Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel




Abstract
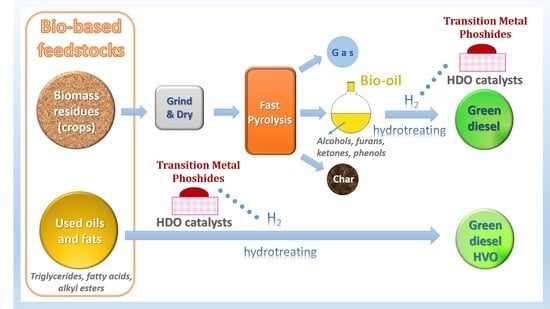
:
1. Introduction
2. Metal Phosphides and Catalysis
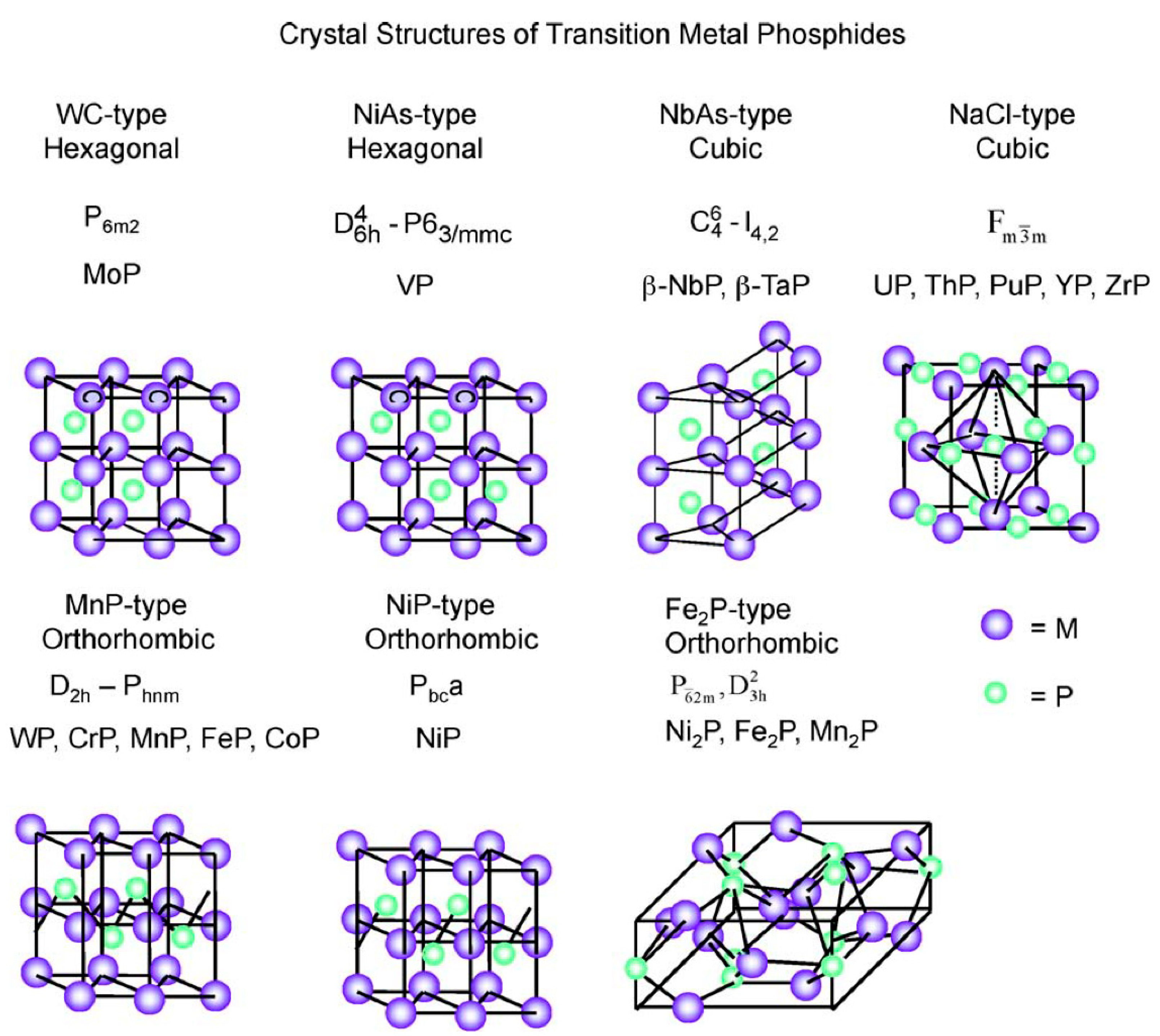
2.1. Metal Phosphide Structures
2.2. Preparation of Transition Metal Phosphides
2.2.1. Solution–Phase Reactions
2.2.2. Gas–Solid Reactions
2.2.3. Solvothermal Reduction Reactions with Phosphorous
2.2.4. Other Synthetic Methods
2.3. Catalytic Applications of Transition Metal Phosphides
2.3.1. Hydrogenation and Hydrotreatments
2.3.2. Electrochemical Performance
2.3.3. Photocatalysis
3. Green Diesel Production through the HDO of Nonedible Oils
3.1. Feed Source and Composition: Model Compounds as Reactants
3.2. Hydrotreatment Reactions
3.3. Catalysts for Green Diesel Production
3.4. HDO Performance Using Transition Metal Phosphide Catalysts
3.5. Reaction Network
3.6. Deactivation and Regeneration of HDO Catalysts
4. Catalysts Based on Metal Phosphides for HDO
4.1. Active Phases
4.2. Influence of Metal–Phosphorus Ratio: Role of Phosphorous
4.3. Support Types and Influence on Reactivity
4.4. Promoters
5. General Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- DIRECTIVE (EU) 2018/2001 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 11 December 2018 on the Promotion of the Use of Energy from Renewable Sources (recast), page L 328/83. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32018L2001&from=EN (accessed on 28 February 2019).
- Alleman, T.L.; McCormick, R.L.; Christensen, E.D.; Fioroni, G.; Moriarty, K.; Yanowitz, J. Biodiesel Handling and Use Guidelines, 5th ed.; DOE/GO-102016-4875; Clean Cities Publications; U.S. Department of Energy: Washington, DC, USA, 2016.
