The Impact of Synthesis Method on the Properties and CO2 Sorption Capacity of UiO-66(Ce)

 ,
,  , , and
, , and 
Abstract
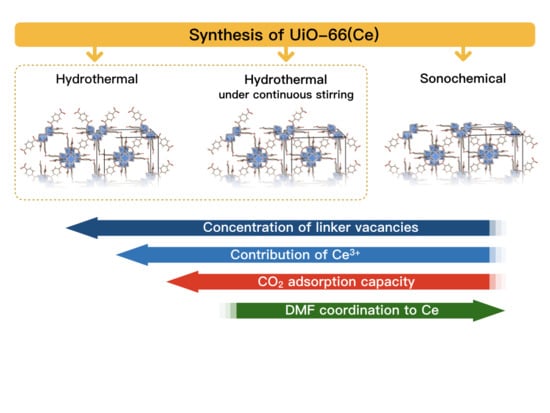
:
1. Introduction
2. Results and Discussion
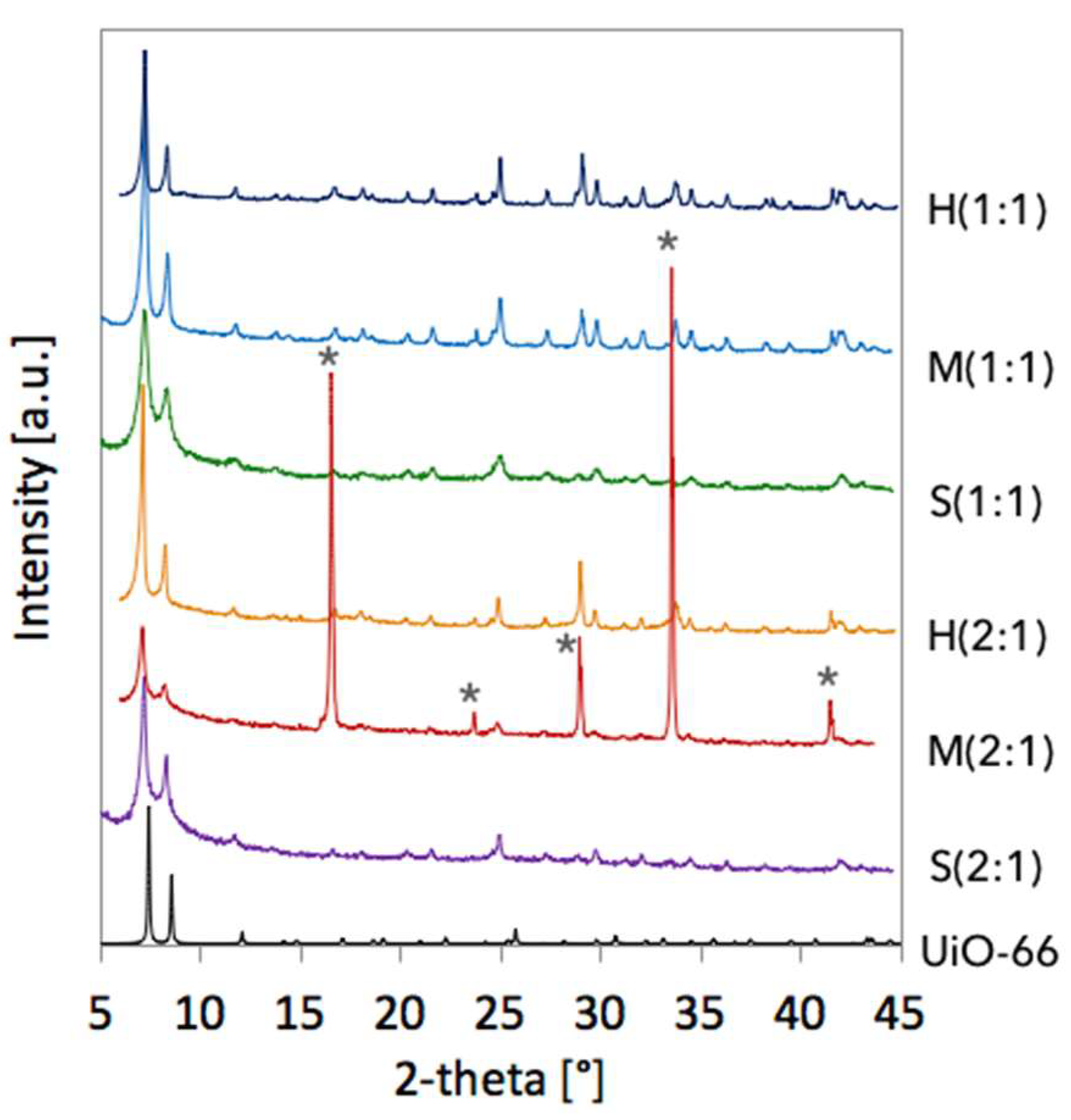
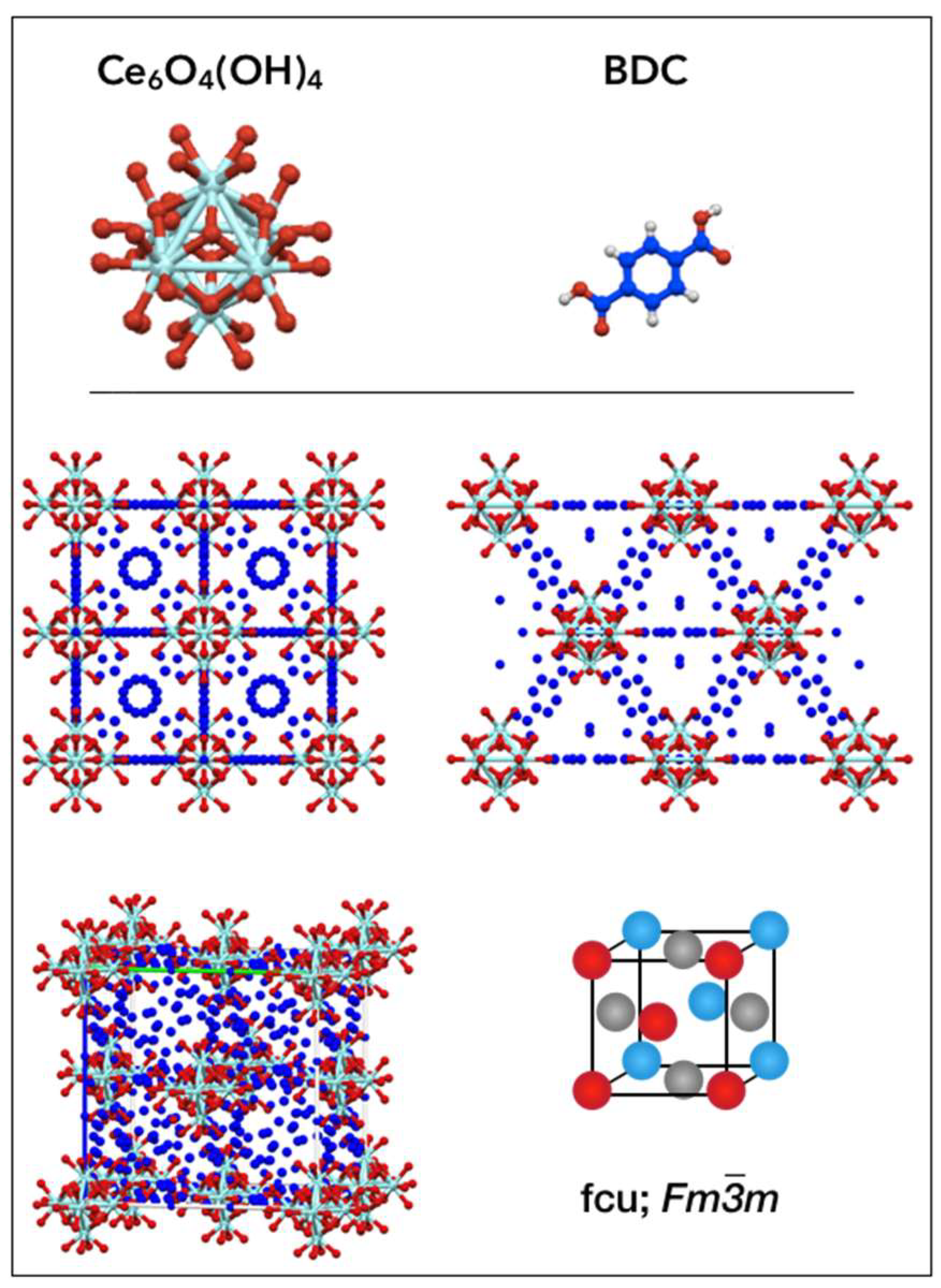
2.1. Crystallographic structure of UiO-66(Ce) materials
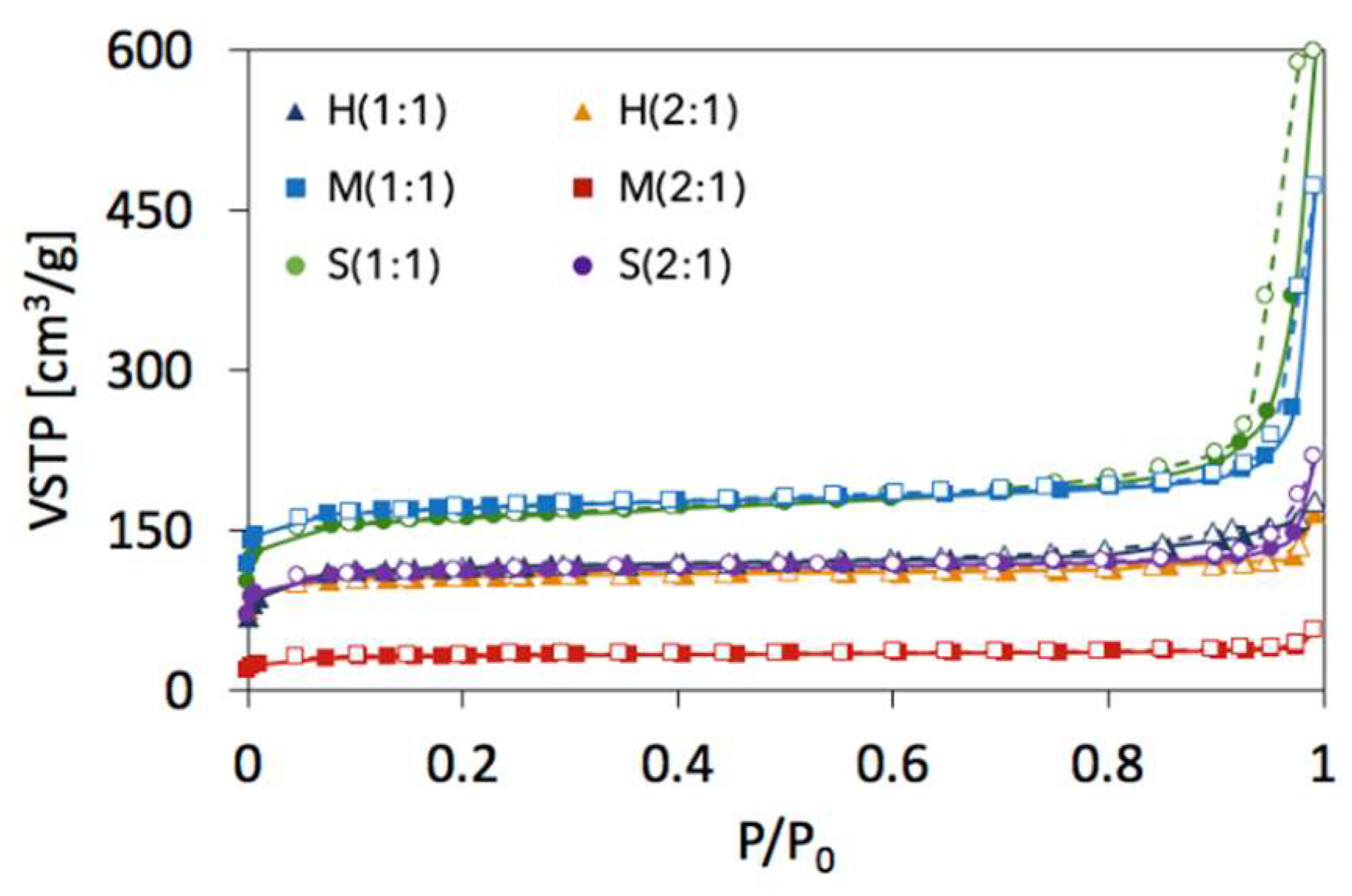
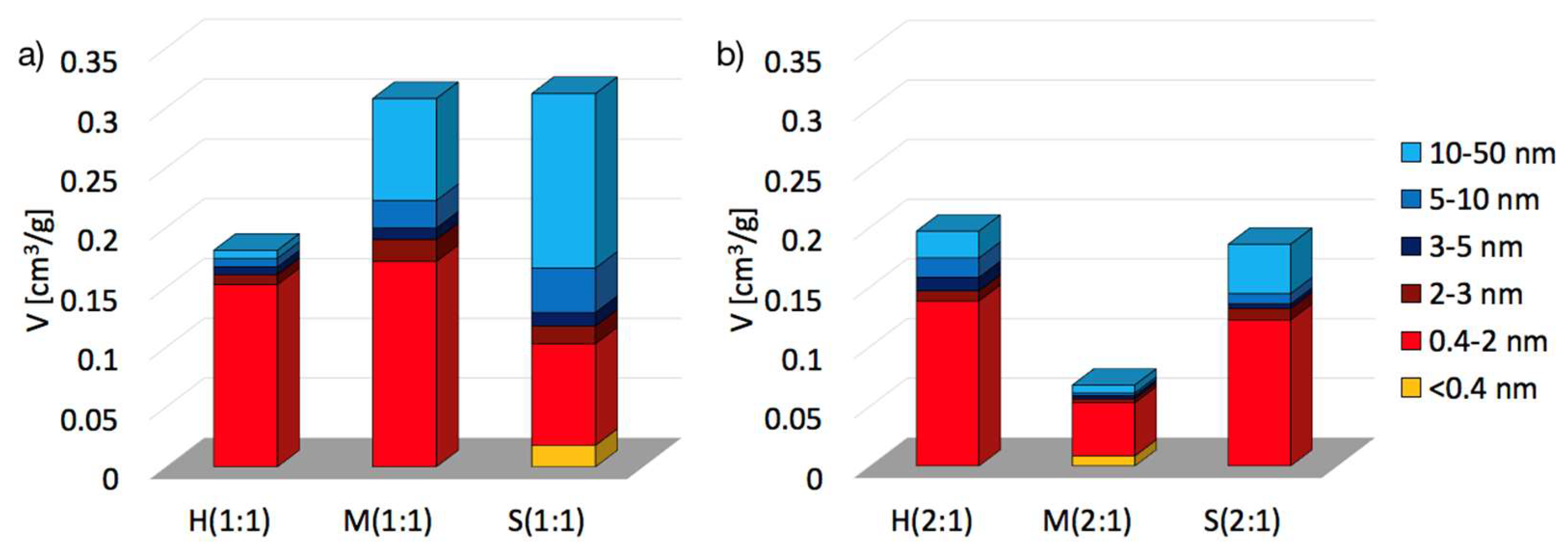
