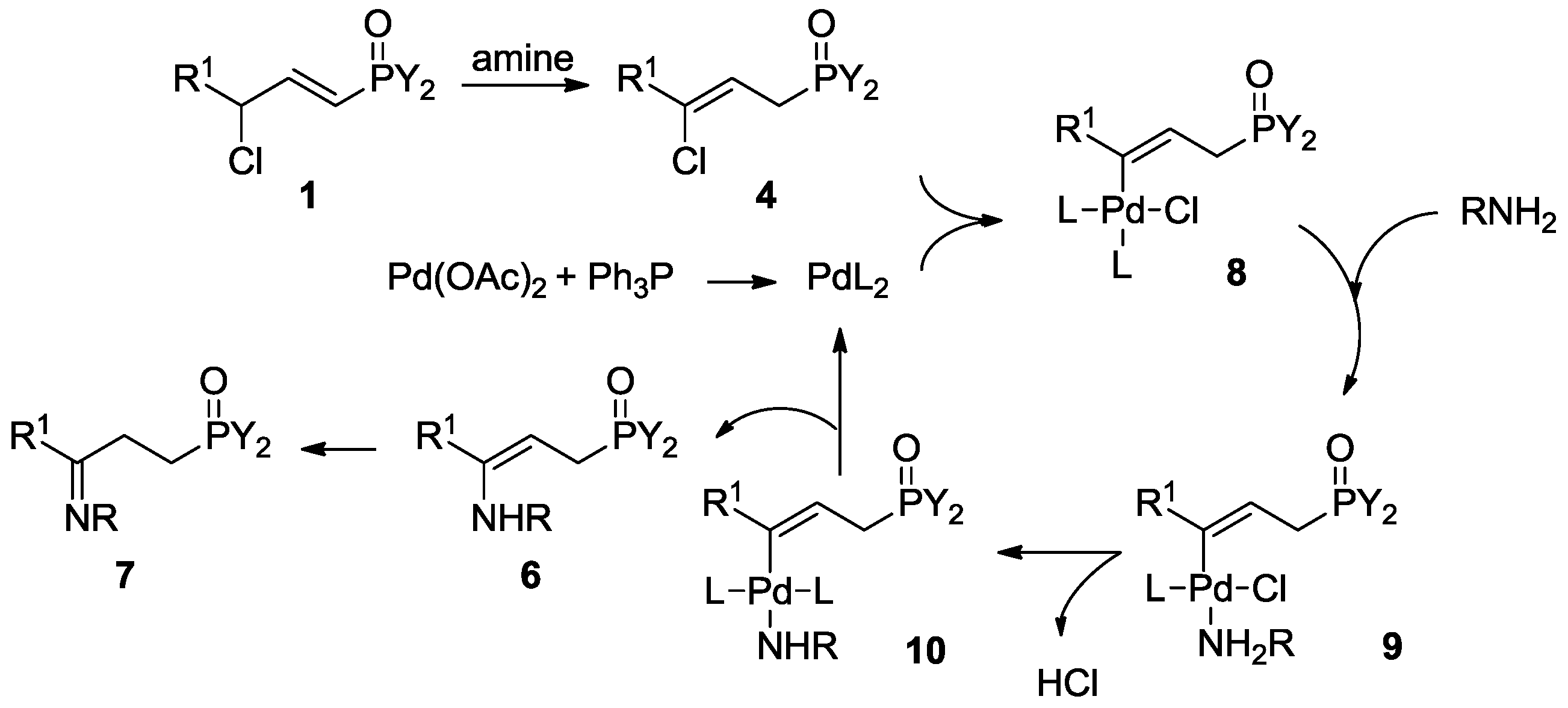
3.2.8. Reaction of 1 with Secondary Amine to Form Functional Phosphorus Derivatives
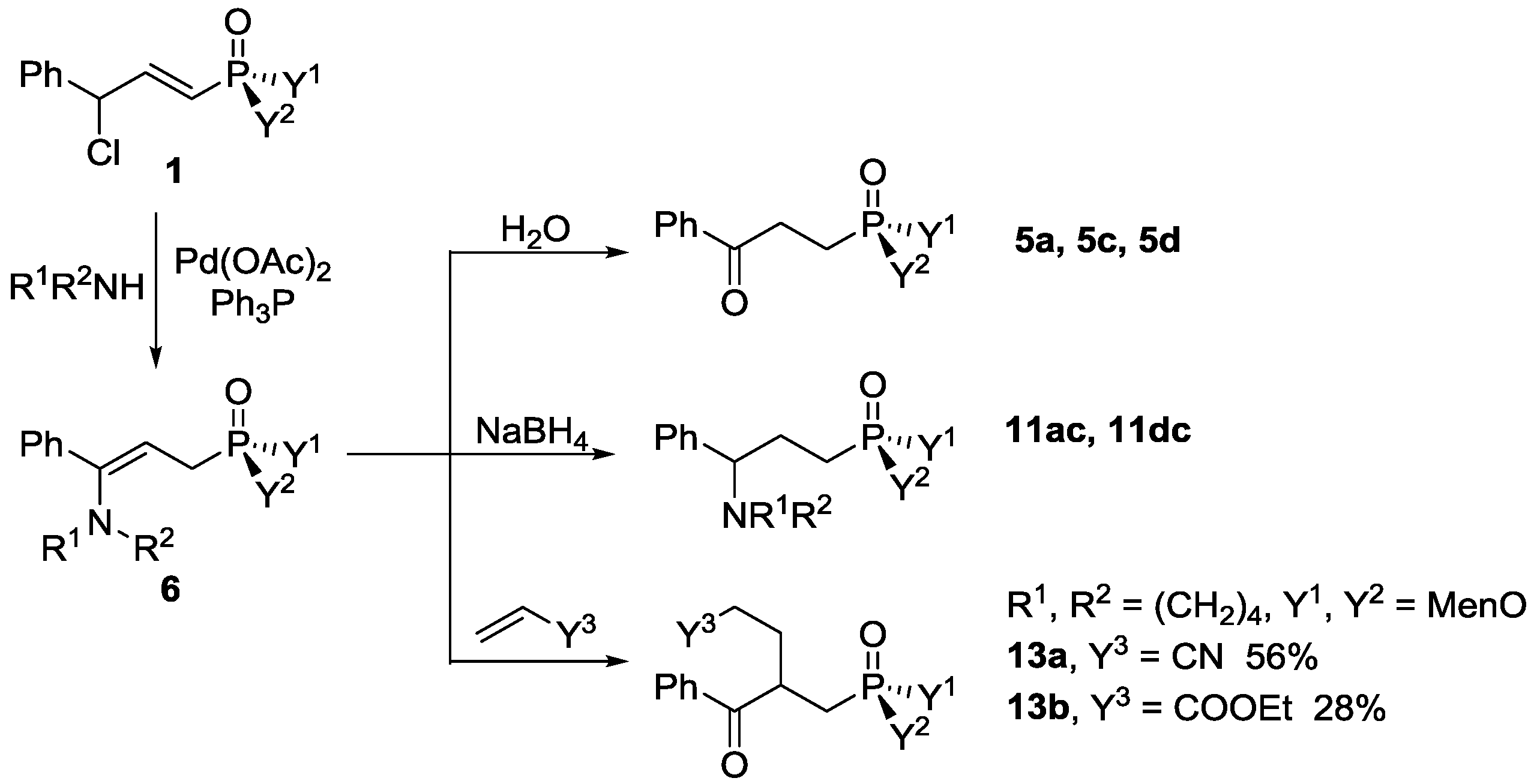
Dimenthyl 2-benzoyl-4-cyanobutylphosphonate, 13a
To a solution of 1d (50 mg, 0.098 mmol), palladium acetate (1.1 mg, 0.005 mmol, 5 mol %), and triphenyl phosphine (5.1 mg, 0.020 mmol, 20 mol %) in toluene (0.5 mL), pyrrolidine (18 µL, 0.216 mmol) was added. The mixture was heated at 120 °C for 24 h. The removal of toluene and excess pyrrolidine in vacuo afforded the crude enamine 6, which was used in situ. The crude 6 was dissolved in 0.5 mL of dry DMF, acrylonitrile (19 µL, 0.294 mmol) was added, and then the mixture was heated at 120 °C for 36 h. After cooling to room temperature, water (2 mL) was added and the mixture was extracted with dichloromethane (3 × 5 mL). The combined organic layer was washed with water, dried over anhydrous magnesium sulfate, and concentrated in vacuo to give the crude product as a brown oil, which was purified with preparative TLC with Rf = 0.34 (silica gel, dichloromethane/ethyl acetate= 5:1), to give pure 13a as a yellow oil. Yield 56% (28 mg). 