- Hilbers, T.J.; Sprakel, L.M.J.; van den Enk, L.B.J.; Zaalberg, B.; van den Berg, H.; van der Ham, L.G.J. Green Diesel from Hydrotreated Vegetable Oil Process Design Study. Chem. Eng. Technol. 2015, 38, 651–657. [Google Scholar] [CrossRef]
- Hannu Aatola, M.L.; Sarjovaara, T.; Mikkonen, S. Hydrotreated Vegetable Oil (HVO) as a Renewable Diesel Fuel: Trade-off between NOx, Particulate Emission, and Fuel Consumption of a Heavy Duty Engine; SAE International: Warrendale, PA, USA, 2008. [Google Scholar]
- Czernik, S.; Bridgwater, A.V. Overview of applications of biomass fast pyrolysis oil. Energy Fuels 2004, 18, 590–598. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Prins, W. Fast pyrolysis technology development. Biofuels Bioprod. Biorefin. 2010, 4, 178–208. [Google Scholar] [CrossRef]
- Wang, Y.X.; He, T.; Liu, K.T.; Wu, J.H.; Fang, Y.M. From biomass to advanced bio-fuel by catalytic pyrolysis/hydro-processing: Hydrodeoxygenation of bio-oil derived from biomass catalytic pyrolysis. Bioresour. Technol. 2012, 108, 280–284. [Google Scholar] [CrossRef]
- Zhang, J.W.; Matsubara, K.; Yun, G.N.; Zheng, H.; Takagaki, A.; Kikuchi, R.; Oyama, S.T. Comparison of phosphide catalysts prepared by temperature-programmed reduction and liquid-phase methods in the hydrodeoxygenation of 2-methylfuran. Appl. Catal. A-Gen. 2017, 548, 39–46. [Google Scholar] [CrossRef]
- Resasco, D.E. What Should We Demand from the Catalysts Responsible for Upgrading Biomass Pyrolysis Oil? J. Phys. Chem. Lett 2011, 2, 2294–2295. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A-Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Arun, N.; Sharma, R.V.; Dalai, A.K. Green diesel synthesis by hydrodeoxygenation of bio-based feedstocks: Strategies for catalyst design and development. Renew. Sustain. Energy Rev. 2015, 48, 240–255. [Google Scholar] [CrossRef]
- Carenco, S.; Portehault, D.; Boissiere, C.; Mezailles, N.; Sanchez, C. Nanoscaled Metal Borides and Phosphides: Recent Developments and Perspectives. Chem. Rev. 2013, 113, 7981–8065. [Google Scholar] [CrossRef]
- Ruddy, D.A.; Schaidle, J.A.; Ferrell, J.R.; Wang, J.; Moens, L.; Hensley, J.E. Recent advances in heterogeneous catalysts for bio-oil upgrading via “ex situ catalytic fast pyrolysis”: Catalyst development through the study of model compounds. Green Chem. 2014, 16, 454–490. [Google Scholar] [CrossRef]
- Oyama, S.T.; Wang, X.; Lee, Y.K.; Chun, W.J. Active phase of Ni2P/SiO2 in hydroprocessing reactions. J. Catal. 2004, 221, 263–273. [Google Scholar] [CrossRef]
- Oyama, S.T.; Wang, X.; Lee, Y.K.; Bando, K.; Requejo, F.G. Effect of phosphorus content in nickel phosphide catalysts studied by XAFS and other techniques. J. Catal. 2002, 210, 207–217. [Google Scholar] [CrossRef]
- Stinner, C.; Prins, R.; Weber, T. Binary and ternary transition-metal phosphides as HDN catalysts. J. Catal. 2001, 202, 187–194. [Google Scholar] [CrossRef]
- Alexander, A.M.; Hargreaves, J.S. Alternative catalytic materials: Carbides, nitrides, phosphides and amorphous boron alloys. Chem. Soc. Rev. 2010, 39, 4388–4401. [Google Scholar] [CrossRef]
- Prins, R.; Bussell, M.E. Metal Phosphides: Preparation, Characterization and Catalytic Reactivity. Catal. Lett. 2012, 142, 1413–1436. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, B. Recent advances in transition metal phosphide nanomaterials: Synthesis and applications in hydrogen evolution reaction. Chem. Soc. Rev. 2016, 45, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Cheng, Y.; Chen, J.; Smith, W.; Dong, P.; Ajayan, P.M.; Ye, M.; Shen, J. Recent developments of transition metal phosphides as catalysts in the energy conversion field. J. Mater. Chem. A 2018, 6, 23220–23243. [Google Scholar] [CrossRef]
- Oyama, S.T.; Gott, T.; Zhao, H.; Lee, Y.-K. Transition metal phosphide hydroprocessing catalysts: A review. Catal. Today 2009, 143, 94–107. [Google Scholar] [CrossRef]
- Oyama, S.T. Novel catalysts for advanced hydroprocessing: Transition metal phosphides. J. Catal. 2003, 216, 343–352. [Google Scholar] [CrossRef]
- Pöttgen, R.; Hönle, W.; von Schnering, H.G. Phosphides: Solid-State Chemistry. Encycl. Inorga. Bioinorg. Chem. 2011. [Google Scholar] [CrossRef]
- Aronsson, B.; Lundstrom, T.; Rundqvist, S. Borides, Silicides and Phosphides: A Critical Review of Their Preparation, Properties and Crystal Chemistry; Wiley: Methuen, MA, USA; New York, NY, USA, 1965. [Google Scholar]
- Habas, S.E.; Baddour, F.G.; Ruddy, D.A.; Nash, C.P.; Wang, J.; Pan, M.; Hensley, J.E.; Schaidle, J.A. A Facile Molecular Precursor Route to Metal Phosphide Nanoparticles and Their Evaluation as Hydrodeoxygenation Catalysts. Chem. Mater. 2015, 27, 7580–7592. [Google Scholar] [CrossRef]