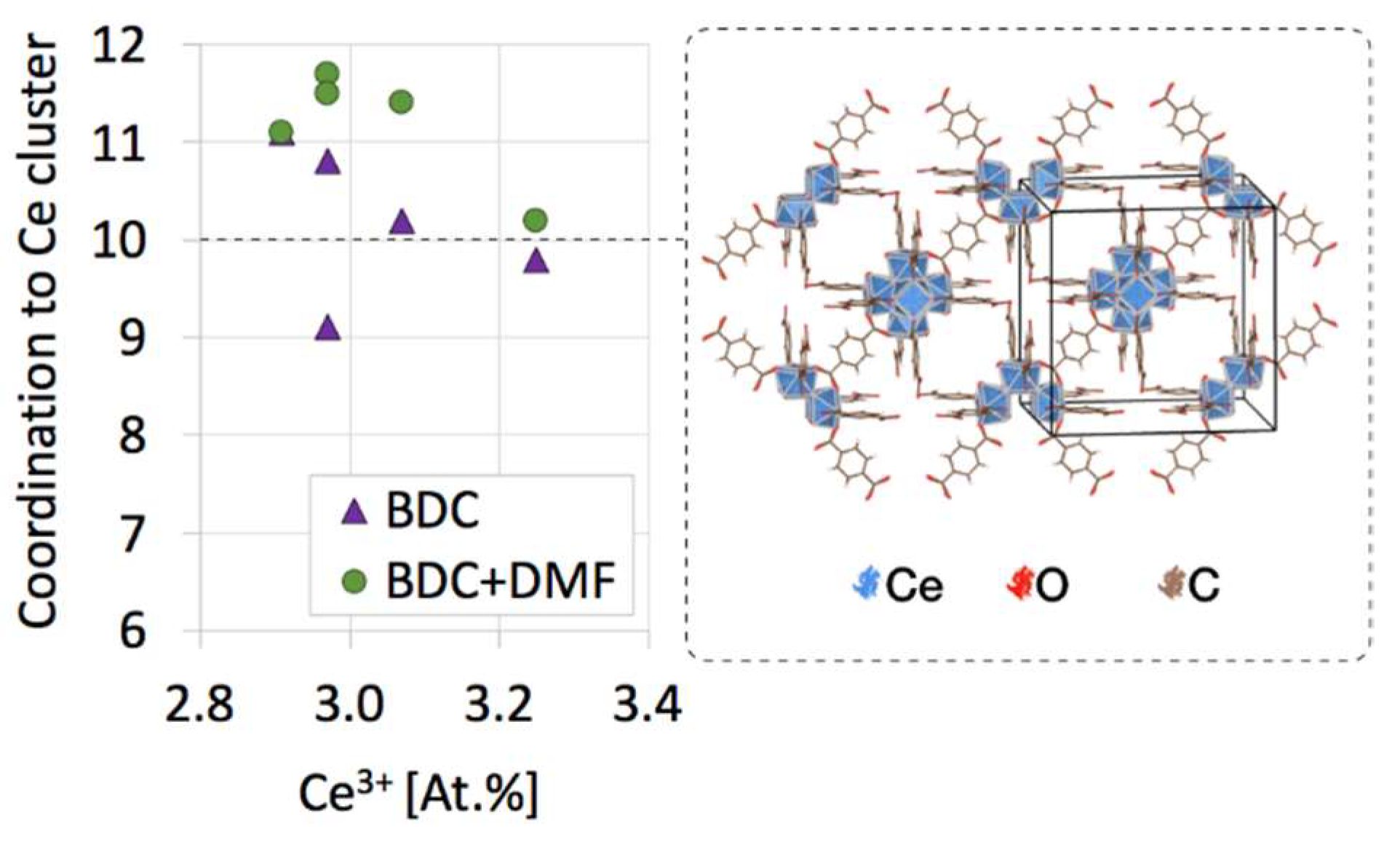
2.2. Textural properties of UiO-66(Ce)
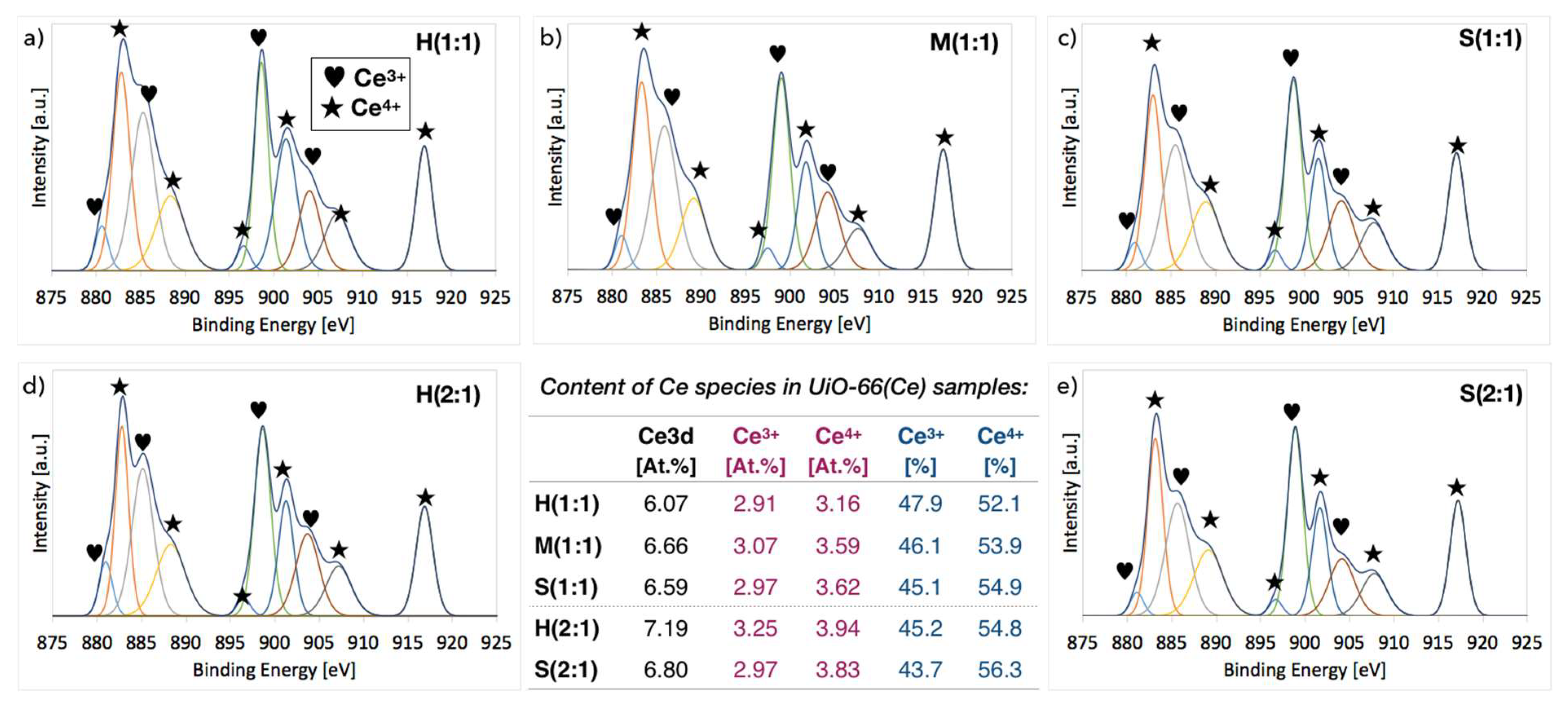
2.3. XPS of UiO-66(Ce)
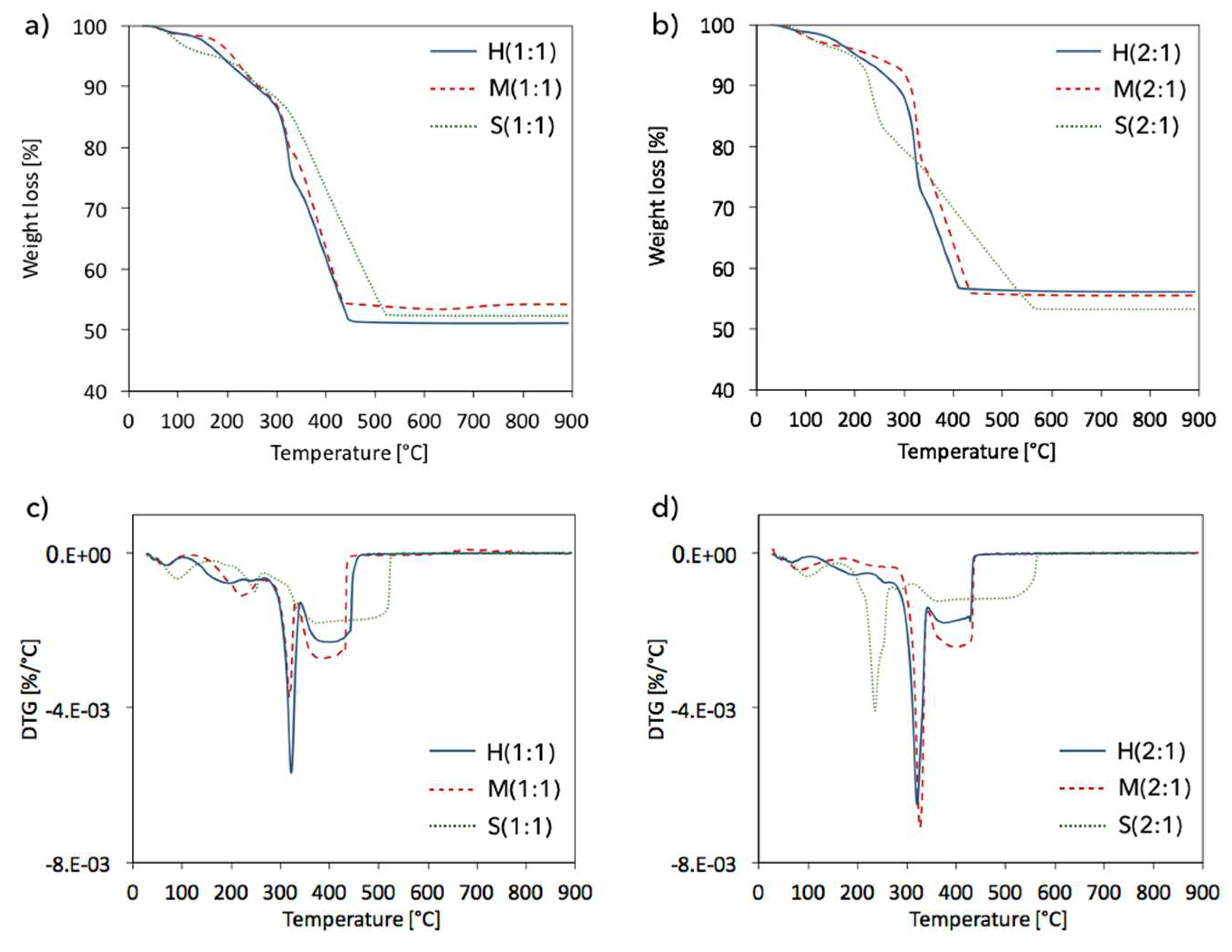
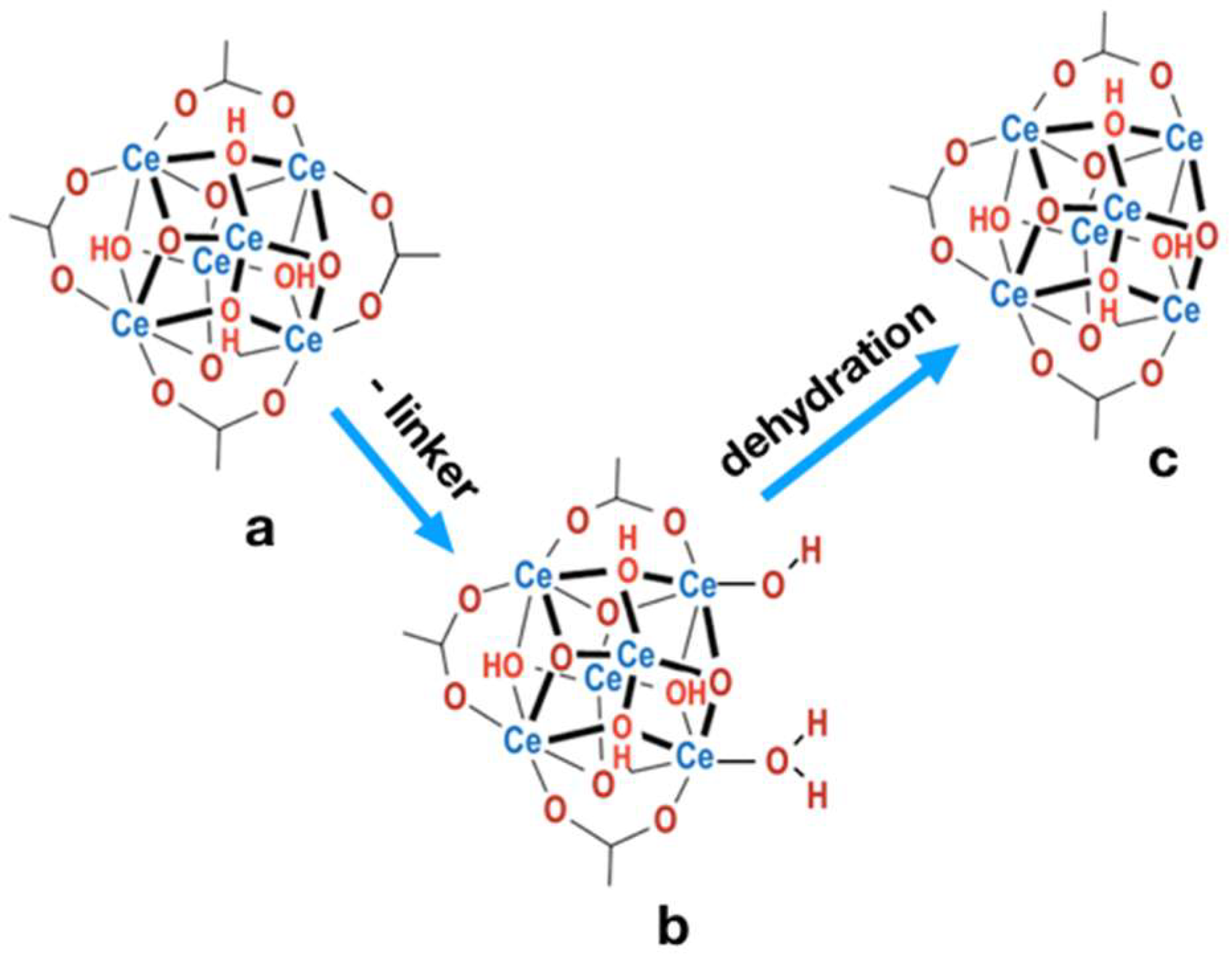
