Ionic, Core-Corona Polymer Microsphere-Immobilized MacMillan Catalyst for Asymmetric Diels-Alder Reaction


Abstract
:
1. Introduction
2. Results and Discussion
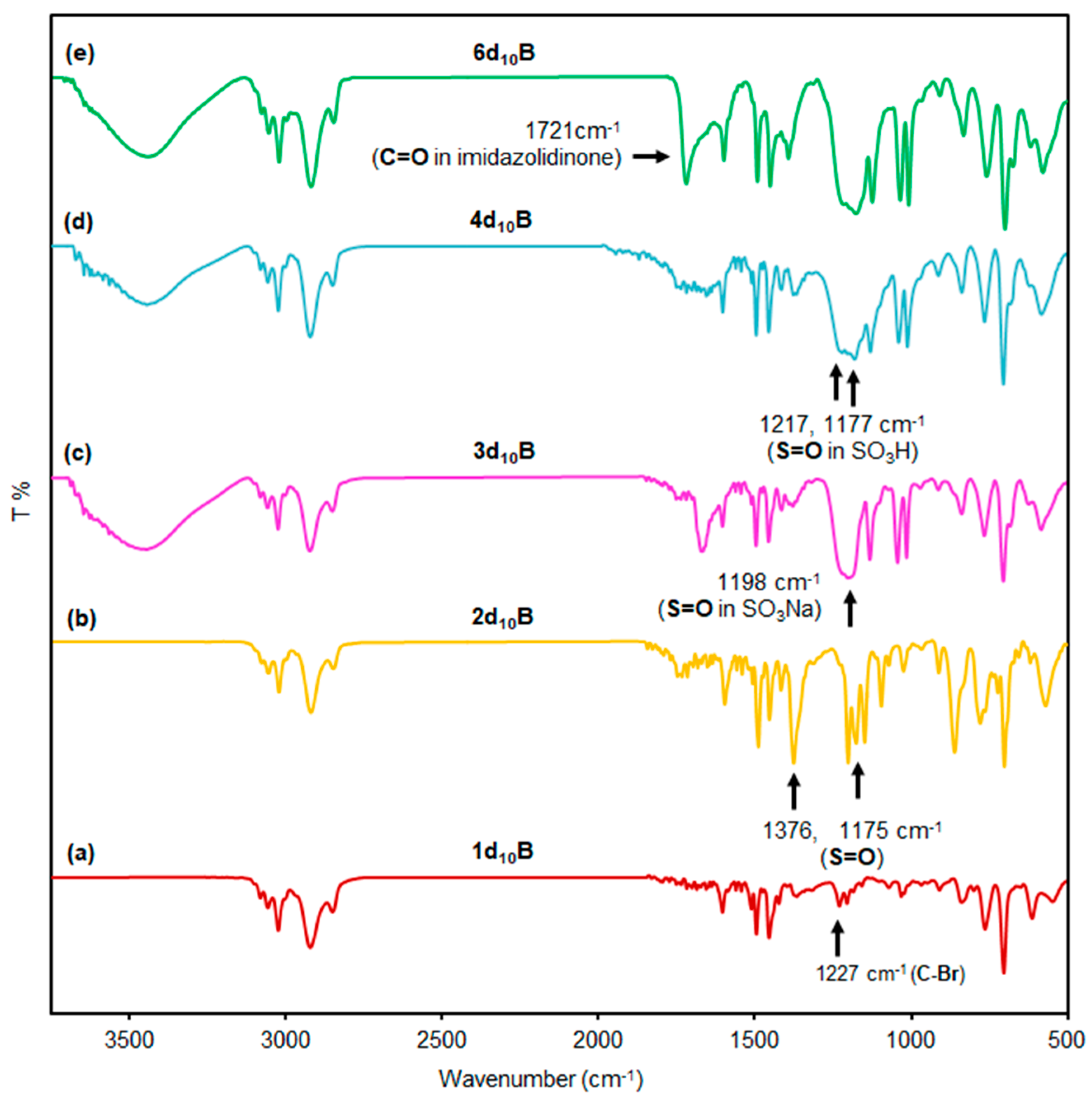
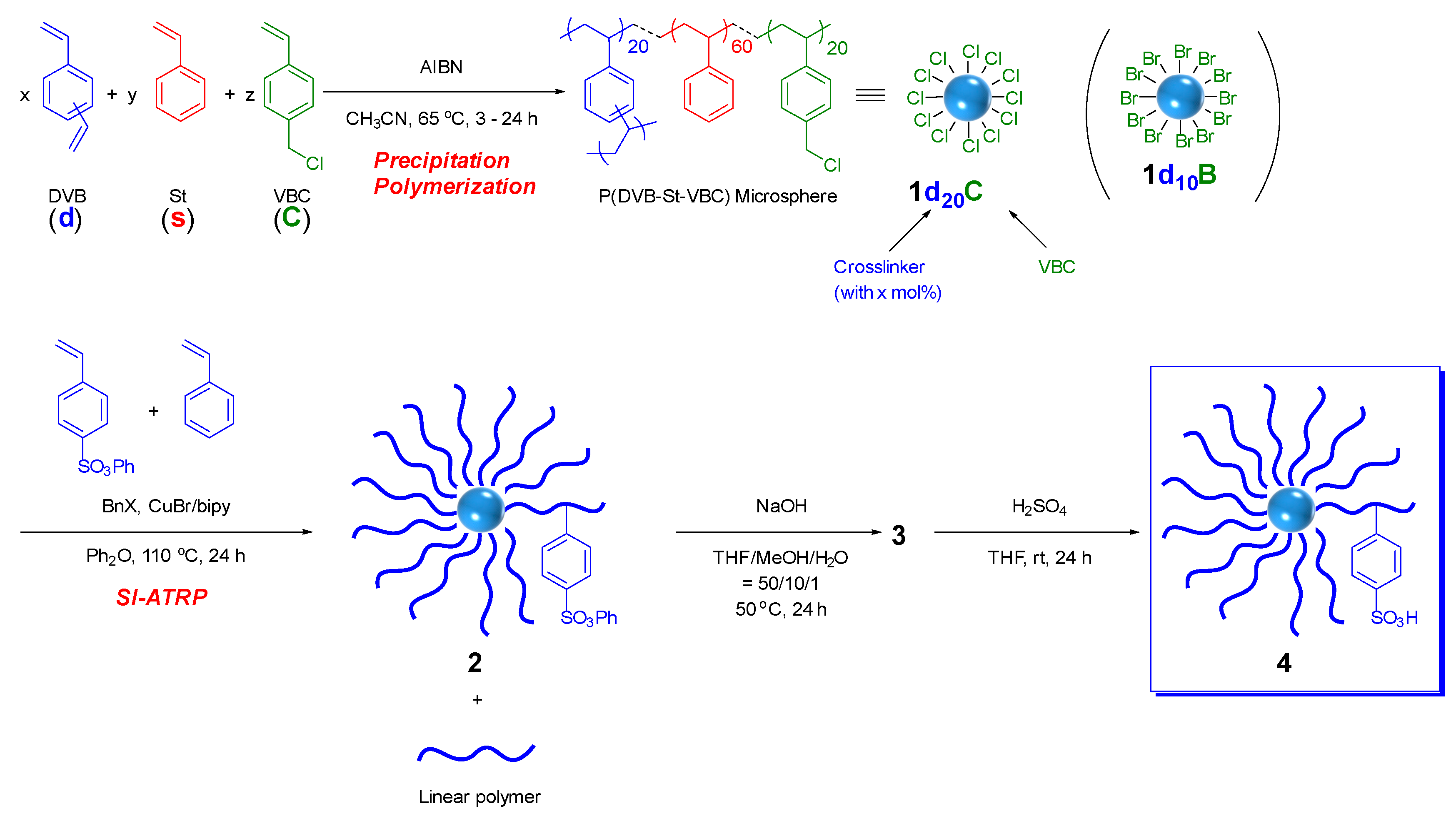
2.1. Preparation of Core-Corona Polymer Microsphere Having Sulfonic Acid (CCM–SO3H)
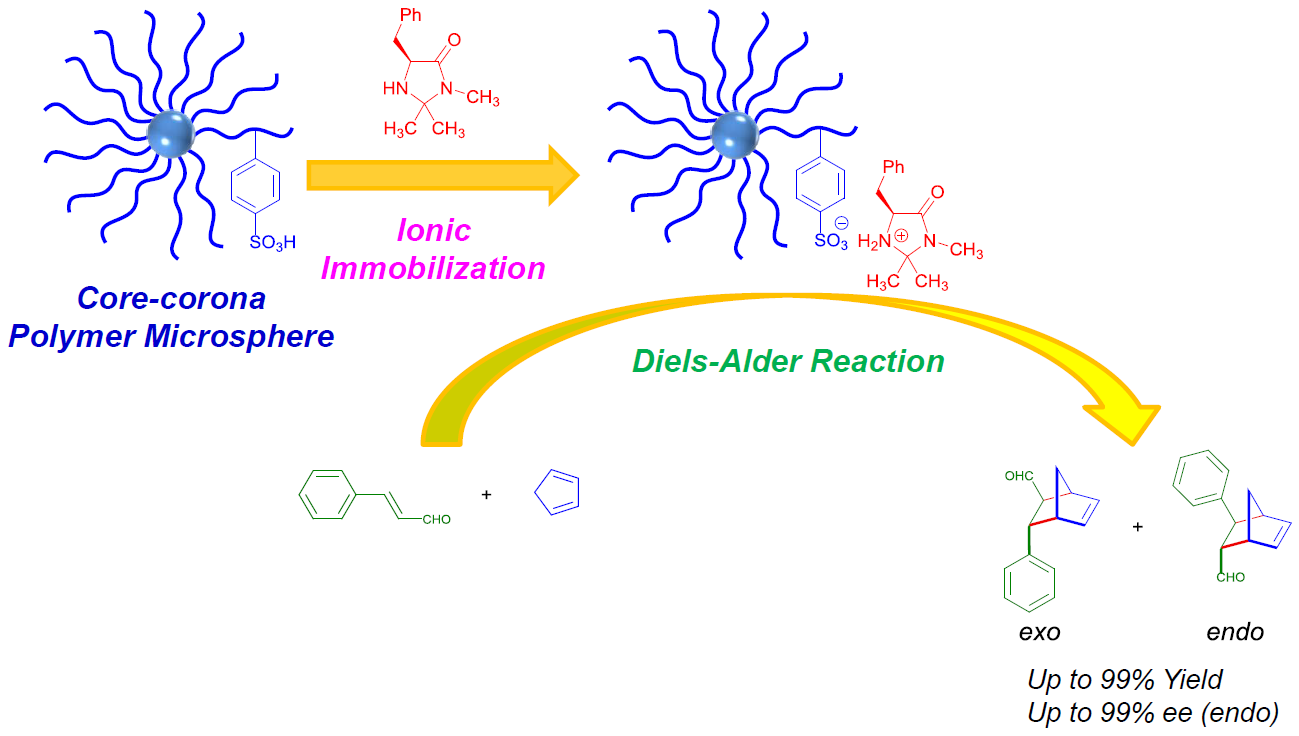
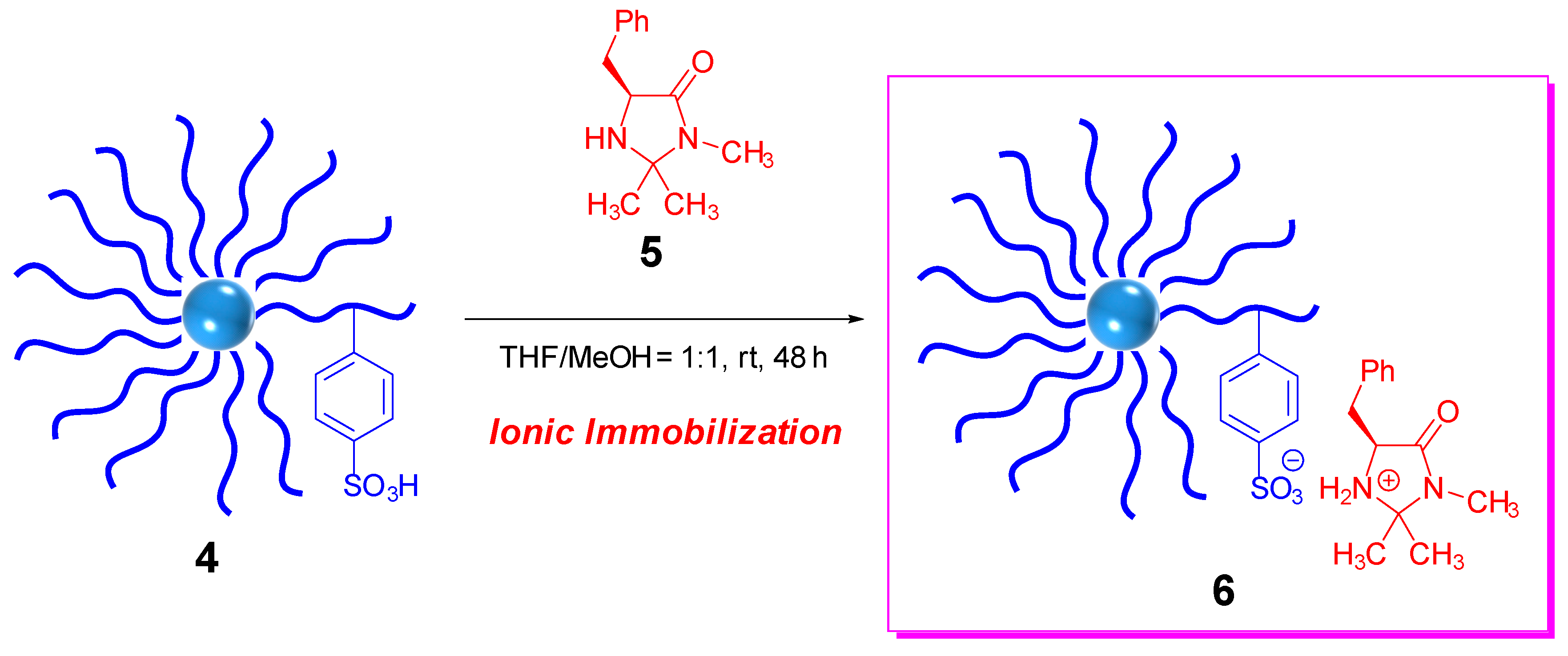
2.2. Synthesis of Core-Corona Polymer, Microsphere-Immobilized Macmillan Catalyst (ICCC)
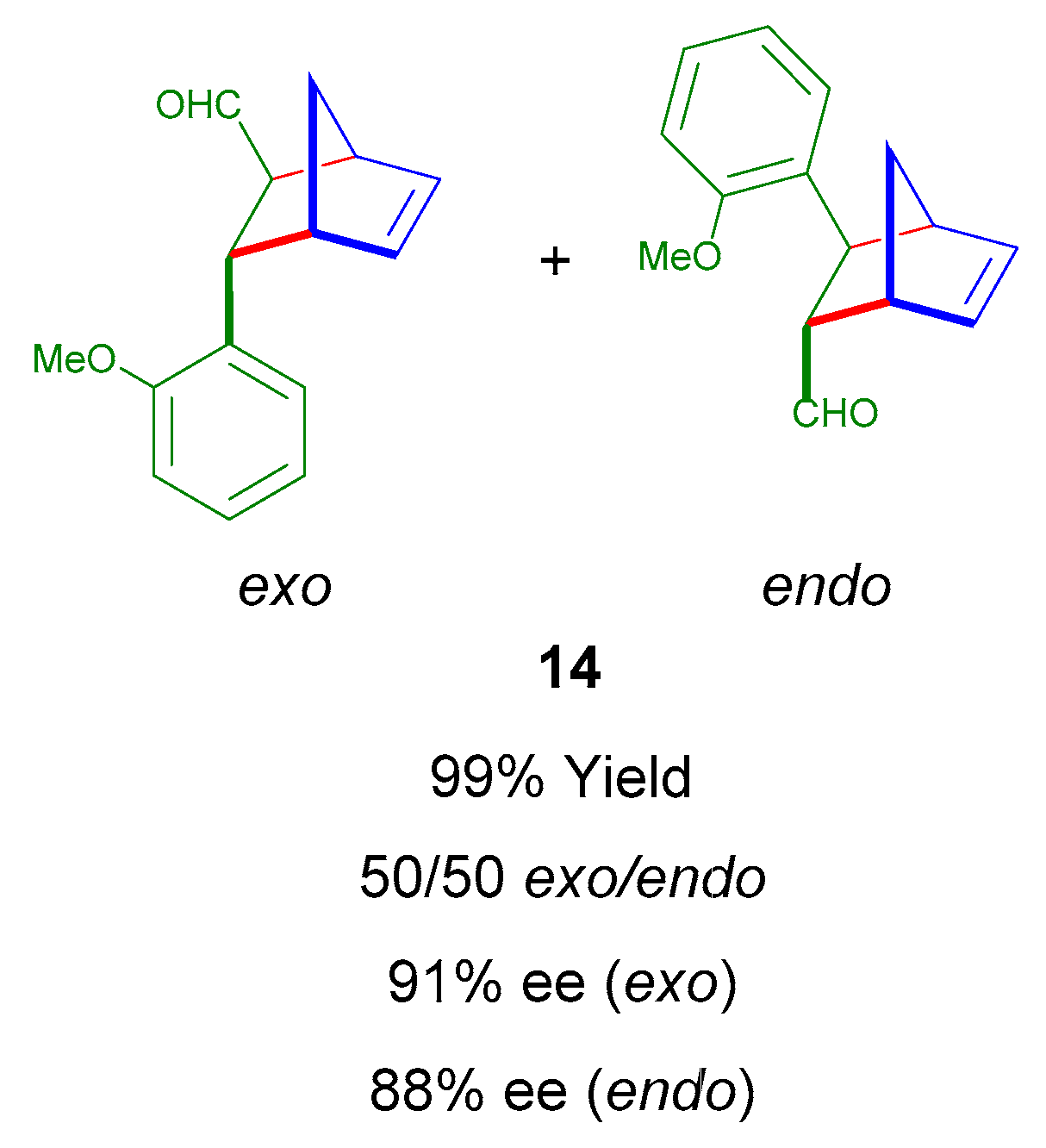
2.3. The Effect of Solvent in an Asymmetric DA Reaction Catalyzed by ICCC 6
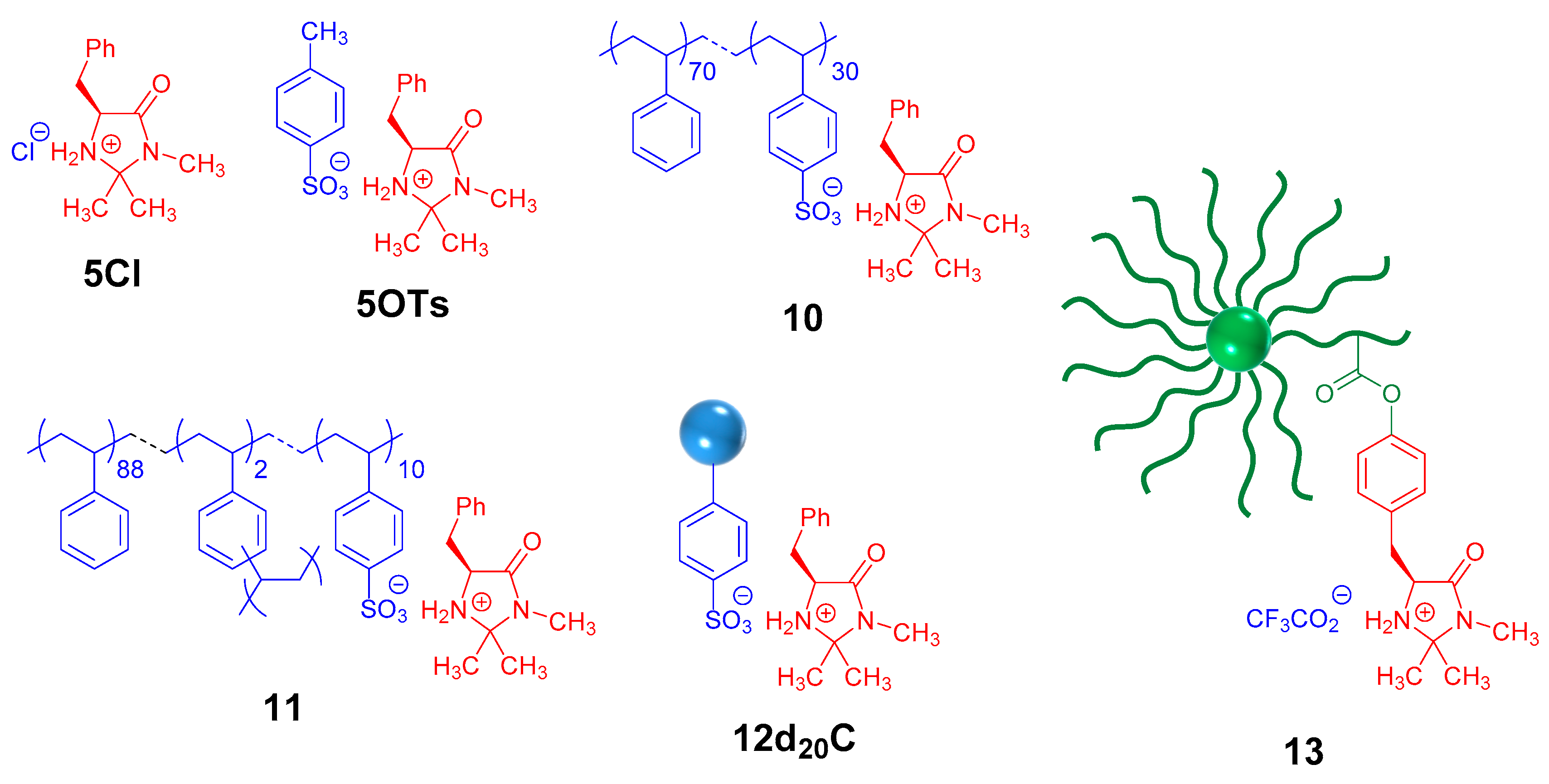
2.4. Comparison of the Catalytic Activeity of a Model and Polymeric Macmillan Catalyst in an Asymmetric DA Reaction
3. Materials and Methods
3.1. General Methods
3.2. General Procedure for the Preparation of CCM–SO3H
3.3. General Procedure for the Preparation of ICCC
3.4. General Procedure for the Asymmetric DA Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis; Berkessel, A.; Groger, H. (Eds.) Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Enantioselective Organocatalysis; Dalko, P.I. (Ed.) Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels-Alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Jen, W.S.; Wiener, J.J.M.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Enantioselective Organocatalytic 1, 3-Dipolar Cycloaddition. J. Am. Chem. Soc. 2000, 122, 9874–9875. [Google Scholar] [CrossRef]
- Pares, N.A.; MacMillan, D.W.C. New strategies in organic catalysis: The first enantioselective organocatalytic Friedel-Crafts alkylation. J. Am. Chem. Soc. 2001, 123, 4370–4371. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.F.; MacMillan, D.W.C. Enantioselective organocatalytic indole alkylations. Design of a new and highly effective chiral amine for iminium catalysis. J. Am. Chem. Soc. 2002, 124, 1172–1173. [Google Scholar] [CrossRef] [PubMed]