- Roberts, E.J.; Habas, S.E.; Wang, L.; Ruddy, D.A.; White, E.A.; Baddour, F.G.; Griffin, M.B.; Schaidle, J.A.; Malmstadt, N.; Brutchey, R.L. High-Throughput Continuous Flow Synthesis of Nickel Nanoparticles for the Catalytic Hydrodeoxygenation of Guaiacol. ACS Sustain. Chem. Eng. 2017, 5, 632–639. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A. A novel method for preparing an active nickel phosphide catalyst for HDS of dibenzothiophene. J. Catal. 2009, 263, 4–15. [Google Scholar] [CrossRef]
- Clark, P.; Li, W.; Oyama, S.T. Synthesis and activity of a new catalyst for hydroprocessing: Tungsten phosphide. J. Catal. 2001, 200, 140–147. [Google Scholar] [CrossRef]
- Bui, P.; Cecilia, J.A.; Oyama, S.T.; Takagaki, A.; Infantes-Molina, A.; Zhao, H.; Li, D.; Rodríguez-Castellón, E.; Jiménez López, A. Studies of the synthesis of transition metal phosphides and their activity in the hydrodeoxygenation of a biofuel model compound. J. Catal. 2012, 294, 184–198. [Google Scholar] [CrossRef]
- Berenguer, A.; Sankaranarayanan, T.M.; Gomez, G.; Moreno, I.; Coronado, J.M.; Pizarro, P.; Serrano, D.P. Evaluation of transition metal phosphides supported on ordered mesoporous materials as catalysts for phenol hydrodeoxygenation. Green Chem. 2016, 18, 1938–1951. [Google Scholar] [CrossRef]
- Qian, X.F.; Zhang, X.M.; Wang, C.; Wang, W.Z.; Qian, Y.T. A New Way to Prepare Nanocrystalline Dinickel Phosphide. Mater. Res. Bull. 1998, 33, 669–672. [Google Scholar] [CrossRef]
- Qian, X.F.; Xie, Y.; Qian, Y.T.; Zhang, X.M.; Wang, W.Z.; Yang, L. Organo-thermal preparation of nanocrystalline cobalt phosphides. Mater. Sci. Eng. B 1997, 49, 135–137. [Google Scholar] [CrossRef]
- Yunle, G.; Fan, G.; Yitai, Q.; Huagui, Z.; Ziping, Y. A solvothermal synthesis of ultra-fine iron phosphide. Mater. Res. Bull. 2002, 37, 1101–1105. [Google Scholar] [CrossRef]
- Barry, B.M.; Gillan, E.G. Low-Temperature Solvothermal Synthesis of Phosphorus-Rich Transition-Metal Phosphides. Chem. Mater. 2008, 20, 2618–2620. [Google Scholar] [CrossRef]
- Ann Aitken, J.; Ganzha-Hazen, V.; Brock, S.L. Solvothermal syntheses of Cu3P via reactions of amorphous red phosphorus with a variety of copper sources. J. Solid State Chem. 2005, 178, 970–975. [Google Scholar] [CrossRef]
- Xie, Y.; Su, H.L.; Qian, X.F.; Liu, X.M.; Qian, Y.T. A Mild One-Step Solvothermal Route to Metal Phosphides (Metal=Co, Ni, Cu). J. Solid State Chem. 2000, 149, 88–91. [Google Scholar] [CrossRef]
- Saadi, F.H.; Carim, A.I.; Verlage, E.; Hemminger, J.C.; Lewis, N.S.; Soriaga, M.P. CoP as an Acid-Stable Active Electrocatalyst for the Hydrogen-Evolution Reaction: Electrochemical Synthesis, Interfacial Characterization and Performance Evaluation. J. Phys. Chem. C 2014, 118, 29294–29300. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Feng, Y.; Zhang, F.; Yang, C.; Yao, Z.; Zhao, W.; Qiu, F.; Yang, L.; Yao, Y.; Zhuang, X.; et al. Metal-Phosphide-Containing Porous Carbons Derived from an Ionic-Polymer Framework and Applied as Highly Efficient Electrochemical Catalysts for Water Splitting. Adv. Funct. Mater. 2015, 25, 3899–3906. [Google Scholar] [CrossRef]
- Zou, X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef]
- Muetterties, E.L.; Sauer, J.C. Catalytic properties of metal phosphides. Qualitative assay of catalytic properties. I. J. Am. Chem. Soc. 1974, 96, 3410–3415. [Google Scholar] [CrossRef]
- Nozaki, F.; Adachi, R. Chemical composition of the catalyst prepared by reduction of nickel orthophosphate in hydrogen and catalytic activity for partial hydrogenation of 1,3-butadiene. J. Catal. 1975, 40, 166–172. [Google Scholar] [CrossRef]
- Nozaki, F.; Kitoh, T.; Sodesawa, T. Promoting effect of oxygen for hydrogenation of butadiene over Ni2P catalyst. J. Catal. 1980, 62, 286–293. [Google Scholar] [CrossRef]
- Yang, P.; Jiang, Z.; Ying, P.; Li, C. Effect of surface composition on the catalytic performance of molybdenum phosphide catalysts in the hydrogenation of acetonitrile. J. Catal. 2008, 253, 66–73. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Q.; Guan, J.; He, D.; Hu, H.; Liang, C. Hydrogenation of naphthalene on nickel phosphide supported on silica. Asia-Pac. J. Chem. Eng. 2009, 4, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Oyama, S.T. Bifunctional nature of a SiO2-supported Ni2P catalyst for hydrotreating: EXAFS and FTIR studies. J. Catal. 2006, 239, 376–389. [Google Scholar] [CrossRef]
- Oyama, S.T.; Lee, Y.K. The active site of nickel phosphide catalysts for the hydrodesulfurization of 4,6-DMDBT. J. Catal. 2008, 258, 393–400. [Google Scholar] [CrossRef]
- Wang, X.; Clark, P.; Oyama, S.T. Synthesis, Characterization, and Hydrotreating Activity of Several Iron Group Transition Metal Phosphides. J. Catal. 2002, 208, 321–331. [Google Scholar] [CrossRef]
- Hansen, M.H.; Stern, L.-A.; Feng, L.; Rossmeisl, J.; Hu, X. Widely available active sites on Ni2P for electrochemical hydrogen evolution—Insights from first principles calculations. Phys. Chem. Chem. Phys. 2015, 17, 10823–10829. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Peng, L.; Qian, Y.; Zhang, X.; Xie, Y.; Cha, J.J.; Yu, G. Dual Tuning of Ni–Co–A (A = P, Se, O) Nanosheets by Anion Substitution and Holey Engineering for Efficient Hydrogen Evolution. J. Am. Chem. Soc. 2018, 140, 5241–5247. [Google Scholar] [CrossRef] [PubMed]