31P-NMR (162 MHz, CDCl3) δ = 25.98 (s, 50%), 25.93 (s, 50%). 1H-NMR (400 MHz, CDCl3) δ = 8.02 (t, J = 7.9 Hz, 2H), 7.59 (dd, J = 7.3, 5.4 Hz, 1H), 7.48 (t, J = 7.5 Hz, 2H), 4.20–4.09 (m, 2H), 3.99 (dd, J = 12.4, 6.2 Hz, 1H), 2.44–2.09 (m, 6H), 2.07–1.68 (m, 4H), 1.61 (d, J = 9.0 Hz, 4H), 1.38 (d, J = 3.1 Hz, 2H), 1.15 (tdd, J = 22.6, 20.4, 11.6 Hz, 3H), 1.00–0.92 (m, 2H), 0.86 (dt, J = 11.4, 6.4 Hz, 11H), 0.79 (d, J = 7.0 Hz, 2H), 0.75 (dd, J = 10.6, 4.4 Hz, 7H). 13C-NMR (101 MHz, CDCl3) δ = 200.5 (dd, J = 19.8, 10.3 Hz), 135.8 (d, J = 5.8 Hz), 133.7 (d, J = 4.3 Hz), 128.8 (dd, J = 22.1, 5.6 Hz), 118.8 (s), 78.1 (d, J = 7.4 Hz), 77.9–76.8 (m), 76.7 (s), 48.7–48.1 (m), 43.6 (d, J = 4.7 Hz), 42.9 (d, J = 13.5 Hz), 39.2 (dd, J = 15.9, 3.9 Hz), 34.0 (d, J = 3.3 Hz), 31.5 (dd, J = 10.7, 3.9 Hz), 30.6 (s), 29.1 (s), 28.3 (dd, J = 9.1, 3.9 Hz), 25.7 (d, J = 9.8 Hz), 25.4 (d, J = 6.3 Hz), 22.7 (t, J = 6.3 Hz), 21.9 (d, J = 3.0 Hz), 20.9 (d, J = 11.1 Hz), 15.9–15.4 (m), 14.8 (d, J = 13.2 Hz). IR (KBr) ν/cm−1: 2247, 1685, 1181, 755, 558. HRMS (ESI+) Calcd. for C32H50NO4P [M + Na+]: 566.3375, Found: 566.3373.
