1. Introduction
Esterases and lipases catalyze the hydrolysis of esters under physiological conditions. However, their popularity as biocatalysts for the chemical industry is also based on their ability to catalyze transesterification reactions in anhydrous organic solvents. While lipases are activated at the interface of water-insoluble substrates like triglycerides, esterases tend to hydrolyze soluble esters. The availability of a large number of esterases and lipases is based on the presence of various high-throughput assays, which enable their discovery in metagenome libraries or optimization via enzyme engineering. These assays target the natural hydrolase activity and are based on e.g., halo formation in agar plates, detection of the carboxylic acid released, or formation of a chromophore or fluorophore [
1,
2]. Furthermore, the large number of characterized esterases and lipases allows for the reliable in silico prediction of novel hydrolases from the steadily growing number of sequenced organisms and metagenomes.
Until now only a few hydrolases, so-called acyltransferases, were shown to catalyze the transesterification of non-activated esters in an aqueous environment. Most of the characterized acyltransferases belong to the group related to
Pseudozyma antarctica lipase A, formerly known as
Candida antarctica lipase A (CAL-A) [
3]. As the hydrolysis reaction is thermodynamically favored, the accumulation of a transesterified product is only transient (
Scheme 1). Acyltransferases enable a higher increase in the reaction rate of the transesterification reaction compared to the hydrolysis reaction. Efficient acyltransferases catalyze transesterification more than 1000 times faster than hydrolysis [
4].
As acyltransferases allow for the direct transesterification reaction in aqueous media, they enable novel reaction concepts in biocatalysis. Transesterification steps can be included in enzyme cascades that demand the use of aqueous media due to the involvement of other enzymes [
5]. Furthermore, transesterifications with highly polar compounds e.g., sugars, that are not sufficiently soluble in the typical non-polar organic solvents, could be established. Toxic organic solvents like toluene, which are frequently used in lipase-catalyzed transesterification reactions, could be replaced by water.
The few currently known acyltransferases were identified from a pool of known hydrolases, as homologs thereof or discovered by accident [
6,
7,
8]. Direct screening for novel acyltransferases from large libraries, e.g., from metagenome libraries, is unfeasible as typical acyltransferase assays are laborious and time-consuming, and thereby limit throughput [
4,
9]. If a collection of pre-selected hydrolases is screened to reduce library size, the identified acyltransferases will by definition have substantial hydrolase activity. This fact complicates the discovery of very efficient acyltransferases, which should exhibit very low or no hydrolase activity, and also explains the limited number of acyltransferases known today. These drawbacks could be overcome by a high-throughput assay suitable for the identification of new acyltransferases from large libraries. Such an acyltransferase assay could enable the identification of acyltransferases with very low or even no detectable hydrolase activity, also leading to the discovery of acyltransferases that are not related to the CAL-A group.
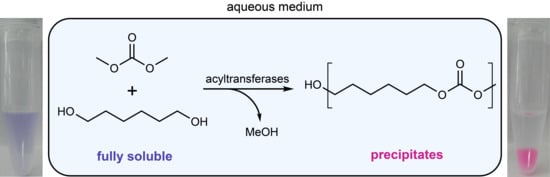
In this contribution, a new high-throughput screening method for acyltransferases, based on the enzymatic synthesis of insoluble poly/oligoesters from water-soluble monomers is envisioned. Acyltransferase activity would lead to product precipitation, which was demonstrated previously and is easily detectable [
10]. When the formation of insoluble oligomers is used as an indicator for acyltransferase activity, the complete solubility of a suitable starting monomer in an aqueous environment is required. The use of a monophasic system is also important as it was shown that typical lipases such as CAL-B can exhibit strong acyltransferase activity in aqueous environments when a second phase is present [
11,
12,
13]. There are several options for monomers to realize the envisioned oligomer formation assay. Besides small lactones for ring-opening-polymerization, also a polycondensation approach with diesters and diols seemed possible. However, a drawback in the formation of oligoesters is that an undesired hydrolysis event would split the growing oligoester chain and generate a carboxylic acid end which can potentially acidify the reaction medium. As this is the thermodynamically favored reaction, the chains will not be joined again. The formation of these ‘dead ends’ can be prevented by the use of a carbonate-based system where highly water-soluble dimethyl carbonate (DMC) is transesterified with a water-soluble diol like 1,6-hexanediol (HD) to form insoluble oligocarbonate particles (
Scheme 2). If a carbonate group within a growing oligomer is hydrolyzed, the carbonic acid monoester formed rapidly decomposes to CO
2 and the alcohol ends. The two terminal alcohol groups (OH ends) can subsequently be joined together by an additional acyl transfer step with another DMC molecule (
Scheme S1). Therefore, this system is more likely to also detect acyltransferases despite some degree of hydrolase activity. The lipase-catalyzed transesterification of DMC with diols has already been demonstrated under anhydrous conditions [
14].
Here, we describe the first high-throughput assay for acyltransferases that is based on the formation of insoluble oligocarbonate particles from a monophasic aqueous system. The applicability is demonstrated by the first identification of a novel acyltransferase capable of forming oligocarbonates in water.