- Mishra, I.K.; Zhou, H.; Sun, J.; Qin, F.; Dahal, K.; Bao, J.; Chen, S.; Ren, Z. Hierarchical CoP/Ni5P4/CoP microsheet arrays as a robust pH-universal electrocatalyst for efficient hydrogen generation. Energy Environ. Sci. 2018, 11, 2246–2252. [Google Scholar] [CrossRef]
- Kim, T.-S.; Song, H.J.; Kim, J.-C.; Ju, B.; Kim, D.-W. 3D Architectures of CoxP Using Silk Fibroin Scaffolds: An Active and Stable Electrocatalyst for Hydrogen Generation in Acidic and Alkaline Media. Small 2018, 14, 1801284. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Feng, L.; Liu, C.; Xing, W. Ni2P Makes Application of the PtRu Catalyst Much Stronger in Direct Methanol Fuel Cells. ChemSusChem 2015, 8, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Feng, L.; Jiang, K.; Xue, H.; Cai, W.-B.; Liu, C.; Xing, W. Pt–CoP/C as an alternative PtRu/C catalyst for direct methanol fuel cells. J. Mater. Chem. A 2016, 4, 18607–18613. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, S.; Key, J.; Nie, S.; Ma, S.; Shen, P.K. Facile synthesis of a molybdenum phosphide (MoP) nanocomposite Pt support for high performance methanol oxidation. Catal. Sci. Technol. 2017, 7, 5974–5981. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Y.; Hu, F.; Wang, Q. Urchin-like CoP Nanocrystals as Hydrogen Evolution Reaction and Oxygen Reduction Reaction Dual-Electrocatalyst with Superior Stability. Nano Lett. 2015, 15, 7616–7620. [Google Scholar] [CrossRef]
- Doan-Nguyen, V.V.T.; Zhang, S.; Trigg, E.B.; Agarwal, R.; Li, J.; Su, D.; Winey, K.I.; Murray, C.B. Synthesis and X-ray Characterization of Cobalt Phosphide (Co2P) Nanorods for the Oxygen Reduction Reaction. ACS Nano 2015, 9, 8108–8115. [Google Scholar] [CrossRef]
- Zhong, X.; Jiang, Y.; Chen, X.; Wang, L.; Zhuang, G.; Li, X.; Wang, J.-G. Integrating cobalt phosphide and cobalt nitride-embedded nitrogen-rich nanocarbons: High-performance bifunctional electrocatalysts for oxygen reduction and evolution. J. Mater. Chem. A 2016, 4, 10575–10584. [Google Scholar] [CrossRef]
- Dong, Y.; Kong, L.; Jiang, P.; Wang, G.; Zhao, N.; Zhang, H.; Tang, B. A General Strategy To Fabricate NixP as Highly Efficient Cocatalyst via Photoreduction Deposition for Hydrogen Evolution. ACS Sustain. Chem. Eng. 2017, 5, 6845–6853. [Google Scholar] [CrossRef]
- Shen, R.; Xie, J.; Lu, X.; Chen, X.; Li, X. Bifunctional Cu3P Decorated g-C3N4 Nanosheets as a Highly Active and Robust Visible-Light Photocatalyst for H2 Production. ACS Sustain. Chem. Eng. 2018, 6, 4026–4036. [Google Scholar] [CrossRef]
- Wang, P.; Zhan, S.; Wang, H.; Xia, Y.; Hou, Q.; Zhou, Q.; Li, Y.; Kumar, R.R. Cobalt phosphide nanowires as efficient co-catalyst for photocatalytic hydrogen evolution over Zn0.5Cd0.5S. Appl. Catal. B Environ. 2018, 230, 210–219. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.; Dong, Y.; Jiang, P. Noble-Metal-Free Iron Phosphide Cocatalyst Loaded Graphitic Carbon Nitride as an Efficient and Robust Photocatalyst for Hydrogen Evolution under Visible Light Irradiation. ACS Sustain. Chem. Eng. 2017, 5, 8053–8060. [Google Scholar] [CrossRef]
- Bi, L.; Gao, X.; Zhang, L.; Wang, D.; Zou, X.; Xie, T. Enhanced Photocatalytic Hydrogen Evolution of NiCoP/g-C3N4 with Improved Separation Efficiency and Charge Transfer Efficiency. ChemSusChem 2018, 11, 276–284. [Google Scholar] [CrossRef]
- Gunstone, F.D.; Harwood, J.L.; Dijkstra, A.J. The Lipid Handbook; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal triacylglycerols as feedstocks for biofuel production: Perspectives and advances. Plant J. 2008, 54, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Austin, A. Boeing planes successfully fly with biofuels. Biodiesel Magazine, 15 January 2009. [Google Scholar]
- Sotelo-Boyás, R.; Trejo-Zárragaand, F.; de Jesús Hernández-Loyo, F. Hydroconversion of Triglycerides into Green Liquid Fuels (Chapter 8) in Hydrogenation (Iyad Karamé ed.); IntechOpen: London, UK, 2012. [Google Scholar]
- Zeman, N. Neste Oil starts construction on Europe’s largest renewable fuels plant. Biodiesel Magazine, 27 May 2009. [Google Scholar]
- Izadifar, M.; Jahromi, M.Z. Application of genetic algorithm for optimization of vegetable oil hydrogenation process. J. Food Eng. 2007, 78, 1–8. [Google Scholar] [CrossRef]
- US Department of Energy. Energy Efficiency and Renewable Energy. Available online: https://afdc.energy.gov/fuels/biodiesel_production.html (accessed on 28 February 2019).
- Alvarez-Galvan, M.C.; Blanco-Brieva, G.; Capel-Sanchez, M.; Morales-delaRosa, S.; Campos-Martin, J.M.; Fierro, J.L.G. Metal phosphide catalysts for the hydrotreatment of non-edible vegetable oils. Catal. Today 2018, 302, 242–249. [Google Scholar] [CrossRef]
- Satyarthi, J.K.; Chiranjeevi, T.; Gokak, D.T.; Viswanathan, P.S. An overview of catalytic conversion of vegetable oils/fats into middle distillates. Catal. Sci. Technol. 2013, 3, 70–80. [Google Scholar] [CrossRef]