Ethyl 4-dimenthylphosphoryl methyl-5-oxo-5-phenylpentanoate, 13b
The enamine 6 was prepared similarly to 13a, which was reacted with ethyl acrylate (31 µL, 0.294 mmol) for 48 h at 120 °C. The crude product was purified with preparative TLC with Rf = 0.41 (silica gel, dichloromethane/ethyl acetate= 20:1) to give pure 13b as a yellow oil. Yield 28% (15 mg). 31P-NMR (162 MHz, CDCl3) δ = 27.98 (s, 50%), 27.85 (s, 50%). 1H-NMR (400 MHz, CDCl3) δ = 8.02 (t, J = 9.1 Hz, 2H), 7.55 (d, J = 6.3 Hz, 1H), 7.46 (t, J = 7.1 Hz, 2H), 4.14–4.04 (m, 4H), 3.96 (s, 1H), 2.44–2.17 (m, 4H), 2.11 (d, J = 7.1 Hz, 2H), 2.05–1.87 (m, 3H), 1.82 (d, J = 17.5 Hz, 1H), 1.72 (s, 1H), 1.59 (s, 4H), 1.36 (s, 2H), 1.19 (t, J = 7.1 Hz, 3H), 1.13–1.01 (m, 2H), 0.98–0.89 (m, 4H), 0.85 (t, J = 6.3 Hz, 8H), 0.80 (d, J = 6.5 Hz, 3H), 0.77 (d, J = 7.5 Hz, 4H), 0.74–0.70 (m, 4H). 13C-NMR (101 MHz, CDCl3) δ = 201.5 (dd, J = 9.3, 6.7 Hz), 172.6 (s), 136.6 (d, J = 5.3 Hz), 133.2 (d, J = 4.0 Hz), 128.6 (dd, J = 7.5, 4.2 Hz), 77.7 (d, J = 7.3 Hz), 77.1 (s), 76.8 (s), 60.4 (s), 48.5 (d, J = 6.7 Hz), 48.4 (d, J = 3.7 Hz), 48.3 (d, J = 7.0 Hz), 43.5 (d, J = 8.3 Hz), 42.8 (d, J = 15.3 Hz), 39.5 (d, J = 3.9 Hz), 39.2 (s), 34.0 (t, J = 3.7 Hz), 31.5 (d, J = 3.5 Hz), 31.4 (s), 31.3 (d, J = 3.0 Hz), 31.2 (s), 30.2 (d, J = 9.7 Hz), 28.8 (dd, J = 26.9, 12.9 Hz), 25.6 (s), 25.5 (s), 25.2 (d, J = 3.0 Hz), 22.7 (d, J = 3.4 Hz), 22.6 (d, J = 4.6 Hz), 21.9 (s), 21.0 (s), 20.9 (s), 15.7 (d, J = 5.0 Hz), 15.6 (s), 14.1 (s). HRMS (ESI+) Calcd. for C34H55NO6P [M + Na+]: 613.3634, Found: 613.3649.
3.2.9. Physical Characterization of the Crystals
Single-crystal X-ray diffraction data for two Schiff base copper complexes, complex 1 and complex 2, were conducted on a Bruker-AXS CCD diffractometer, which was equipped with graphite-monochromated Mo-Ka radiation (λ = 0.71073 Å), at 298 K. All absorption corrections were applied using a multi-scan technique. The structures were solved by the direct method and refined through the full-matrix least-squares method on F2 using the SHELXTL 97 crystallographic software package. The FT-IR spectra were recorded with KBr as pellets in the range 4000–400 cm−1 on a Nicolet 170 SXFT/IR spectrometer (Nicolet, Madison, WI, USA).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