- Brochu, M.P.; Brown, S.P.; MacMillan, D.W.C. Direct and enantioselective organocatalytic α-chlorination of aldehydes. J. Am. Chem. Soc. 2004, 126, 4108–4109. [Google Scholar] [CrossRef]
- Beeson, T.D.; MacMillan, D.W.C. Enantioselective organocatalytic α-fluorination of aldehydes. J. Am. Chem. Soc. 2005, 127, 8826–8828. [Google Scholar] [CrossRef]
- Northrup, A.B.; MacMillan, D.W.C. The first direct and enantioselective cross-aldol reaction of aldehydes. J. Am. Chem. Soc. 2002, 124, 6798–6799. [Google Scholar] [CrossRef]
- Fonseca, M.T.H.; List, B. Catalytic asymmetric intramolecular Michael reaction of aldehydes. Angew. Chem. Int. Ed. 2004, 43, 3958–3960. [Google Scholar] [CrossRef]
- Lee, S.; MacMillan, D.W.C. Enantioselective organocatalytic epoxidation using hypervalent iodine reagents. Tetrahedron 2006, 62, 11413–11424. [Google Scholar] [CrossRef]
- Beeson, T.D.; Mastracchio, A.; Homg, J.B.; Ashton, K.; MacMillan, D.W.C. Enantioselective Organocatalysis Using SOMO Activation. Science 2007, 316, 582–585. [Google Scholar] [CrossRef]
- Nicewicz, D.A.; MacMillan, D.W.C. Merging photoredox catalysis with organocatalysis: The direct asymmetric alkylation of aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Devery, J.J., III; Conrad, J.C.; MacMillan, D.W.C.; Flowers, R.A., II. Mechanistic Complexity in Organo-SOMO Activation. Angew. Chem. Int. Ed. 2010, 49, 6106–6110. [Google Scholar] [CrossRef] [PubMed]
- Mastracchio, A.; Warkentin, A.A.; Walji, A.M.; MacMillan, D.W.C. Direct and enantioselective α-allylation of ketones via singly occupied molecular orbital (SOMO) catalysis. Proc. Natl. Acd. Sci. USA 2010, 107, 20648–20651. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.V.; Ashton, K.; MacMillan, D.W.C. The intramolecular asymmetric allylation of aldehydes via organo-SOMO catalysis: A novel approach to ring construction. Chem. Sci. 2011, 2, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angew. Chem. Int. Ed. 2011, 50, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.W.; Vander Wal, M.N.; Grange, R.L.; MacMillan, D.W.C. Enantioselective α-Benzylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc. 2010, 132, 13600–13603. [Google Scholar] [CrossRef]