- Ali, M.F.; el Ali, B.M.; Speight, J.G. Handbook of Industrial Chemistry: Organic Chemicals; Mc Graw-Hill: New York, NY, USA, 2005. [Google Scholar]
- Ancheyta, J.; Trejo, F.; Rana, M.S. Asphaltenes: Chemical Transformation during Hydroprocessing of Heavy Oils; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Sotelo-Boyas, R.; Liu, Y.Y.; Minowa, T. Renewable Diesel Production from the Hydrotreating of Rapeseed Oil with Pt/Zeolite and NiMo/Al2O3 Catalysts. Ind. Eng. Chem. Res. 2011, 50, 2791–2799. [Google Scholar] [CrossRef]
- Li, K.; Wang, R.; Chen, J. Hydrodeoxygenation of Anisole over Silica-Supported Ni2P, MoP, and NiMoP Catalysts. Energy Fuels 2011, 25, 854–863. [Google Scholar] [CrossRef]
- Duan, X.P.; Teng, Y.; Wang, A.J.; Kogan, V.M.; Li, X.; Wang, Y. Role of sulfur in hydrotreating catalysis over nickel phosphide. J. Catal. 2009, 261, 232–240. [Google Scholar] [CrossRef]
- He, Z.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Chem. 2012, 28–52. [Google Scholar] [CrossRef]
- Bowker, R.H.; Smith, M.C.; Pease, M.L.; Slenkamp, K.M.; Kovarik, L.; Bussell, M.E. Synthesis and Hydrodeoxygenation Properties of Ruthenium Phosphide Catalysts. ACS Catal. 2011, 1, 917–922. [Google Scholar] [CrossRef]
- Hicks, J.C. Advances in C-O Bond Transformations in Lignin-Derived Compounds for Biofuels Production. J. Phys. Chem. Lett. 2011, 2, 2280–2287. [Google Scholar] [CrossRef]
- Talukdar, A.K.; Bhattacharyya, K.G.; Sivasanker, S. Hydrogenation of phenol over supported platinum and palladium catalysts. Appl. Catal. A-Gen. 1993, 96, 229–239. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Li, D.; Bui, P.; Oyama, S.T. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts. Appl. Catal. A Gen. 2011, 391, 305–310. [Google Scholar] [CrossRef]
- Chen, J.; Shi, H.; Li, L.; Li, K. Deoxygenation of methyl laurate as a model compound to hydrocarbons on transition metal phosphide catalysts. Appl. Catal. B Environ. 2014, 144, 870–884. [Google Scholar] [CrossRef]
- Yang, Y.; Ochoa-Hernández, C.; Pizarro, P.; de la Peña O’Shea, V.A.; Coronado, J.M.; Serrano, D.P. Influence of the Ni/P ratio and metal loading on the performance of NixPy/SBA-15 catalysts for the hydrodeoxygenation of methyl oleate. Fuel 2015, 144, 60–70. [Google Scholar] [CrossRef]
- Stinner, C.; Tang, Z.; Haouas, M.; Weber, T.; Prins, R. Preparation and P-31 NMR characterization of nickel phosphides on silica. J. Catal. 2002, 208, 456–466. [Google Scholar] [CrossRef]
- Shi, H.; Chen, J.X.; Yang, Y.; Tian, S.S. Catalytic deoxygenation of methyl laurate as a model compound to hydrocarbons on nickel phosphide catalysts: Remarkable support effect. Fuel Process. Technol. 2014, 118, 161–170. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.X.; Shi, H. Deoxygenation of Methyl Laurate as a Model Compound to Hydrocarbons on Ni2P/SiO2, Ni2P/MCM-41, and Ni2P/SBA-15 Catalysts with Different Dispersions. Energy Fuels 2013, 27, 3400–3409. [Google Scholar] [CrossRef]
- Deliy, I.; Shamanaev, I.; Gerasimov, E.; Pakharukova, V.; Yakovlev, I.; Lapina, O.; Aleksandrov, P.; Bukhtiyarova, G. HDO of Methyl Palmitate over Silica-Supported Ni Phosphides: Insight into Ni/P Effect. Catalysts 2017, 7, 298. [Google Scholar] [CrossRef]
- Ruinart de Brimont, M.; Dupont, C.; Daudin, A.; Geantet, C.; Raybaud, P. Deoxygenation mechanisms on Ni-promoted MoS2 bulk catalysts: A combined experimental and theoretical study. J. Catal. 2012, 286, 153–164. [Google Scholar] [CrossRef]
- Guan, Q.; Wan, F.; Han, F.; Liu, Z.; Li, W. Hydrodeoxygenation of methyl palmitate over MCM-41 supported nickel phosphide catalysts. Catal. Today 2016, 259, 467–473. [Google Scholar] [CrossRef]
- Shamanaev, I.V.; Deliy, I.V.; Aleksandrov, P.V.; Gerasimov, E.Y.; Pakharukova, V.P.; Kodenev, E.G.; Ayupov, A.B.; Andreev, A.S.; Lapina, O.B.; Bukhtiyarova, G.A. Effect of precursor on the catalytic properties of Ni2P/SiO2 in methyl palmitate hydrodeoxygenation. RSC Adv. 2016, 6, 30372–30383. [Google Scholar] [CrossRef]
- Cheng, S.; Wei, L.; Zhao, X.; Julson, J. Application, Deactivation, and Regeneration of Heterogeneous Catalysts in Bio-Oil Upgrading. Catalysts 2016, 6, 195. [Google Scholar] [CrossRef]
- Twaiq, F.A.; Zabidi, N.A.M.; Bhatia, S. Catalytic Conversion of Palm Oil to Hydrocarbons: Performance of Various Zeolite Catalysts. Ind. Eng. Chem. Res. 1999, 38, 3230–3237. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, X.; Bai, T.; Chen, T.; Fan, W. Coking kinetics and influence of reaction-regeneration on acidity, activity and deactivation of Zn/HZSM-5 catalyst during methanol aromatization. J. Energy Chem. 2015, 24, 108–118. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A.; Takahashi, Y.; Nakamura, K. Water–gas-shift reaction on a Ni2P(001) catalyst: Formation of oxy-phosphides and highly active reaction sites. J. Catal. 2009, 262, 294–303. [Google Scholar] [CrossRef]