2.4. Structure defects and thermal stability of UiO-66(Ce)
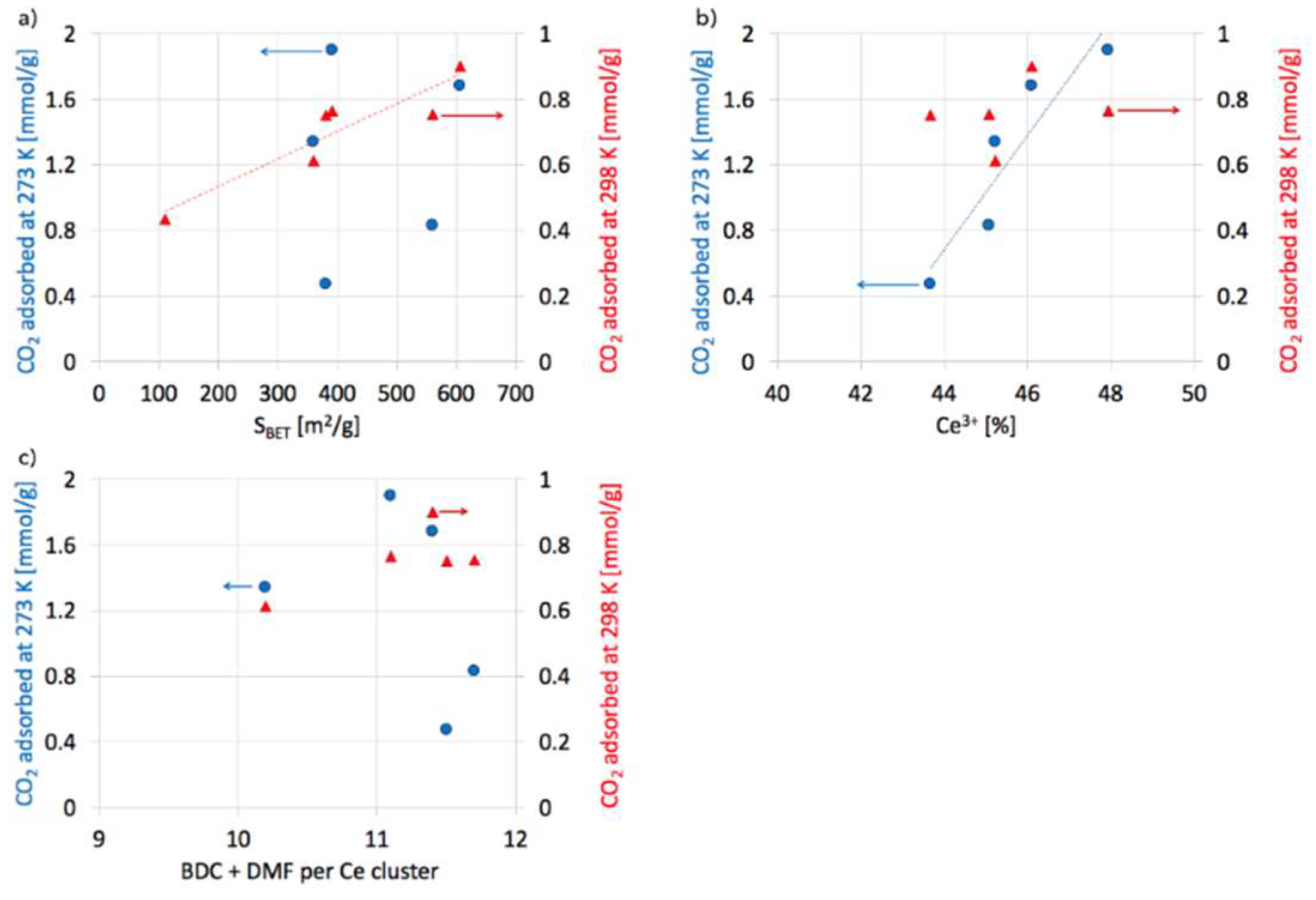
2.5. CO2 adsorption on UiO-66(Ce)
3. Materials and Methods
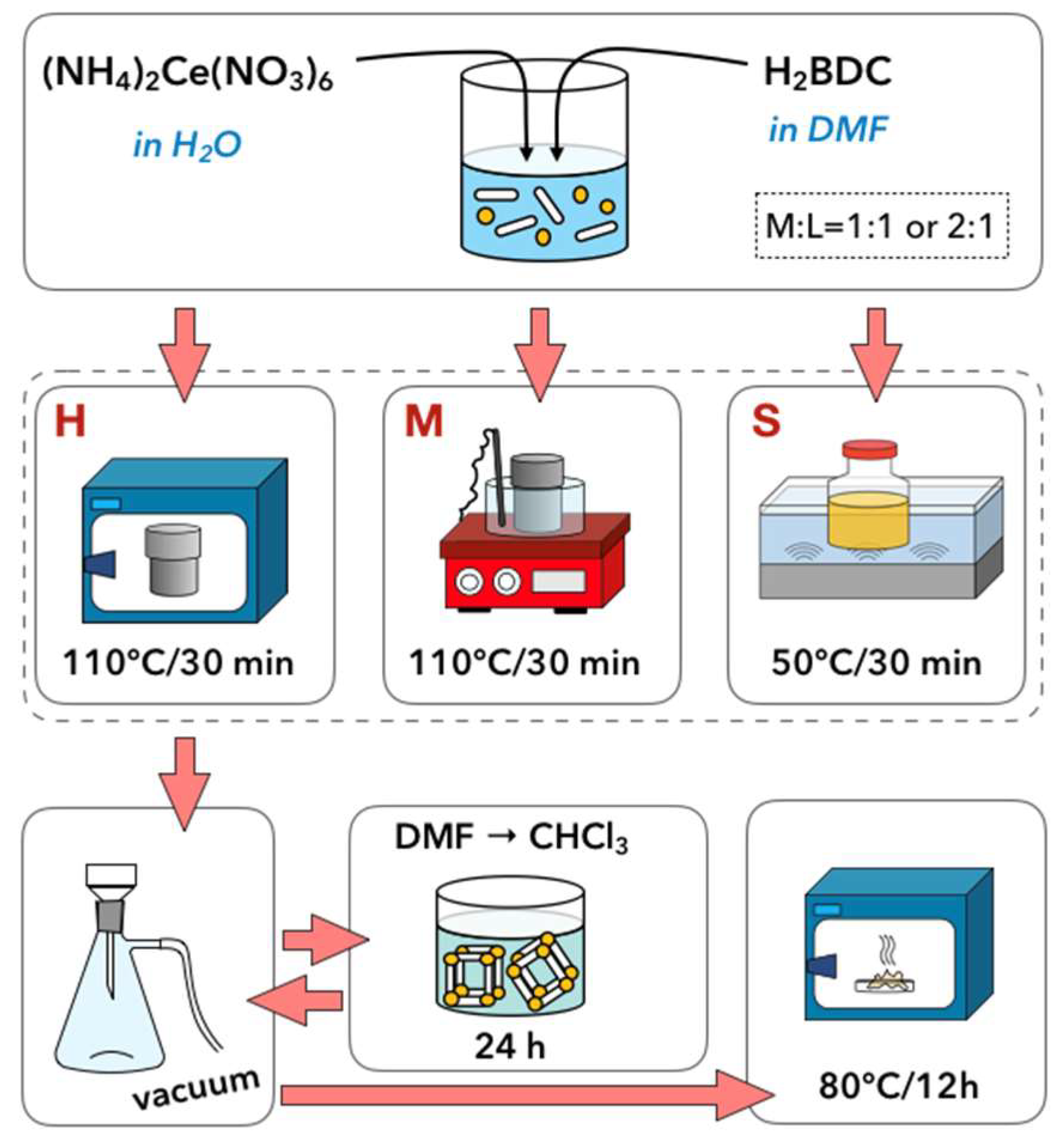
3.1. Synthesis of UiO-66(Ce)
- Hydrothermal synthesis (H): The mixture of Ce precursor and linker was placed in a Teflon lined steel autoclave and heated up to 110 °C in a dryer. After 30 min, the autoclave was slowly cooled down to room temperature.
