Correlation of the Photocatalytic Activities of Cu, Ce and/or Pt-Modified Titania Particles with their Bulk and Surface Structures Studied by Reversed Double-Beam Photoacoustic Spectroscopy



Abstract
:1. Introduction
2. Results and Discussion
2.1. Physical and Structural Characteristics of Samples
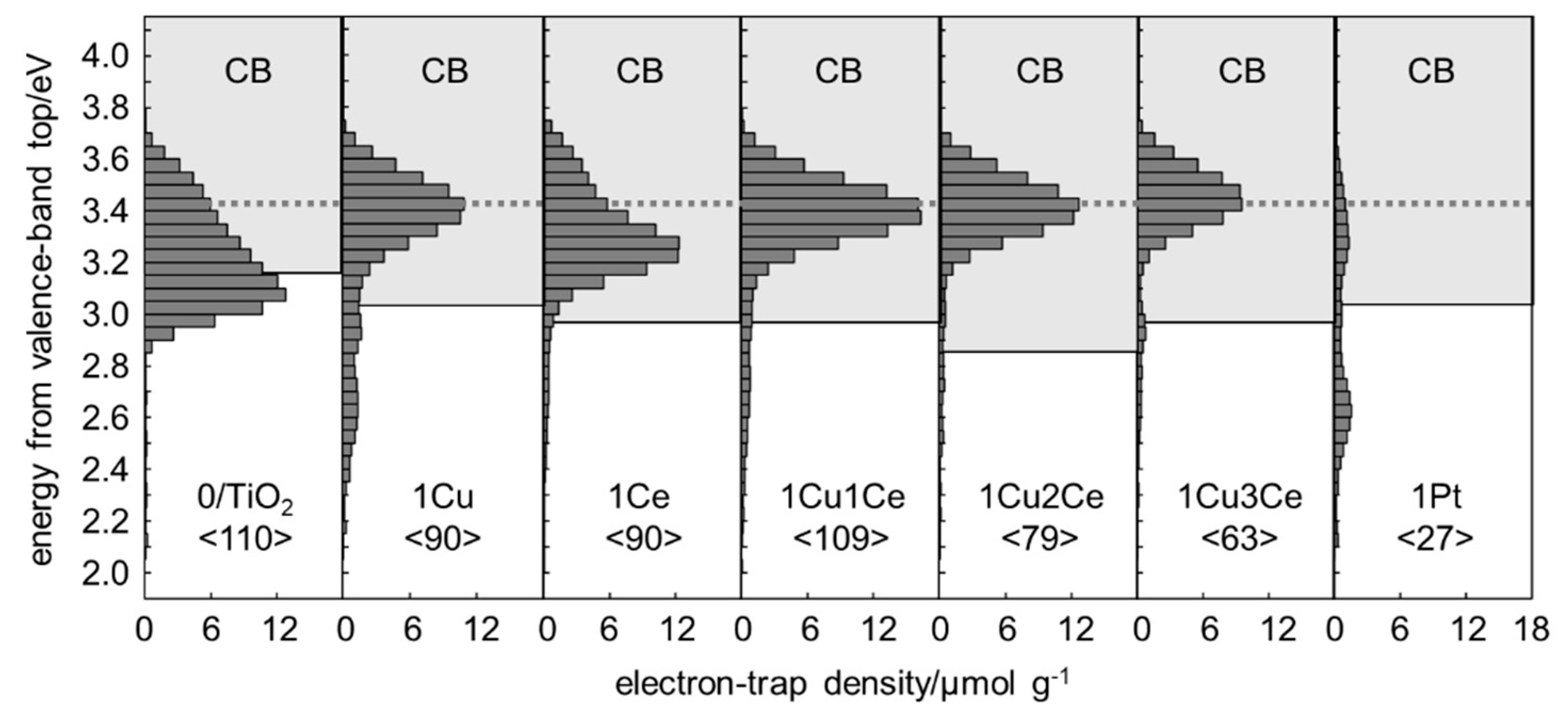
2.2. Energy-Resolved Distribution of Electron Traps of Samples
2.3. Photocatalytic Activity of Samples
3. Materials and Methods
3.1. Preparation of Titania Photocatalyst Samples
3.2. Characterization
3.3. Photocatalytic Activity Tests
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nakata, K.; Fujishima, A. TiO2 photocatalysis: Design and applications. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 169–189. [Google Scholar] [CrossRef]
- Ochiai, T.; Fujishima, A. Photoelectrochemical properties of TiO2 photocatalyst and its applications for environmental purification. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 247–262. [Google Scholar] [CrossRef]
- Ohtani, B. Photocatalysis A to Z―What We Know and What We Don’t Know. J. Photochem. Photobiol. C Photochem. Rev. 2010, 11, 157–178. [Google Scholar] [CrossRef]
- Ohtani, B. Revisiting the Fundamental Physical Chemistry in Heterogeneous Photocatalysis: Its Thermodynamics and Kinetics. Phys. Chem. Chem. Phys. 2014, 16, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Nitta, A.; Takase, M.; Takashima, M.; Murakami, N.; Ohtani, B. A Fingerprint of Metal-oxide Powders: Energy-resolved Distribution of Electron Traps. Chem. Commun. 2016, 52, 12096–12099. [Google Scholar] [CrossRef]
- Nitta, A.; Takashima, M.; Murakami, N.; Takase, M.; Ohtani, B. Reversed double-beam photoacoustic spectroscopy of metal-oxide powders for estimation of their energy-resolved distribution of electron traps and electronic-band structure. Electrochim. Acta 2018, 264, 83–90. [Google Scholar] [CrossRef]
- Unwiset, P.; Makdee, A.; Chanapattharapol, K.C.; Kidkhunthod, P. Effect of Cu addition on TiO2 surface properties and photocatalytic performance: X-ray Absorption Spectroscopy analysis. J. Phys.Chem. Solids 2018, 120, 231–240. [Google Scholar] [CrossRef]
- Shafei, A.; Salarpour, M.E.; Sheibani, S. Effect of intermediate ball milling on the synthesis of Cu-doped TiO2 nano-photocatalyst by sol-gel method. J. Sol-Gel Sci. Tech. 2019, 92, 173–185. [Google Scholar] [CrossRef]
- Makdee, A.; Unwiset, P.; Chanapattharapol, K.C.; Kidkhunthod, P. Effects of Ce addition on the properties and photocatalytic activity of TiO2, investigated by X-ray absorption spectroscopy. Mater. Chem. Phys. 2018, 213, 431–443. [Google Scholar] [CrossRef]