- Zhu, X.; Lobban, L.L.; Mallinson, R.G.; Resasco, D.E. Bifunctional transalkylation and hydrodeoxygenation of anisole over a Pt/HBeta catalyst. J. Catal. 2011, 281, 21–29. [Google Scholar] [CrossRef]
- Song, W.; Liu, Y.; Baráth, E.; Zhao, C.; Lercher, J.A. Synergistic effects of Ni and acid sites for hydrogenation and C–O bond cleavage of substituted phenols. Green Chem. 2015, 17, 1204–1218. [Google Scholar] [CrossRef]
- Taufiqurrahmi, N.; Mohamed, A.R.; Bhatia, S. Nanocrystalline zeolite beta and zeolite Y as catalysts in used palm oil cracking for the production of biofuel. J. Nanopart. Res. 2011, 13, 3177–3189. [Google Scholar] [CrossRef]
- Josl, R.; Klingmann, R.; Traa, Y.; Gläser, R.; Weitkamp, J. Regeneration of zeolite catalysts deactivated in isobutane/butene alkylation: An in situ FTIR investigation at elevated H2 pressure. Catal. Commun. 2004, 5, 239–241. [Google Scholar] [CrossRef]
- Ivanov, D.P.; Sobolev, V.I.; Panov, G.I. Deactivation by coking and regeneration of zeolite catalysts for benzene-to-phenol oxidation. Appl. Catal. A Gen. 2003, 241, 113–121. [Google Scholar] [CrossRef]
- Si, Z.; Zhang, X.; Wang, C.; Ma, L.; Dong, R. An Overview on Catalytic Hydrodeoxygenation of Pyrolysis Oil and Its Model Compounds. Catalysts 2017, 7, 169. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Wang, A.J.; Liu, S.; Yao, Y.L.; Sun, Z.C.; Li, X.; Liu, Y.Y.; Wang, Y.; Camaioni, D.M.; Lercher, J.A. Hydrodeoxygenation of phenolic compounds to cycloalkanes over supported nickel phosphides. Catal. Today 2019, 319, 48–56. [Google Scholar] [CrossRef]
- Guo, T.; Chen, J.X.; Li, K.L. Promotion Effect of Steam Treatment on Activity of Ni2P/SiO2 for Hydrodechlorination of Chlorobenzene. Chin. J. Catal. 2012, 33, 1080–1085. [Google Scholar] [CrossRef]
- Li, D.; Bui, P.; Zhao, H.Y.; Oyama, S.T.; Dou, T.; Shen, Z.H. Rake mechanism for the deoxygenation of ethanol over a supported Ni2P/SiO2 catalyst. J. Catal. 2012, 290, 1–12. [Google Scholar] [CrossRef]
- Iino, A.; Cho, A.; Takagaki, A.; Kikuchi, R.; Oyama, S.T. Kinetic studies of hydrodeoxygenation of 2-methyltetrahydrofuran on a Ni2P/SiO2 catalyst at medium pressure. J. Catal. 2014, 311, 17–27. [Google Scholar] [CrossRef]
- Shafaghat, H.; Rezaei, P.S.; Daud, W. Effective parameters on selective catalytic hydrodeoxygenation of phenolic compounds of pyrolysis bio-oil to high-value hydrocarbons. RSC Adv. 2015, 5, 103999–104042. [Google Scholar] [CrossRef]
- Robinson, A.M.; Hensley, J.E.; Medlin, J.W. Bifunctional Catalysts for Upgrading of Biomass-Derived Oxygenates: A Review. ACS Catal. 2016, 6, 5026–5043. [Google Scholar] [CrossRef]
- Peroni, M.; Lee, I.; Huang, X.; Barath, E.; Gutierrez, O.Y.; Lercher, J.A. Deoxygenation of Palmitic Acid on Unsupported Transition-Metal Phosphides. ACS Catal. 2017, 7, 6331–6341. [Google Scholar] [CrossRef]
- Li, X.; Luo, X.Y.; Jin, Y.B.; Li, J.Y.; Zhang, H.D.; Zhang, A.P.; Xie, J. Heterogeneous sulfur-free hydrodeoxygenation catalysts for selectively upgrading the renewable bio-oils to second generation biofuels. Renew. Sustain. Energy Rev. 2018, 82, 3762–3797. [Google Scholar] [CrossRef]
- Jain, V.; Bonita, Y.; Brown, A.; Taconi, A.; Hicks, J.C.; Rai, N. Mechanistic insights into hydrodeoxygenation of phenol on bimetallic phosphide catalysts. Catal. Sci. Technol. 2018, 8, 4083–4096. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A.; Oyama, S.T. Oxygen-removal of dibenzofuran as a model compound in biomass derived bio-oil on nickel phosphide catalysts: Role of phosphorus. Appl. Catal. B Environ. 2013, 136–137, 140–149. [Google Scholar] [CrossRef]
- Zhang, Z.N.; Tang, M.X.; Chen, J.X. Effects of P/Ni ratio and Ni content on performance of gamma-Al2O3-supported nickel phosphides for deoxygenation of methyl laurate to hydrocarbons. Appl. Surf. Sci. 2016, 360, 353–364. [Google Scholar] [CrossRef]
- Liu, Y.H.; Yao, L.; Xin, H.; Wang, G.S.; Li, D.; Hu, C.W. The production of diesel-like hydrocarbons from palmitic acid over HZSM-22 supported nickel phosphide catalysts. Appl. Catal. B-Environ. 2015, 174, 504–514. [Google Scholar] [CrossRef]
- Goncalves, V.O.O.; de Souza, P.M.; Cabioc’h, T.; da Silva, V.T.; Noronha, F.B.; Richard, F. Effect of P/Ni ratio on the performance of nickel phosphide phases supported on zirconia for the hydrodeoxygenation of m-cresol. Catal. Commun. 2019, 119, 33–38. [Google Scholar] [CrossRef]
- Koranyi, T.I.; Vit, Z.; Poduval, D.G.; Ryoo, R.; Kim, H.S.; Hensen, E.J.M. SBA-15-supported nickel phosphide hydrotreating catalysts. J. Catal. 2008, 253, 119–131. [Google Scholar] [CrossRef]
- Xin, H.; Guo, K.; Li, D.; Yang, H.Q.; Hu, C.W. Production of high-grade diesel from palmitic acid over activated carbon-supported nickel phosphide catalysts. Appl. Catal. B-Environ. 2016, 187, 375–385. [Google Scholar] [CrossRef]
- Wu, S.K.; Lai, P.C.; Lin, Y.C. Atmospheric Hydrodeoxygenation of Guaiacol over Nickel Phosphide Catalysts: Effect of Phosphorus Composition. Catal. Lett. 2014, 144, 878–889. [Google Scholar] [CrossRef]