3. Discussion
While previously described acyltransferase assays demand time-consuming gas chromatography (GC) measurements to detect the carboxylic acid esters formed [
22] or depend on the use of an additional enzyme reporter system [
9], the herein described screening method can be applied in microtiter plates and active enzymes can be identified by the naked eye or by absorbance and fluorescence measurements. This new concept enables the direct detection of acyltransferase activity from larger libraries. The assay is based on the formation of oligocarbonates which precipitate from an aqueous solution and can also be easily adapted to fluorescence readout by staining of the oligocarbonates with Nile Red. The reaction conditions were optimized with the newly identified acyltransferase Est8 as a model enzyme. This enzyme was previously only known for its esterase activity [
16,
23] which is in agreement with the observed disappearance of the transesterification product after longer incubation times and the formation of CO
2 in these reactions. As the presented assay allows continuous measurement of product formation, in contrast to previously reported end-point measurements, e.g. by GC, the chance of missing the potentially short time span of transient product accumulation is greatly reduced. Finally, it was demonstrated that the assay is also applicable to other acyltransferases.
While previously described acyltransferase assays utilize carboxylic acid esters [
4,
19], this study presents the first example of an aqueous acyltransferase reaction involving a carbonate moiety. Besides extending the scope of acyltransferase-generated products, the use of carbonates has advantages for screening approaches. An undesired hydrolysis step during the oligocarbonate synthesis results in the formation of CO
2, which easily escapes the reaction, instead of forming a carboxylic acid, which would strongly acidify the medium, potentially leading to enzyme inactivation. Another advantage is the possibility to again combine two OH ends generated by an unwanted hydrolysis event within the carbonate chain (
Scheme S1). These characteristics of the oligocarbonate-based assay allow for the identification of acyltransferases spanning a range of residual hydrolase activity. This is desired for an initial screening where only a few hits are expected and identified acyltransferases will be analyzed in detail afterwards.
As the new assay allows for the direct screening of large libraries for acyltransferase activity, it has the potential to identify novel enzymes e.g., from metagenomes. Since the enzymes identified from metagenome libraries would not have been previously identified by hydrolase activity assays, it might be possible to identify very efficient acyltransferases with very low hydrolase activity.
It has been shown that our new assay is also applicable to other acyltransferases as MsAcT also led to oligocarbonate formation. Interestingly, none of the CAL-A-related enzymes were active (as judged by precipitate formation) under the conditions tested. The reason for this could be their typical activity on large hydrophobic substrates such as fatty acid esters or triglycerides [
4,
9]. As these substrates are poorly soluble in aqueous media, they form a second organic phase that is known to strongly promote acyltransferase activity [
11,
13]. This potential activating phase was intentionally avoided in our screening, perhaps explaining the lack of activity using the CAL-A-related enzymes. Furthermore, in our assay the comparably small acyl donor DMC is transesterified with the larger alcohol HD, which is different from the typical reaction system for CAL-A-related acyltransferases where larger acyl donors (fatty acid esters) are combined with smaller alcohols (like methanol or ethanol) [
4]. On this background it is not surprising that for MsAcT, which was previously demonstrated to utilize smaller acyl donors like ethyl acetate [
19], activity could indeed be detected in our assay.
The enzymatic formation of oligocarbonates in aqueous environments could also have broader applications in other research areas. As the oligomers precipitate, they are easily separated from the bulk aqueous phase. These oligomers represent a prime starting material for further water-free, enzymatic polymerization which is described in the literature [
18]. This pre-polymerization will remove most of the co-product methanol that would reduce the molecular weight of the following polymerization step. This could replace the typically used two-step polymerization where the high-boiling oligomers need to be produced in an initial step before the final polymerization under lower pressure can be performed.
4. Materials and Methods
4.1. Chemicals
Extra dry dimethyl carbonate (>99%) was obtained from Acros (Schwerte, Germany), 1,6-hexanediol (99%) was bought from Aldrich (Darmstadt, Germany) and Nile Red (pure) was bought from Roth (Karlsruhe, Germany). Solvents used were HPLC grade. CAL-A and CAL-B lyophilizate were obtained from c-LEcta (Leipzig, Germany).
4.2. Acyltransferase Assay
For the initial screening, the enzyme preparations were dissolved at a concentration of around 10 g L−1 in 50 mM NaPi, pH 7.4. Insoluble particles were removed by centrifugation at 12,000 g. Fifty microliters of the enzyme solutions were added to a 200 µL substrate solution which consisted of 10% (v/v) DMC and 10% (w/v) HD in the same buffer. The plates were sealed with a transparent plastic film and incubated at room temperature. Controls either without DMC or without HD were conducted.
Later, the formation of insoluble oligocarbonate was monitored by absorbance measurement at 600 nm. All reactions were carried out in 50 mM NaPi, pH 7.4 at room temperature if not stated otherwise. Continuous absorbance or fluorescence measurements in a plate reader could only be performed between 24 °C and 28 °C due to the heat generated by the reader during the measurement. Reactions in 96-well plates consisted of 270 µL reaction mixture to which 30 µL of enzyme solution (10 g L−1) was added to start the reaction. The given substrate concentrations refer to the concentrations in the substrate mixture before enzyme addition if not stated otherwise.