- Pirnot, M.T.; Rankic, D.A.; Martin, D.B.C.; MacMillan, D.W.C. Photoredox Activation for the Direct β-Arylation of Ketones and Aldehydes. Science 2013, 339, 1593–1596. [Google Scholar] [CrossRef]
- Terrett, J.A.; Clift, M.D.; MacMillan, D.W.C. Direct β-Alkylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc. 2014, 136, 6858–6861. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Benaglia, M.; Celentano, G.; Cinquini, M.; Puglisi, A.; Cozzi, F. Poly (ethylene glycol)-supported chiral imidazolidin-4-one: An efficient organic catalyst for the enantioselective Diels-Alder cycloaddition. Adv. Synth. Catal. 2002, 344, 149–152. [Google Scholar] [CrossRef]
- Selkälä, S.A.; Tois, J.; Pihko, P.M.; Koskinen, A.M.P. Asymmetric organocatalytic Diels-Alder reactions on solid support. Adv. Synth. Catal. 2002, 344, 941–945. [Google Scholar] [CrossRef]
- Puglisi, A.; Benaglia, M.; Cinquini, M.; Cozzi, F.; Celentano, G. Enantioselective 1, 3-dipolar cycloadditions of unsaturated aldehydes promoted by a poly (ethylene glycol)-supported organic catalyst. Eur. J. Org. Chem. 2004, 567–573. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Zhao, L.; Lee, S.S.; Ying, J.Y. Enantioselective catalysis over chiral imidazolidin-4-one immobilized on siliceous and polymer-coated mesocellular foams. Adv. Synth. Catal. 2006, 348, 2027–2032. [Google Scholar] [CrossRef]
- Mason, B.P.; Bogdan, A.R.; Goswami, A.; McQuade, D.T. A general approach to creating soluble catalytic polymers heterogenized in microcapsules. Org. Lett. 2007, 9, 3449–3451. [Google Scholar] [CrossRef]
- Kristensen, T.E.; Vestli, K.; Jakobsen, M.G.; Hansen, F.K.; Hansen, T. A general approach for preparation of polymer-supported chiral organocatalysts via acrylic copolymerization. J. Org. Chem. 2010, 75, 1620–1629. [Google Scholar] [CrossRef]