- Hydrothermal synthesis with continuous mixing (M): The reagents mixture was placed in a Teflon lined steel autoclave and heated up to 110 °C in an oil bath that was placed on a magnetic stirrer. After 30 min, the autoclave was slowly cooled down to room temperature. The mixture was stirred throughout the whole synthesis, including heating and cooling steps.
- Sonochemical synthesis (S): The mixture of Ce precursor and linker was placed in a glass vessel and heated up to 50 °C in an ultrasound bath (35 kHz). After 30 min, the mixture was cooled down to room temperature.
3.2. Characterization of UiO-66(Ce)
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hong, D.H.; Suh, M.P. Selective CO2 adsorption in a metal–organic framework constructed from an organic ligand with flexible joints. Chem. Commun. 2012, 48, 9168–9170. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, H.; Wang, S.; Xie, Z.; Li, J.; Lin, W. A high connectivity metal–organic framework with exceptional hydrogen and methane uptake capacities. Chem. Sci. 2012, 3, 3032–3037. [Google Scholar] [CrossRef]
- Nagarkar, S.S.; Joarder, B.; Chaudhari, A.K.; Mukherjee, S.; Ghosh, S.K. Highly selective detection of nitro explosives by a luminescent metal-organic framework. Angew. Chem. Int. Ed. 2013, 52, 2881–2885. [Google Scholar] [CrossRef]
- Rojas, S.; Wheatley, P.S.; Quartapelle-Procopio, E.; Gil, B.; Marszalek, B.; Morris, R.E.; Barea, E. Metal-organic frameworks as potential multi-carriers of drugs. CrystEngComm 2013, 15, 9364–9367. [Google Scholar] [CrossRef]
- Horike, S.; Dincǎ, M.; Tamaki, K.; Long, J.R. Size-Selective Lewis Acid Catalysis in a Microporous Metal-Organic Framework with Exposed Mn2+ Coordination Sites. J. Am. Chem. Soc. 2008, 130, 5854–5855. [Google Scholar] [CrossRef] [PubMed]
- Hafizovic Cavka, J.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Wang, B.; Yang, Q.; Guo, C.; Sun, Y.X.; Xie, L.H.; Li, J.R. Stable Zr(IV)-Based Metal-Organic Frameworks with Predesigned Functionalized Ligands for Highly Selective Detection of Fe(III) Ions in Water. ACS Appl. Mater. Interfaces 2017, 9, 10286–10295. [Google Scholar] [CrossRef]
- Hafizovic Cavka, J.; Grande, C.A.; Mondino, G.; Blom, R. High Pressure Adsorption of CO2 and CH4 on Zr-MOFs. Ind. Eng. Chem. Res. 2014, 53, 15500–15507. [Google Scholar] [CrossRef]
- Biswas, S.; Van Der Voort, P. A General Strategy for the Synthesis of Functionalised UiO-66 Frameworks: Characterisation, Stability and CO2 Adsorption Properties. Eur. J. Inorg. Chem. 2013, 2013, 2154–2160. [Google Scholar] [CrossRef]
- Jakobsen, S.; Gianolio, D.; Wragg, D.S.; Nilsen, M.H.; Emerich, H.; Bordiga, S.; Lamberti, C.; Olsbye, U.; Tilset, M.; Lillerud, K.P. Structural determination of a highly stable metal-organic framework with possible application to interim radioactive waste scavenging: Hf-UiO-66. Phys. Rev. B Condens. Matter Mater. Phys. 2012, 86, 125429–125439. [Google Scholar] [CrossRef]
- Smith, S.J.D.; Ladewig, B.P.; Hill, A.J.; Lau, C.H.; Hill, M.R. Post-synthetic Ti Exchanged UiO-66 Metal-Organic Frameworks that Deliver Exceptional Gas Permeability in Mixed Matrix Membranes. Sci. Rep. 2015, 5, 7823. [Google Scholar] [CrossRef] [Green Version]
- Lammert, M.; Wharmby, M.T.; Smolders, S.; Bueken, B.; Lieb, A.; Lomachenko, K.A.; Vos, D.D.; Stock, N. Cerium-based metal organic frameworks with UiO-66 architecture. Chem. Commun. 2015, 51, 12578–12581. [Google Scholar] [CrossRef]
- Lammert, M.; Glißmann, C.; Reinsch, H.; Stock, N. Synthesis and Characterization of New Ce(IV)-MOFs Exhibiting Various Framework Topologies. Cryst. Growth Des. 2017, 17, 1125–1131. [Google Scholar] [CrossRef]
- Dalapati, R.; Sakthivel, B.; Dhakshinamoorthy, A.; Buragohain, A.; Bhunia, A.; Janiak, C.; Biswas, S. A highly stable dimethyl-functionalized Ce(IV)-based UiO-66 metal-organic framework material for gas sorption and redox catalysis. CrystEngComm 2016, 18, 7855–7864. [Google Scholar] [CrossRef]
- Smolders, S.; Lomachenko, K.A.; Bueken, B.; Struyf, A.; Bugaev, A.L.; Atzori, C.; Stock, N.; Lamberti, C.; Roeffaers, M.B.J.; De Vos, D.E. Unravelling the redox-catalytic behavior of Ce4+-MOFs: A XAS study. ChemPhysChem 2018, 19, 373–378. [Google Scholar] [CrossRef]
- Nouar, F.; Breeze, M.; Campo, B.C.; Clet, G.; Daturi, M.; Devic, T.; Vimont, A.; Walton, R.I.; Serre, C. Tuning the properties of the UiO-66 metal organic framework by Ce substitution. Chem. Commun. 2015, 4, 14458–14461. [Google Scholar] [CrossRef]
- Sridharan, V.; Menéndez, J.C. Cerium(IV) Ammonium Nitrate as a Catalyst in Organic Synthesis. Chem. Rev. 2010, 110, 3805–3849. [Google Scholar] [CrossRef] [PubMed]
- Esch, F.; Fabris, S.; Zhou, L.; Montini, T.; Africh, C.; Fornasiero, P.; Comelli, G.; Rosei, R. Electron localization determines defect formation on ceria substrates. Science 2005, 309, 752–755. [Google Scholar] [CrossRef]
- Campbell, C.T.; Peden, C.H.F. Chemistry. Oxygen vacancies and catalysis on ceria surfaces. Science 2005, 309, 713–714. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Yin, J.; Ma, Y.; Zhang, P.; Li, J.; Ding, Y.; Zhang, J.; Zhao, Y.; Chai, Z.; et al. Acquired superoxide-scavenging ability of ceria nanoparticles. Angew. Chem. Int. Ed. 2015, 54, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, C.; Zhang, X.; Wei, M.; Evans, D.G.; Duan, X. Catalytic behavior of supported Ru nanoparticles on the {100}. {110}, and {111} facet of CeO2. J. Catal. 2015, 329, 177–186. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Sanz, J.F.; et al. Catalysis. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wei, M.; Evans, D.G.; Duan, X. CeO2-based heterogeneous catalysts toward catalytic conversion of CO2. J. Mater. Chem. A 2016, 4, 5773–5783. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Bonura, G.; Arena, F.; Mezzatesta, G.; Cannilla, C.; Spadaro, L.; Frusteri, F. Role of the ceria promoter and carrier on the functionality of Cu-based catalysts in the CO2-to-methanol hydrogenation reaction. Catal. Today 2011, 171, 251–256. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Zafarana, G.; Trunfio, G.; Frusteri, F.; Spadaro, L. Effects of oxide carriers on surface functionality and process performance of the Cu-ZnO system in the synthesis of methanol via CO2 hydrogenation. J. Catal. 2013, 300, 141–151. [Google Scholar] [CrossRef]