- Lee, J.; Choi, J. Sonochemical Synthesis of Ce-doped TiO2 Nanostructure: A Visible-Light-Driven Photocatalyst for Degradation of Toluene and O-Xylene. Materials 2019, 12, 1265. [Google Scholar] [CrossRef]
- Matsubara, K.; Inoue, M.; Hagiwara, H.; Abe, T. Photocatalytic water splitting over Pt-loaded TiO2 (Pt/TiO2) catalysts prepared by the polygonal barrel-sputtering method. Appl. Catal. B Envion. 2019, 254, 7–14. [Google Scholar] [CrossRef]
- Nishimoto, S.-I.; Ohtani, B.; Kajiwara, H.; Kagiya, T. Correlation of the Crystal Structure of Titanium Dioxide Prepared from Titanium Tetra-2-propoxide with the Photocatalytic Activity for Redox Reactions in Aqueous Propan-2-ol and Silver Salt Solutions. J. Chem. Soc. Faraday Trans. 1 1985, 81, 61–68. [Google Scholar] [CrossRef]
- Gleń, M.; Grzmil, B. Photostability and optical properties of modified titanium dioxide. Pure Appl. Chem. 2012, 84, 2531–2547. [Google Scholar] [CrossRef]
- Grzmil, B.; Gleń, M.; Kic, B.; Lubkowski, K. Study of the anatase to rutile transformation kinetics of the modified TiO2. Pol. J. Chem. Technol. 2013, 15, 73–80. [Google Scholar] [CrossRef]
- Nitta, A.; Takashima, M.; Takase, M.; Ohtani, B. Identification and characterization of titania photocatalyst powders using their energy-resolved density of electron traps as a fingerprint. Catal. Today 2019, 321–322, 2–8. [Google Scholar] [CrossRef]
- Ohtani, B.; Nitta, A.; Takase, M.; Takashima, M. Design of heterogeneous photocatalysis based on energy-resolved distribution of electron traps. In Proceedings of the 8th Asia-Pacific Congress on Catalysis (APCAT-8), Bangkok, Thailand, 4–7 August 2019. Unpublished results. [Google Scholar]
- Higashimoto, S. Titanium-Dioxide-Based Visible-Light-Sensitive Photocatalysis: Mechanistic Insight and Applications. Catalysts. 2019, 9, 201. [Google Scholar] [CrossRef]
- Higashimoto, S.; Ushiroda, Y.; Azuma, M.; Ohue, H. Synthesis, characterization and photocatalytic activity of N-doped TiO2 modified by platinum chloride. Catal. Today 2008, 132, 165–169. [Google Scholar] [CrossRef]
- Ahmed, L.M.; Ivanova, I.; Hussein, F.H.; Bahnemann, D.W. Role of Platinum Deposited on TiO2 in Photocatalytic Methanol Oxidation and Dehydrogenation Reactions. Int. J. Photoenergy 2014, 2014, 503516. [Google Scholar] [CrossRef]
- Wicaksana, Y.; Liu, S.; Scott, J.; Amal, R. Tungsten Trioxide as a Visible Light Photocatalyst for Volatile Organic Carbon Removal. Molecules 2014, 19, 17747–17762. [Google Scholar] [CrossRef]