- Guizzetti, S.; Benaglia, M.; Siegel, J.S. Poly (methylhydrosiloxane)-supported chiral imidazolinones: New versatile, highly efficient and recyclable organocatalysts for stereoselective Diels-Alder cycloaddition reactions. Chem. Commun. 2012, 48, 3188–3190. [Google Scholar] [CrossRef]
- Riente, P.; Yadav, J.; Pericàs, M.A. A click strategy for the immobilization of MacMillan organocatalysts onto polymers and magnetic nanoparticles. Org. Lett. 2012, 14, 3668–3671. [Google Scholar] [CrossRef]
- Moore, B.L.; Lu, A.; Longbottom, D.A.; O’Reilly, R.K. Immobilization of MacMillan catalyst via controlled radical polymerization: Catalytic activity and reuse. Polym. Chem. 2013, 4, 2304–2312. [Google Scholar] [CrossRef]
- Chiroli, V.; Benaglia, M.; Cozzi, F.; Puglisi, A.; Annuziata, R.; Cwlwntano, G. Continuous-flow stereoselective organocatalyzed Diels-Alder reactions in a chiral catalytic “Homemade” HPLC column. Org. Lett. 2013, 15, 3590–3593. [Google Scholar] [CrossRef]
- Wang, C.A.; Zhang, Y.; Shi, J.Y.; Wang, W. A self-supported polymeric MacMillan catalyst for homogeneous organocatalysis and heterogeneous recycling. Chem. Asian J. 2013, 8, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Chiroli, V.; Benaglia, M.; Puglisi, A.; Porta, R.; Jumde, R.P.; Mandoli, A. A chiral organocatalytic polymer-based monolithic reactor. Green Chem. 2014, 16, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Salvo, A.M.P.; Giacalone, F.; Nato, R.; Gruttadauria, M. Recyclable heterogeneous and low-loading homogeneous chiral imidazolidinone catalysts for α-alkylation of aldehydes. ChemPlusChem 2014, 79, 857–862. [Google Scholar] [CrossRef]
- Takata, L.M.S.; Iida, H.; Shimomura, K.; Hayashi, K.; Santos, A.A.D.; Yashima, E. Helical poly (phenylacetylene) bearing chiral and achiral imidazolidinone-based pendants that catalyze asymmetric reactions due to catalytically active achiral pendants assisted by macromolecular helicity. Macromol. Rapid Commun. 2015, 36, 2047–2054. [Google Scholar] [CrossRef]