4.3. Production of Enzymes
Est8 was heterologously expressed in
Escherichia coli BL21(DE3) from pET26_Est8. The overnight culture was grown at 37 °C in LB with 50 mg L
−1 kanamycin and 1% (
w/
v) glucose. TB medium supplemented with 50 mg L
−1 kanamycin was inoculated from the overnight culture and incubated at 37 °C at 180 rpm until the culture reached an OD
600 of 1.0. Then the culture was cooled to 20 °C and induced with 0.5 mM IPTG for 24 h. Cells were harvested by centrifugation at 4,500
g and 4 °C for 1 h. Cells were washed with 50 mM NaPi, pH 7.4 before they were resuspended in the same buffer containing 5 mg L
−1 DNase and lysed by one passage through a French press. Insolubles were removed by centrifugation at 10,000
g and 4 °C for 30 min, and the lysate was passed through a 0.45 µm filter before it was frozen and lyophilized. MsAcT was expressed as described in the literature [
19]. The clarified and lyophilized lysate was stored at 4 °C.
The expression of the CAL-A-related acyltransferases was performed according to the literature [
4]. Briefly, sequences of CaLIP4, CalLAc5 and CalLAc8 were obtained using accession numbers AF191317, XP_717001.1 and XP_711685, respectively. For cloning into pPICZαB (via FastCloning), natural signal peptides were replaced by
Saccharomyces cerevisiae α-mating factor provided in the plasmid. After transformation into
Pichia pastoris X-33, colonies expressing active enzyme were selected via lipase activity indicator plates. Five milliliters of YPD medium containing 100 mg L
−1 zeocin were inoculated with a
Pichia pastoris strain harboring the expression plasmid. After 24 h at 30 °C, 50 mL BMGY medium with 100 mg L
−1 zeocin was inoculated and incubated for 24 h at 180 rpm and 30 °C. The cell cultures were then centrifuged (10,000
g, 4 °C, 15 min) and the cell pellets were used to innoculate 50 mL BMMY media. The cells were cultured at 30 °C at 180 rpm for 120 h. One percent (
v/
v) methanol was added every 24 h to induce protein expression. Later, cell cultures were centrifuged at 10,000
g and 4 °C for 15 min to separate the cells from the supernatant. The supernatant, rich in the secreted enzyme, was filtered through a 0.2 μm filter, frozen at −80 °C, and lyophilized.
4.4. Preparative Production of Oligocarbonate
To obtain the enzyme-generated oligocarbonates, the reaction was scaled up to 50 mL. DMC (50 mmol) and HD (50 mmol) were dissolved in 50 mM NaPi, pH 7.4. Est8 lyophilizate was added to a final concentration of 3.2 g L−1, and the reaction was performed for 23.5 h at room temperature. After cooling at 4 °C for 3 h, the precipitate was collected by centrifugation. The raw product was extracted four times with 4 mL dichloromethane, and the combined organic phases were dried over anhydrous Na2SO4. After evaporation of the solvent, 443 mg of oligocarbonate was obtained as a slightly viscous liquid which solidified at 4 °C.
The masses of the oligomers were determined using the Advion expressionL CMS. The direct analysis probe, otherwise known as ASAP® (Atmospheric Solid Analysis Probe, Advion Ltd., Harlow, UK) mode was used for the measurement. The analysis was performed under “high temperature and low fragmentation” conditions to minimize the fragmentation of the oligomers. Vaporized methanol was used as an additional running solvent to carry the organic compound. Firstly, the empty ASAP® probe capillary was injected into the CMS to record the blank or background noise for 30–60 s. Subsequently, the probe was immersed into the sample and injected into the CMS. The data was analyzed using the Data Express software provided by Advion. The 1H-NMR spectrum of the isolated oligocarbonates was recorded on a 300 MHz Avance II (Bruker Daltonik GmbH, Bremen, Germany) in CDCl3.
4.5. Fluorescent Staining
Nile Red was used to fluorescently stain the enzymatically formed oligocarbonate particles. As the emission and excitation wavelengths of Nile Red are highly variable depending on the surrounding environment [
17], spectral scans were performed to identify the optimal wavelengths for oligocarbonate-bound Nile Red. Nile Red was added from a 1 mg mL
−1 stock solution in DMSO. In the final reaction, the concentration was 1 µg mL
−1. Excitation was realized at 560 nm, and emission was recorded at 630 nm. Particles stained with Nile Red were also visualized by fluorescence microscopy using a ZEISS Axio Vert.A1 inverted microscope with a 20x objective. For excitation, a 540-580 nm LED module was used with a 545/25 nm bandpass filter, and emitted light was passed through a 605/70 nm bandpass filter. This combination of an LED module and filter set resembled the optics used for fluorescence measurements in 96-well plates.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