- Aminian, M.K.; Ye, J. Morphology influence on photocatalytic activity of tungsten oxide loaded by platinum nanoparticles. J. Mater. Res. 2010, 25, 141–148. [Google Scholar] [CrossRef]
- Pellegrino, F.; Sordello, F.; Minella, M.; Minero, C.; Maurino, V. The Role of Surface Texture on the Photocatalytic H2 Production on TiO2. Catalysts 2019, 9, 32. [Google Scholar] [CrossRef]
- Jovic, V.; Al-Azri, Z.H.N.; Chen, W.-T.; Sun-Waterhouse, D.; Idriss, H.; Waterhouse, G.I.N. Photocatalytic H2 Production from Ethanol–Water Mixtures Over Pt/TiO2 and Au/TiO2 Photocatalysts: A Comparative Study. Top Catal 2013, 56, 1139–1151. [Google Scholar] [CrossRef]
- Nakabayashi, S.; Fujishima, A.; Honda, K. Experimental-Evidence for the Hydrogen Evolution Site in Photocatalytic Process on Pt/TiO2. Chem. Phys. Lett. 1983, 102, 464–465. [Google Scholar] [CrossRef]
- Kohtani, S.; Kawashima, A.; Miyabe, H. Reactivity of Trapped and Accumulated Electrons in Titanium Dioxide Photocatalysis. Catalysts 2017, 7, 303. [Google Scholar] [CrossRef]
- Bard, A.J.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution; Marcel Dekker: New York, NJ, USA; Basel, Switzerland, 1985. [Google Scholar]
- Abe, R.; Takami, H.; Murakami, N.; Ohtani, B. Pristine Simple Oxide Photocatalysts as Visible Light Driven Photocatalysts: Highly Efficient Decomposition of Organic Compounds over Platinum-loaded Tungsten Oxide. J. Am. Chem. Soc. 2008, 130, 7780–7781. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, B.; Prieto-Mahaney, O.O.; Li, D.; Abe, R. What is Degussa (Evonik) P25? Crystalline composition analysis, reconstruction from isolated pure particles and photocatalytic activity test. J. Photochem. Photobiol. A Chem. 2010, 216, 179–182. [Google Scholar] [CrossRef]
- Naderi, M. Surface Area: Brunauer–Emmett–Teller (BET). In Progress in Filtration and Separation; Academic Press: Cambridge, MA, USA, 2015; pp. 585–608. [Google Scholar]
- Ikeda, S.; Sugiyama, N.; Murakami, S.-Y.; Kominami, H.; Kera, Y.; Noguchi, H.; Uosaki, K.; Torimoto, T.; Ohtani, B. Quantitative Analysis of Defective Sites in Titanium(IV) Oxide Photocatalyst Powders. Phys. Chem. Chem. Phys. 2003, 5, 778–783. [Google Scholar] [CrossRef]
- Yan, X.; Ohno, T.; Nishijima, K.; Abe, R.; Ohtani, B. Is methylene blue an appropriate substrate for a photocatalytic activity test? A study with visible-light responsive titania. Chem. Phys. Lett. 2006, 429, 606–610. [Google Scholar] [CrossRef]