- Ranjbar, S.; Riente, P.; Rodriguez-Escrich, C.; Yadav, J.; Ramineni, K.; Pericàs, M.A. Polystyrene or magnetic nanoparticles as support in enantioselective organocatalysis? A case study in Friedel-Crafts chemistry. Org. Lett. 2016, 18, 1602–1605. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, B.; Zhang, S.; Jia, X.; Hu, Z. Facile synthesis of hairy microparticle-/nanoparticle-supported MacMillan and its application to Diels-Alder reaction in water. Colloid. Polym. Sci. 2017, 295, 573–582. [Google Scholar] [CrossRef]
- Arakawa, Y.; Haraguchi, N.; Itsuno, S. An immobilization method of chiral quaternary ammonium salts onto polymer supports. Angew. Chem. Int. Ed. 2008, 47, 8232–8235. [Google Scholar] [CrossRef]
- Haraguchi, N.; Takemura, Y.; Itsuno, S. Novel polymer-supported organocatalyst via ion exchange reaction: Facile immobilization of chiral imidazolidin-4-one and its application to Diels-Alder reaction. Tetrahedron Lett. 2010, 51, 1205–1208. [Google Scholar] [CrossRef]
- Itsuno, S.; Paul, D.K.; Salam, M.A.; Haraguchi, N. Main-chain ionic chiral polymers: Synthesis of optically active quaternary ammonium sulfonate polymers and their application in asymmetric catalysis. J. Am. Chem. Soc. 2010, 132, 2864–2865. [Google Scholar] [CrossRef]
- Haraguchi, N.; Kiyono, H.; Takemura, Y.; Itsuno, S. Design of main-chain polymers of chiral imidazolidinone for asymmetric organocatalysis application. Chem. Commun. 2012, 48, 4011–4013. [Google Scholar] [CrossRef] [PubMed]
- Itsuno, S.; Oonami, T.; Takenaka, N.; Haraguchi, N. Synthesis of Chiral Polyethers Containing Imidazolidinone Repeating Units and Application as Catalyst in Asymmetric Diels–Alder Reaction. Adv. Synth. Catal. 2015, 357, 3995–4002. [Google Scholar] [CrossRef]