- Guo, C.; Rao, K.T.V.; Yuan, Z.S.; He, S.; Rohani, S.; Xu, C.B. Hydrodeoxygenation of fast pyrolysis oil with novel activated carbon-supported NiP and CoP catalysts. Chem. Eng. Sci. 2018, 178, 248–259. [Google Scholar] [CrossRef]
- Pham, L.K.H.; Tran, T.T.V.; Kongparakul, S.; Reubroycharoen, P.; Kamjanakom, S.; Guan, G.Q.; Samart, C. Formation and activity of activated carbon supported Ni2P catalysts for atmospheric deoxygenation of waste cooking oil. Fuel Process. Technol. 2019, 185, 117–125. [Google Scholar] [CrossRef]
- Dierks, M.; Cao, Z.W.; Manayil, J.C.; Akilavasan, J.; Wilson, K.; Schuth, F.; Rinaldi, R. Impact of Hydrophobic Organohybrid Silicas on the Stability of Ni2P Catalyst Phase in the Hydrodeoxygenation of Biophenols. ChemCatChem 2018, 10, 2219–2231. [Google Scholar] [CrossRef]
- Ochoa-Hernández, C.; Yang, Y.; Pizarro, P.; de la Peña O’Shea, V.A.; Coronado, J.M.; Serrano, D.P. Hydrocarbons production through hydrotreating of methyl esters over Ni and Co supported on SBA-15 and Al-SBA-15. Catal. Today 2013, 210, 81–88. [Google Scholar] [CrossRef]
- Deliy, I.V.; Shamanaev, I.V.; Aleksandrov, P.V.; Gerasimov, E.Y.; Pakharukova, V.P.; Kodenev, E.G.; Yakovlev, I.V.; Lapina, O.B.; Bukhtiyarova, G.A. Support Effect on the Performance of Ni2P Catalysts in the Hydrodeoxygenation of Methyl Palmitate. Catalysts 2018, 8, 515. [Google Scholar] [CrossRef]
- Yang, Y.; Ochoa-Hernández, C.; de la Peña O’Shea, V.A.; Coronado, J.M.; Serrano, D.P. Ni2P/SBA-15 As a Hydrodeoxygenation Catalyst with Enhanced Selectivity for the Conversion of Methyl Oleate Inton-Octadecane. ACS Catal. 2012, 2, 592–598. [Google Scholar] [CrossRef]
- Zhu, T.H.; Song, H.; Dai, X.Y.; Song, H.L. Preparation of Ni2P/Al-SBA-15 catalyst and its performance for benzofuran hydrodeoxygenation. Chin. J. Chem. Eng. 2017, 25, 1784–1790. [Google Scholar] [CrossRef]
- Berenguer, A.; Bennett, J.A.; Hunns, J.; Moreno, I.; Coronado, J.M.; Lee, A.F.; Pizarro, P.; Wilson, K.; Serrano, D.P. Catalytic hydrodeoxygenation of m-cresol over Ni2P/hierarchical ZSM-5. Catal. Today 2018, 304, 72–79. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Sanmartin-Donoso, J.; Rodriguez-Aguado, E.; Ballesteros-Plata, D.; Rodriguez-Castellon, E. Enhanced HDO activity of Ni2P promoted with noble metals. Catal. Sci. Technol. 2016, 6, 7323–7333. [Google Scholar] [CrossRef]
- Liu, X.G.; Li, Z.Y.; Zhang, B.Q.; Hu, M.C. Improvement of hydrodeoxygenation stability of nickel phosphide based catalysts by silica modification as structural promoter. Fuel 2017, 204, 144–151. [Google Scholar] [CrossRef]
- Cheng, R.H.; Shu, Y.Y.; Li, L.; Zheng, M.Y.; Wang, X.D.; Wang, A.Q.; Zhang, T. Synthesis and characterization of high surface area molybdenum phosphide. Appl. Catal. A-Gen. 2007, 316, 160–168. [Google Scholar] [CrossRef]
- Whiffen, V.M.L.; Smith, K.J.; Straus, S.K. The influence of citric acid on the synthesis and activity of high surface area MoP for the hydrodeoxygenation of 4-methylphenol. Appl. Catal. A-Gen. 2012, 419, 111–125. [Google Scholar] [CrossRef]
- Yang, Y.X.; Ochoa-Hernandez, C.; O’Shea, V.A.D.; Pizarro, P.; Coronado, J.M.; Serrano, D.P. Transition Metal Phosphide Nanoparticles Supported on SBA-15 as Highly Selective Hydrodeoxygenation Catalysts for the Production of Advanced Biofuels. J. Nanosci. Nanotechnol. 2015, 15, 6642–6650. [Google Scholar] [CrossRef]
- Shamanaev, I.V.; Deliy, I.V.; Gerasimov, E.Y.; Pakharukova, V.P.; Kodenev, E.G.; Aleksandrov, P.V.; Bukhtiyarova, G.A. Synergetic Effect of Ni2P/SiO2 and gamma-Al2O3 Physical Mixture in Hydrodeoxygenation of Methyl Palmitate. Catalysts 2017, 7, 329. [Google Scholar] [CrossRef]
- Shamanaev, I.V.; Deliy, I.V.; Pakharukova, V.P.; Gerasimov, E.Y.; Rogov, V.A.; Bukhtiyarova, G.A. Effect of the preparation conditions on the physicochemical and catalytic properties of Ni2P/SiO2 catalysts. Russ. Chem. Bull. 2015, 64, 2361–2370. [Google Scholar] [CrossRef]
- Peroni, M.; Mancino, G.; Baráth, E.; Gutiérrez, O.Y.; Lercher, J.A. Bulk and γ-Al2O3-supported Ni2P and MoP for hydrodeoxygenation of palmitic acid. Appl. Catal. B Environ. 2016, 180, 301–311. [Google Scholar] [CrossRef]
- Liu, S.Y.; Zhu, Q.Q.; Guan, Q.X.; He, L.N.; Li, W. Bio-aviation fuel production from hydroprocessing castor oil promoted by the nickel-based bifunctional catalysts. Bioresour. Technol. 2015, 183, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Phimsen, S.; Kiatkittipong, W.; Yamada, H.; Tagawa, T.; Kiatkittipong, K.; Laosiripojana, N.; Assabumrungrat, S. Oil extracted from spent coffee grounds for bio-hydrotreated diesel production. Energy Convers. Manag. 2016, 126, 1028–1036. [Google Scholar] [CrossRef]
- Zarchin, R.; Rabaev, M.; Vidruk-Nehemya, R.; Landau, M.V.; Herskowitz, M. Hydroprocessing of soybean oil on nickel-phosphide supported catalysts. Fuel 2015, 139, 684–691. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