- Demessence, A.; D’Alessandro, D.M.; Foo, M.L.; Long, J.R. Strong CO2 Binding in a Water-Stable. Triazolate-Bridged Metal-Organic Framework Functionalized with Ethylenediamine. J. Am. Chem. Soc. 2009, 131, 8784–8786. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Kim, J.; Ahn, W.S. Synthesis of metal-organic frameworks: A mini review. Korean J. Chem. Eng. 2013, 30, 1667–1680. [Google Scholar] [CrossRef]
- Kim, S.N.; Lee, Y.R.; Hong, S.H.; Jang, M.S.; Ahn, W.S. Pilot-scale synthesis of a zirconium-benzenedicarboxylate UiO-66 forCO2 adsorption and catalysis. Catal. Today 2015, 245, 54–60. [Google Scholar] [CrossRef]
- Safarifard, V.; Morsali, A. Applications of ultrasound to the synthesis of nanoscale metal-organic coordination polymers. Coord. Chem. Rev. 2015, 292, 1–14. [Google Scholar] [CrossRef]
- Waitschat, S.; Fröhlich, D.; Reinsch, H.; Terraschke, H.; Lomachenko, K.A.; Lamberti, C.; Kummer, H.; Helling, T.; Baumgartner, M.; Henninger, S.; et al. Synthesis of M-UiO-66 (M = Zr. Ce or Hf) employing 2,5-pyridinedicarboxylic acid as linker: Defect chemistry, framework hydrophilisation and sorption properties. Dalton Trans. 2018, 47, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Øien, S.; Wragg, D.; Reinsch, H.; Svelle, S.; Bordiga, S.; Lamberti, C.; Lillerud, K.P. Detailed structure analysis of atomic positions and defects in zirconium metal-organic frameworks. Cryst. Growth Des. 2014, 14, 5370–5372. [Google Scholar] [CrossRef]
- Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef] [Green Version]
- Henning, C.; Ikeda-Ohno, A.; Kraus, W.; Weiss, S.; Pattison, P.; Emerich, H.; Abdala, P.M.; Scheinost, A. Crystal structure and solution species of Ce(III) and Ce(IV) formates-from mononuclear to hexanuclear complexes. Inorg. Chem. 2013, 52, 11734–11743. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, J.M.M.; Freitas, L. Study of Water and Dimethylformamide Interaction by Computer Simulation. Z. Nat. A 1999, 54, 110–116. [Google Scholar] [CrossRef]
- García-Ruiz, J.M.; Otálora, F.; García-Caballero, A. The role of mass transport in protein crystallization. Acta Cryst. F Struct. Biol. Commun. 2016, 72, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, R.J.; Schroeder, S.L.; Horst, J.H. Nucleation of organic crystals--a molecular perspective. Angew. Chem. Int. Ed. Engl. 2013, 52, 2166–2179. [Google Scholar] [CrossRef]
- Dodds, J.A.; Espitalier, F.; Louisnard, O.; Grossier, R.; David, R.; Hassoun, M.; Baillon, F.; Gatumel, C.; Lyczko, N. The effect of ultrasound on crystallisation-precipitation processes: Some examples and a new segregation model. Particle and Particle Systems Characterization. Part. Part. Syst. Charact. 2007, 24, 18–28. [Google Scholar] [CrossRef]
- Enomoto, N.; Sung, T.H.; Nakagawa, Z.E.; Lee, S.C. Effect of ultrasonic waves on crystallization from a supersaturated solution of alum. J. Mater. Sci. 1992, 27, 5239–5243. [Google Scholar] [CrossRef]
- Furukawa, H.; Gándara, F.; Zhang, Y.B.; Jiang, J.; Queen, W.L.; Hudson, M.R.; Yaghi, O.M. Water Adsorption in Porous Metal-Organic Frameworks and Related Materials. J. Am. Chem. Soc. 2014, 136, 4369–4381. [Google Scholar] [CrossRef]
- Aguado, J.; Serrano, D.P.; Escola, J.M.; Rodríguez, J.M. Low temperature synthesis and properties of ZSM-5 aggregates formed by ultra-small nanocrystals. Microporous Mesoporous Mater. 2004, 75, 41–49. [Google Scholar] [CrossRef]
- De Coste, J.B.; Peterson, G.W.; Jasuja, H.; Glover, T.G.; Huang, Y.; Walton, K.S. Stability and degradation mechanisms of metal-organic frameworks containing the Zr6O4(OH)4 secondary building unit. J. Mater. Chem. A 2013, 1, 5642–5650. [Google Scholar] [CrossRef]
- Mitchell, D.F.; Sproule, G.I.; Graham, M.J. Sputter reduction of oxides by ion bombardment during Auger depth profile analysis. Surf. Interface Anal. 1990, 15, 487–497. [Google Scholar] [CrossRef]
- Holgado, J.P.; Alvarez, R.; Munuera, G. Study of CeO XPS spectra by factor analysis: Reduction of CeO2. Appl. Surf. Sci. 2000, 161, 301–315. [Google Scholar] [CrossRef]
- Hattori, T.; Hattori, M.; Ozawa, M. Preparation and Surface Reduction Behavior of CeO2 Nanoparticle Layer on Al2O3(0001) Crystal Substrate. J. Surf. Sci. Nanotechnol. 2018, 16, 172–176. [Google Scholar] [CrossRef]
- De Vos, A.; Hendrickx, K.; Van Der Voort, P.; Van Speybroeck, V.; Lejaeghere, K. Missing Linkers: An Alternative Pathway to UiO-66 Electronic Structure Engineering. Chem. Mater. 2017, 29, 3006–3019. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.J.; Brown, Z.J.; Colón, Y.J.; Siu, P.W.; Scheidt, K.A.; Snurr, R.Q.; Hupp, J.T.; Farha, O.K. A facile synthesis of UiO-66. UiO-67 and their derivatives. Chem. Commun. 2013, 49, 9449–9451. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, L.; Civalleri, B.; Chavan, S.; Bordiga, S.; Nilsen, M.H.; Jakobsen, S.; Lillerud, K.P.; Lamberti, C. Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory. Chem. Mater. 2011, 23, 1700–1718. [Google Scholar] [CrossRef]
- Bristow, J.K.; Svane, K.L.; Tiana, D.; Skelton, J.M.; Gale, J.D.; Walsh, A. Free Energy of Ligand Removal in the Metal-Organic Framework UiO-66. J. Phys. Chem. C 2016, 120, 9276–9281. [Google Scholar] [CrossRef]