- Hassan, M.M.; Haraguchi, N.; Itsuno, S. Highly active polymeric organocatalyst: Chiral ionic polymers prepared from 10,11-didehydrogenated cinchonidinium salt. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 621–627. [Google Scholar] [CrossRef]
- Haraguchi, N.; Nguyen, T.L.; Itsuno, S. Polyesters containing chiral imidazolidinone salts in polymer main chain: Heterogeneous organocatalysts for the asymmetric Diels-Alder reaction. CHEMCATCHEM 2017, 9, 3786–3794. [Google Scholar] [CrossRef]
- Haraguchi, N.; Takenaka, N.; Najwa, A.; Takahara, Y.; Kar Mun, M.; Itsuno, S. Synthesis of main-chain ionic polymers of chiral imidazolidinone organocatalysts and their application to asymmetric Diels-Alder reactions. Adv. Synth. Catal. 2018, 360, 112–123. [Google Scholar] [CrossRef]
- Xia, Y.; Gates, B.; Yin, Y.; Lu, Y. Monodispersed colloidal spheres: Old materials with new applications. Adv. Mater. 2000, 12, 693–713. [Google Scholar] [CrossRef]
- Wang, J.; Cormack, P.A.G.; Sherrington, D.C.; Khoshdel, E. Monodisperse, molecularly imprinted polymer microspheres prepared by precipitation polymerization for affinity separation applications. Angew. Chem. Int. Ed. 2003, 42, 5336–5338. [Google Scholar] [CrossRef]
- Barner, L. Synthesis of microspheres as versatile functional scaffolds for materials science applications. Adv. Mater. 2009, 21, 2547–2553. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, W.; Wang, Y.; Zhang, M. Palladium-iminodiacetic acid immobilized on pH-responsive polymeric microspheres: Efficient quasi-homogeneous catalyst for Suzuki and Heck reactions in aqueous solution. Adv. Synth. Catal. 2008, 350, 2065–2076. [Google Scholar] [CrossRef]
- Haraguchi, N.; Nishiyama, A.; Itsuno, S. Synthesis of polymer microspheres functionalized with chiral ligand by precipitation polymerization and their application to asymmetric transfer hydrogenation. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 3340–3349. [Google Scholar] [CrossRef]