- Ohtani, B. Hidden but Possibly Fatal Misconceptions in Photocatalysis Studies: A Short Critical Review. Catalysts 2016, 6, 192–197. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Composition a (%) | SSA b | Pore Volume c | dETd | dET/SSA | ECBBe | Photocatalytic Activity f | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | R | NC | (a) CO2 | (b1) H2 | (b2) H2 (Pt) g | ||||||
| 0/TiO2 | 83.5 | 2.2 | 14.3 | 124 | 0.291 | 110 | 0.90 | 3.16 | 22 | 3 | 442 |
| 1Cu | 81.0 | 0.0 | 19.0 | 130 | 0.328 | 90 | 0.69 | 3.05 | 29 | 41 | 76 |
| 1Ce | 90.0 | 1.4 | 8.6 | 140 | 0.355 | 90 | 0.64 | 2.95 | 20 | 5 | 301 |
| 1Cu/1Ce | 84.9 | 4.8 | 10.3 | 148 | 0.400 | 109 | 0.72 | 3.00 | 22 | 24 | 68 |
| 1Cu/2Ce | 79.3 | 1.0 | 19.7 | 154 | 0.414 | 79 | 0.51 | 2.88 | 20 | 17 | 55 |
| 1Cu/3Ce | 81.6 | 0.1 | 18.3 | 152 | 0.403 | 63 | 0.41 | 2.95 | 16 | 23 | 39 |
| 1Pt | 79.7 | 5.5 | 14.8 | 116 | 0.287 | 27 | 0.23 | 3.08 | 120 | 352 | 316 |
| Samples | Atomic Percentage of Each Element (%) | ||||
|---|---|---|---|---|---|
| O | Ti | Cu | Ce | Pt | |
| 0/TiO2 | 81.46 | 18.54 | - | - | - |
| 1Cu | 77.30 | 21.20 | 1.50 | - | - |
| 1Ce | 80.18 | 19.71 | - | 0.11 | - |
| 1Cu/1Ce | 80.94 | 17.15 | 1.79 | 0.11 | - |
| 1Cu/2Ce | 80.54 | 15.85 | 3.48 | 0.14 | - |
| 1Cu/3Ce | 80.45 | 16.31 | 3.00 | 0.25 | - |
| 1Pt | 80.17 | 19.76 | - | - | 0.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unwiset, P.; Chen, G.; Ohtani, B.; Chanapattharapol, K.C. Correlation of the Photocatalytic Activities of Cu, Ce and/or Pt-Modified Titania Particles with their Bulk and Surface Structures Studied by Reversed Double-Beam Photoacoustic Spectroscopy. Catalysts 2019, 9, 1010. https://doi.org/10.3390/catal9121010
Unwiset P, Chen G, Ohtani B, Chanapattharapol KC. Correlation of the Photocatalytic Activities of Cu, Ce and/or Pt-Modified Titania Particles with their Bulk and Surface Structures Studied by Reversed Double-Beam Photoacoustic Spectroscopy. Catalysts. 2019; 9(12):1010. https://doi.org/10.3390/catal9121010
Chicago/Turabian StyleUnwiset, Preeya, Guangyi Chen, Bunsho Ohtani, and Kingkaew Chayakul Chanapattharapol. 2019. "Correlation of the Photocatalytic Activities of Cu, Ce and/or Pt-Modified Titania Particles with their Bulk and Surface Structures Studied by Reversed Double-Beam Photoacoustic Spectroscopy" Catalysts 9, no. 12: 1010. https://doi.org/10.3390/catal9121010
APA StyleUnwiset, P., Chen, G., Ohtani, B., & Chanapattharapol, K. C. (2019). Correlation of the Photocatalytic Activities of Cu, Ce and/or Pt-Modified Titania Particles with their Bulk and Surface Structures Studied by Reversed Double-Beam Photoacoustic Spectroscopy. Catalysts, 9(12), 1010. https://doi.org/10.3390/catal9121010