- Vermoortele, F.; Bueken, B.; le Bars, G.; van de Voorde, B.; Vandichel, M.; Houthoofd, K.; Vimont, A.; Daturi, M.; Waroquier, M.; van Speybroeck, V. Synthesis Modulation as a Tool To Increase the Catalytic Activity of Metal-Organic Frameworks: The Unique Case of UiO-66 (Zr). J. Am. Chem. Soc. 2013, 135, 11465–11468. [Google Scholar] [CrossRef]
- Fang, Z.; Bueken, B.; de Vos, D.E.; Fischer, R.A. Defect-Engineered Metal–Organic Frameworks. Angew. Chem. Int. Ed. Engl. 2015, 54, 7234–7254. [Google Scholar] [CrossRef] [Green Version]
- Vermoortele, F.; Vandichel, M.; Van de Voorde, B.; Ameloot, R.; Waroquier, M.; Van Speybroeck, V.; De Vos, D.E. Electronic Effects of Linker Substitution on Lewis Acid Catalysis with Metal-Organic Frameworks. Angew. Chem. Int. Ed. 2012, 51, 4887–4890. [Google Scholar] [CrossRef]
- Cirujano, F.G.; Corma, A.; Llabrés i Xamena, F.X. Zirconium-containing metal organic frameworks as solid acid catalysts for the esterification of free fatty acids: Synthesis of biodiesel and other compounds of interest. Catal. Today 2015, 257, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Caratelli, C.; Hajek, J.; Cirujano, F.G.; Waroquier, M.; Llabrés i Xamena, F.X.; Van Speybroeck, V. Nature of active sites on UiO-66 and beneficial influence of water in the catalysis of Fischer esterification. J. Catal. 2017, 352, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Ethiraj, J.; Albanese, E.; Civalleri, B.; Vitillo, J.G.; Bonino, F.; Chavan, S.; Shearer, G.C.; Lillerud, K.P.; Bordiga, S. Carbon Dioxide Adsorption in Amine-Functionalized Mixed-Ligand Metal–Organic Frameworks of UiO-66 Topology. ChemSusChem 2014, 7, 3382–3388. [Google Scholar] [CrossRef]
- D’Amato, R.; Donnadio, A.; Carta, M.; Sangregorio, C.; Vivani, R.; Taddei, M.; Costantino, F. Green Synthesis and Enhanced CO2 Capture Performance of Perfluorinated Cerium-Based Metal-Organic Frameworks with UiO-66 and MIL-140 Topology. ChemRxiv 2018. [Google Scholar] [CrossRef]
- Bucci, A.; Menendez Rodriguez, G.; Bellachioma, G.; Zuccaccia, C.; Poater, A.; Cavallo, L.; Macchioni, A. An Alternative Reaction Pathway for Iridium Catalyzed Water Oxidation Driven by CAN. ACS Catal. 2016, 6, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Kimoto, A.; Yamauchi, K.; Yoshida, M.; Masaoka, S.; Sakai, K. Kinetics and DFT studies on water oxidation by Ce4+ catalyzed by [Ru(terpy)(bpy)(OH2)]2+. Chem. Commun. 2012, 48, 239–241. [Google Scholar] [CrossRef]
- Wu, H.; Shen Chua, Y.; Krungleviciute, V.; Tyagi, M.; Chen, P.; Yildirim, T.; Zhou, W. Unusual and Highly Tunable Missing-Linker Defects in Zirconium Metal-Organic Framework UiO-66 and Their Important Effects on Gas Adsorption. J. Am. Chem. Soc. 2013, 135, 10525–10532. [Google Scholar] [CrossRef]
- Thornton, A.W.; Babarao, R.; Jain, A.; Trousseletc, F.; Coudert, F.X. Defects in metal–organic frameworks: A compromise between adsorption and stability. Dalton Trans. 2016, 45, 4352–4359. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a [Å] | Volume [Å3] | D111 [nm] |
|---|---|---|---|
| H(1:1) | 21.0469 | 9323.2 | 83.5 |
| M(1:1) | 21.2110 | 9543.0 | 65.8 |
| S(1:1) | 21.2797 | 9636.0 | 43.4 |
| H(2:1) | 21.4095 | 9813.4 | 83.7 |
| M(2:1) | 21.4544 | 9875.4 | 59.7 |
| S(2:1) | 21.3468 | 9727.4 | 46.9 |
| UiO-66 [6] | 20.7004 | 8870.3 | - |
| UiO-66(Ce) [12] | 21.4727 | 9900.6 | - |
| Formula: [Ce6O4(OH)4(BDC)6] Space group: Fm-3m | |||
| SBET [m2/g] | Vmic+Vmes [cm3/g] | dmes [nm] | Ømic | Ømes | |
|---|---|---|---|---|---|
| H(1:1) | 391 | 0.1810 | 4.37 | 0.84 | 0.16 |
| M(1:1) | 606 | 0.3079 | 7.94 | 0.56 | 0.44 |
| S(1:1) | 560 | 0.3120 | 10.42 | 0.33 | 0.67 |
| H(2:1) | 360 | 0.1970 | 5.98 | 0.70 | 0.30 |
| M(2:1) | 111 | 0.0679 | 5.42 | 0.78 | 0.22 |
| S(2:1) | 381 | 0.1860 | 7.99 | 0.66 | 0.34 |
| UiO-66 | 1187 [6] 1086 [9] 1290 [41] 1080 [43] | ||||
| UiO-66(Ce) | 1282 [12] |
| Sample | Molecules per Ce Cluster | N [At.%] (from XPS) | ||
|---|---|---|---|---|
| BDC | DMF | BDC+DMF | ||
| H(1:1) | 11.1 | 0.0 | 11.1 | 1.2 |
| M(1:1) | 10.2 | 1.2 | 11.4 | 2.1 |
| S(1:1) | 10.8 | 0.8 | 11.7 | 4.1 |
| H(2:1) | 9.8 | 0.4 | 10.2 | 1.4 |
| M(2:1) | 10.3 | 0.0 | 10.3 | - |
| S(2:1) | 9.1 | 2.4 | 11.5 | 6.5 |
| Sample | CO2 Adsorption at 273 K | CO2 Adsorption at 298 K | ||
|---|---|---|---|---|
| [mmol/g] | [mmol/g] | Reversible [%] | Irreversible [%] | |
| H(1:1) | 1.90 | 0.7640 | 83.6 | 16.4 |
| H(2:1) | 1.34 | 0.6125 | 84.6 | 15.4 |
| M(1:1) | 1.68 | 0.9011 | 92.9 | 7.1 |
| M(2:1) | - | 0.4328 | 58.5 | 41.5 |
| S(1:1) | 0.83 | 0.7545 | 74.9 | 25.1 |
| S(2:1) | 0.47 | 0.7498 | 75.1 | 24.9 |
| CeO2 | - | 0.6528 | 47.8 | 52.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stawowy, M.; Róziewicz, M.; Szczepańska, E.; Silvestre-Albero, J.; Zawadzki, M.; Musioł, M.; Łuzny, R.; Kaczmarczyk, J.; Trawczyński, J.; Łamacz, A. The Impact of Synthesis Method on the Properties and CO2 Sorption Capacity of UiO-66(Ce). Catalysts 2019, 9, 309. https://doi.org/10.3390/catal9040309
Stawowy M, Róziewicz M, Szczepańska E, Silvestre-Albero J, Zawadzki M, Musioł M, Łuzny R, Kaczmarczyk J, Trawczyński J, Łamacz A. The Impact of Synthesis Method on the Properties and CO2 Sorption Capacity of UiO-66(Ce). Catalysts. 2019; 9(4):309. https://doi.org/10.3390/catal9040309
Chicago/Turabian StyleStawowy, Michalina, Maciej Róziewicz, Ewa Szczepańska, Joaquin Silvestre-Albero, Mirosław Zawadzki, Marta Musioł, Rafał Łuzny, Jan Kaczmarczyk, Janusz Trawczyński, and Agata Łamacz. 2019. "The Impact of Synthesis Method on the Properties and CO2 Sorption Capacity of UiO-66(Ce)" Catalysts 9, no. 4: 309. https://doi.org/10.3390/catal9040309
APA StyleStawowy, M., Róziewicz, M., Szczepańska, E., Silvestre-Albero, J., Zawadzki, M., Musioł, M., Łuzny, R., Kaczmarczyk, J., Trawczyński, J., & Łamacz, A. (2019). The Impact of Synthesis Method on the Properties and CO2 Sorption Capacity of UiO-66(Ce). Catalysts, 9(4), 309. https://doi.org/10.3390/catal9040309