- Ullah, M.W.; Thao, N.T.P.; Sugimoto, T.; Haraguchi, N. Synthesis of core-corona polymer microsphere-supported cinchonidinium salt and its application to asymmetric synthesis. Mol. Catal. 2019, 473. [Google Scholar] [CrossRef]
- Li, W.-H.; Li, K.; Stöver, H.D.H. Monodisperse poly (chloromethylstyrene-co-divinylbenzene) microspheres by precipitation polymerization. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 2295–2303. [Google Scholar] [CrossRef]
- Haraguchi, N.; Sugimoto, T.; Ullah, M.W. Synthesis of monodispersed polymer microsphere functionalized with benzyl bromides by chemical scission of cleavable crosslinker. 2019; under preparation. [Google Scholar]
- Ullah, M.W.; Haraguchi, N. Synthesis of well-defined hairy polymer microspheres by precipitation polymerization and surface-initiated atom transfer radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1296–1304. [Google Scholar] [CrossRef]
- Brazier, J.B.; Jones, K.M.; Platts, J.A.; Tomkinson, N.C.O. On the roles of protic solvents in imidazolidinone-catalyzed transformations. Angew. Chem. Int. Ed. 2011, 50, 1613–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 4 | Dn (Core) (μm) a | Corona | Dn (Core-Corona) (μm) a | U (4) a | S Content (mmol g−1) c | |
|---|---|---|---|---|---|---|---|
| Mnb | Mw/Mnb | ||||||
| 1 | 4d20C | 1.14 | 7630 | 1.18 | 1.34 | 1.00 | 1.00 |
| 2 | 4d10B | 1.08 | 11,000 | 1.32 | 1.40 | 1.23 | 1.61 |
| 3 | 4d10B-200 | 1.08 | 30,400 | 1.57 | 1.95 | 1.20 | 1.48 |
| Entry | 6 | Solvent | Degree of Immobilization (%) | Catalyst Content (mmol g−1) |
|---|---|---|---|---|
| 1 | 6d20C | 1/1 THF/CH3OH | 61 | 0.533 |
| 2 | 6d10B | 1/1 THF/CH3OH | 67 | 0.581 |
| 3 | 6d10B | 1/1 CH2Cl2/CH3OH | 86 | 1.06 |
| 4 | 6d10B-200 | 1/1 CH2Cl2/CH3OH | 98 | 1.10 |
| Entry | Solvent | Time (h) | 9 | |||
|---|---|---|---|---|---|---|
| Yield (%) b | Exo/Endob | ee (Exo) (%) c | ee (Endo) (%) c | |||
| 1 | THF | 60 | 18 | 51/49 | 57 | 67 |