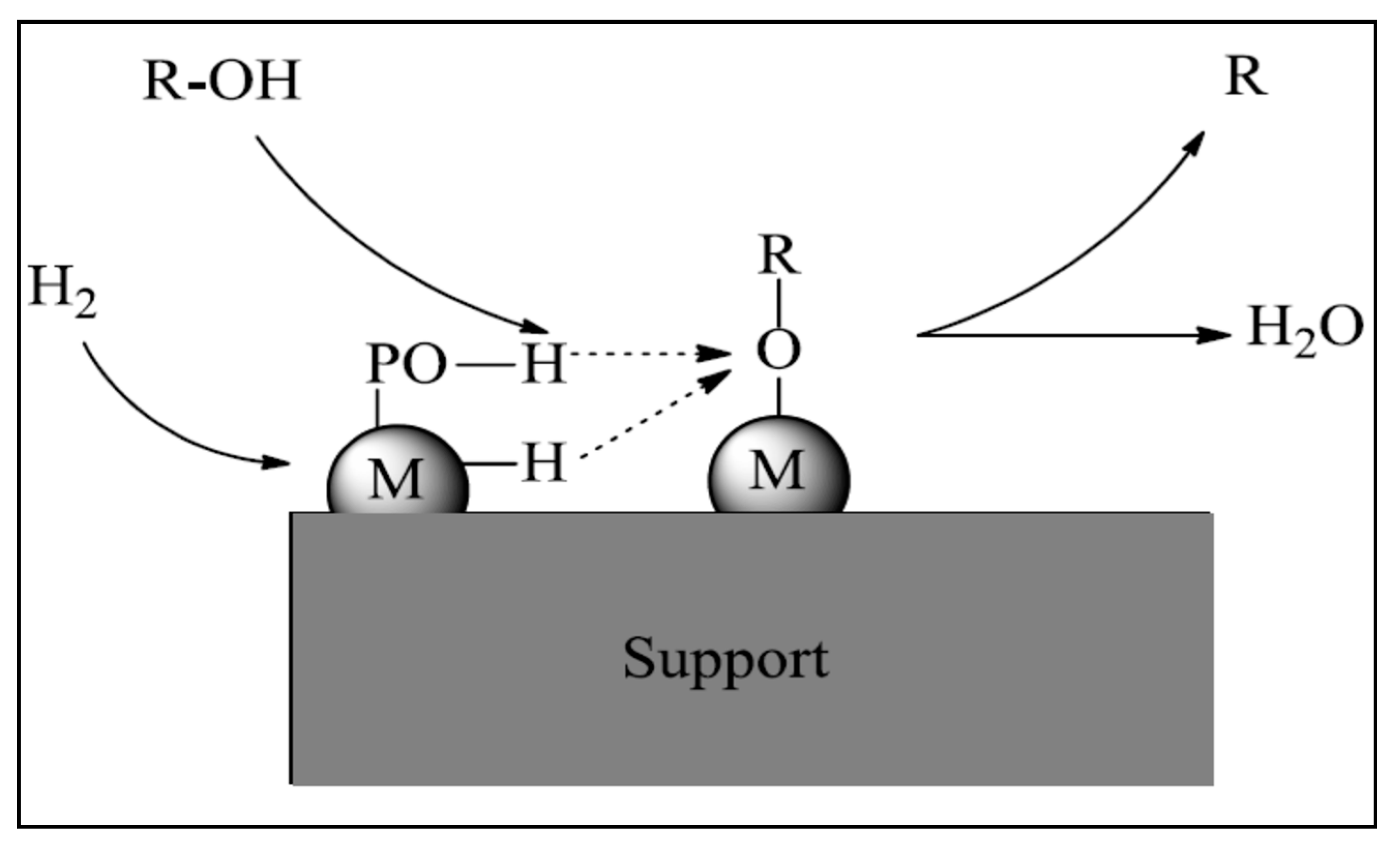{kind=link}
{kind=link}
{kind=link}
| Catalysts | Catalytic Application | Specific Process | Reactants | Ref. |
|---|---|---|---|---|
| TMPs/alumina | hydrogenation | acetylene | [40] | |
| Ni phosphide | butadiene | [41,42] | ||
| Mo phosphide | acetonitrile | [43] | ||
| Ni phosphide/silica | naphtalene | [44] | ||
| Ni2P/SiO2 | hydrotreatment | HDS, HDN, HDO | dibenzothiophene, quinoline, benzofuran | [14] |
| Co2P, Ni2P, MoP, WP, CoMoP, NiMoP, | HDN | [16] | ||
| TMPs | HDS/HDN | review | [18] | |
| TMPs | HDS/HDN | review | [21] | |
| Fe2P, CoP, MoP, WP, Ni2P supported on SiO2 | HDS/HDN | dibenzotiophene/quinoline | [22] | |
| WP | HDS/HDN | dibenzotiophene/quinoline | [28] | |
| Ni2P/SiO2 | HDS/HDN | dimethyldibenzotiophene, dimethylsulfide, quinoline | [45] | |
| Ni2P/SiO2 or MCM-41 | HDS | dimethyldibenzotiophene | [46] | |
| Fe2P, CoP, Ni2P/SiO2 | HDS/HDN | quinoline/dibenzotiophene/benzofuran/tetralin/tetradecane | [47] | |
| TMPs | electrochemistry | (HER, OER, methanol oxidation, photocatalysts for H2O splitting) | Review—energy conversion | [20] |
| Ni2P | HER | hydrogen | [48] | |
| Ni–Co–P | [49] | |||
| CoP/Ni5P4 | [50] | |||
| CoxP | [51] | |||
| CoP | [55] | |||
| CoP | OER | oxygen | [55] | |
| Co2P | [56] | |||
| Co phosphide | [57] | |||
| Ni2P | Methanol oxidation | methanol | [52] | |
| Pt–CoP/C | [53] | |||
| MoP | [54] | |||
| NixP | cocatalysts in photocatalysis | H2O splitting | H2O | [58] |
| Cu3P/g-C3N4 | [59] | |||
| Co phosphide/Zn0.5Cd0.5S | [60] | |||
| Fe phosphide/g-C3N4 | [61] | |||
| NiCoP/g-C3N4 | [62] |
| Reactant | Catalysts | Preparation Method | Reactor | Temp (°C) | P (bar) | Conversion/Select. | Ref. |
|---|---|---|---|---|---|---|---|
| Alkyl esters | |||||||
| Methyl laurate | Ni2P/SiO2, Ni2P/MCM-41, Ni2P/SBA-15 | phosphate | flow | 300–340 | 20 | Conv > 97% and select to C11+C12 > 99%, | [86] |
| Methyl laurate | Ni phosphide (initial Ni/P = 1)/SiO2, CeO2, TiO2, and SAPO-11, | phosphate | flow | 300–340 | 20 | Conv and select to C11+C12 alkanes close to 100% for Ni2P/SiO2 (340 °C) | [85] |
| Methyl oleate | NixPy/SBA-15 | phosphate | flow | 250–340 | 3–40 | For Ni/P = 1, 80% Conv, (30 bar, 290 °C) | [83] |
| Methyl oleate (70%) | (Fe, Co, Ni, and Mo) phosphides (M/P = 1, initial) dispersed on SBA-15 | phosphate | flow | 250 | 3–40 | Higher conversion for Ni2P/SBA-15 HDO selectivity with MoP/SBA-15 > 90% | [129] |
| Methyl palmitate | Ni2P/SiO2 catalyst, and in mixture with inert (SiC or SiO2) or acidic (γ-Al2O3) | phosphate | flow | 270–330 | 30 | Conv = 100% (310 °C) Ni2P/SiO2 (γ-Al2O3) | [130] |
| Methyl palmitate | Ni2P/SiO2 | phosphate | flow | 290 | 30 | Catalyst reduced at 600 °C, 6 h: 15 mol MP/h·mol Ni Select. C16 ~62% | [131] |
| Methyl palmitate | NiP/silica (Ni/P molar ratio 2/1, 1/1 and 1/2) | phosphate | flow | 290 | 30 | Ni2P: Conv close to 100% at the beginning (fast deactivation in 6 h) | [87] |
| Methyl palmitate | Ni2P/γ-Al2O3; Ni2P/SiO2 | phosphate and phosphite | flow | 250–330 | 30 | Conv = 95% (320 °C) with Ni2P/Al2O3 (phosphate) | [121] |
| Carboxylic acids | |||||||
| Palmitic acid | Ni2P and MoP bulk and supported on γ-Al2O3 | phosphate | flow | 180–300 | 40 | Conversion: 80% and 60% for supported Ni2P and MoP, respectively (300 °C) | [132] |
| Feed | Catalyst | Preparation Method | Reactor | Temp (°C) | P (bar) | Conv/Select. | Ref. |
|---|---|---|---|---|---|---|---|
| Castor oil | Ni2P/SAPO-11 | phosphite | Flow | 300 | 30 | Conv = 99% yield to C16-C19 = 92.3% | [133] |
| (Spent) coffee oil | NiP/alumina | phosphate | Batch | 375–425 | 20–40 | 77.4% conv. Yield to gasoline = 31.2%, Yield to diesel 33.8% (400 °C and 40 bar) | [134] |
| Fast pyrolysis oil (Commercial Alcohol Inc. Hardwood sawdust) | Ni or Co phosphide/activated carbon | phosphate | Batch | 300 | 50 | Greater yields for M/P = 3/2 | [117] |
| Soybean oil | Ni2P–Pd–SiO2/α-Al2O3 | Hydrothermal + phosphate method | Flow | 340 | Nearly 100% conv | [126] | |
| Soybean oil | Ni2P/SiO2 and Ni2P/HY | Phosphate method | Flow | 340–370 | 30 | Organic liquid yield, wt% >82% | [135] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-Galvan, M.C.; Campos-Martin, J.M.; Fierro, J.L.G. Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel. Catalysts 2019, 9, 293. https://doi.org/10.3390/catal9030293
Alvarez-Galvan MC, Campos-Martin JM, Fierro JLG. Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel. Catalysts. 2019; 9(3):293. https://doi.org/10.3390/catal9030293
Chicago/Turabian StyleAlvarez-Galvan, M. Consuelo, Jose M. Campos-Martin, and Jose L. G. Fierro. 2019. "Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel" Catalysts 9, no. 3: 293. https://doi.org/10.3390/catal9030293
APA StyleAlvarez-Galvan, M. C., Campos-Martin, J. M., & Fierro, J. L. G. (2019). Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel. Catalysts, 9(3), 293. https://doi.org/10.3390/catal9030293