| 2 | CH2Cl2 | 60 | 25 | 50/50 | 61 | 68 |
| 3 | CH3CN | 60 | 29 | 52/48 | 66 | 74 |
| 4 | MeOH | 38 | 91 | 56/44 | 88 | >99 |
| 5 | H2O | 60 | 97 | 52/48 | 82 | >99 |
| 6 | Dry MeOH | 38 | 92 | 44/56 | 85 | 94 |
| 7 | 95/5 THF/H2O | 60 | 87 | 52/48 | 85 | 97 |
| 8 | 95/5 CHCN/H2O | 60 | 96 | 53/47 | 81 | 92 |
| 9 | 95/5 MeOH/H2O | 38 | 99 | 54/46 | 92 | >99 |
| 10 | 85/15 MeOH/H2O | 50 | 99 | 56/44 | 91 | 99 |
| 11 | 75/25 MeOH/H2O | 62 | 99 | 50/50 | 89 | 98 |
| Entry | Catalyst | Time (h) | 9 | |||
|---|---|---|---|---|---|---|
| Yield (%) b | Exo/Endob | ee (Exo) (%) c | ee (Endo) (%) c | |||
| 1 | 5Cl | 8 | 99 | 57/43 | 93 | 93 |
| 2 | 5OTs | 24 | 94 | 55/45 | 88 | 92 |
| 3 | 10 | 36 | 89 | 60/40 | 94 | 96 |
| 4 | 11 | 24 | 99 | 55/45 | 84 | 83 |
| 5 | 12d20C | 44 | 97 | 55/45 | 90 | >99 |
| 6 | 6d20C | 36 | 94 | 54/46 | 92 | >99 |
| 7 | 6d10B | 38 | 99 (97) d | 54/46 | 92 | >99 |
| 8 | 6d10B-200 | 60 | 64 | 51/49 | 91 | 95 |
| 9 e | 13 | 21 | 78 | 36/64 | 92 | 88 |
| 10 f | 6d10B | 40 | 99 | 54/46 | 92 | >99 |
| 11 g | 6d10B | 43 | 99 | 55/45 | 91 | >99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, M.W.; Haraguchi, N. Ionic, Core-Corona Polymer Microsphere-Immobilized MacMillan Catalyst for Asymmetric Diels-Alder Reaction. Catalysts 2019, 9, 960. https://doi.org/10.3390/catal9110960
Ullah MW, Haraguchi N. Ionic, Core-Corona Polymer Microsphere-Immobilized MacMillan Catalyst for Asymmetric Diels-Alder Reaction. Catalysts. 2019; 9(11):960. https://doi.org/10.3390/catal9110960
Chicago/Turabian StyleUllah, Md. Wali, and Naoki Haraguchi. 2019. "Ionic, Core-Corona Polymer Microsphere-Immobilized MacMillan Catalyst for Asymmetric Diels-Alder Reaction" Catalysts 9, no. 11: 960. https://doi.org/10.3390/catal9110960
APA StyleUllah, M. W., & Haraguchi, N. (2019). Ionic, Core-Corona Polymer Microsphere-Immobilized MacMillan Catalyst for Asymmetric Diels-Alder Reaction. Catalysts, 9(11), 960. https://doi.org/10.3390/catal9110960