2.1. Characterization
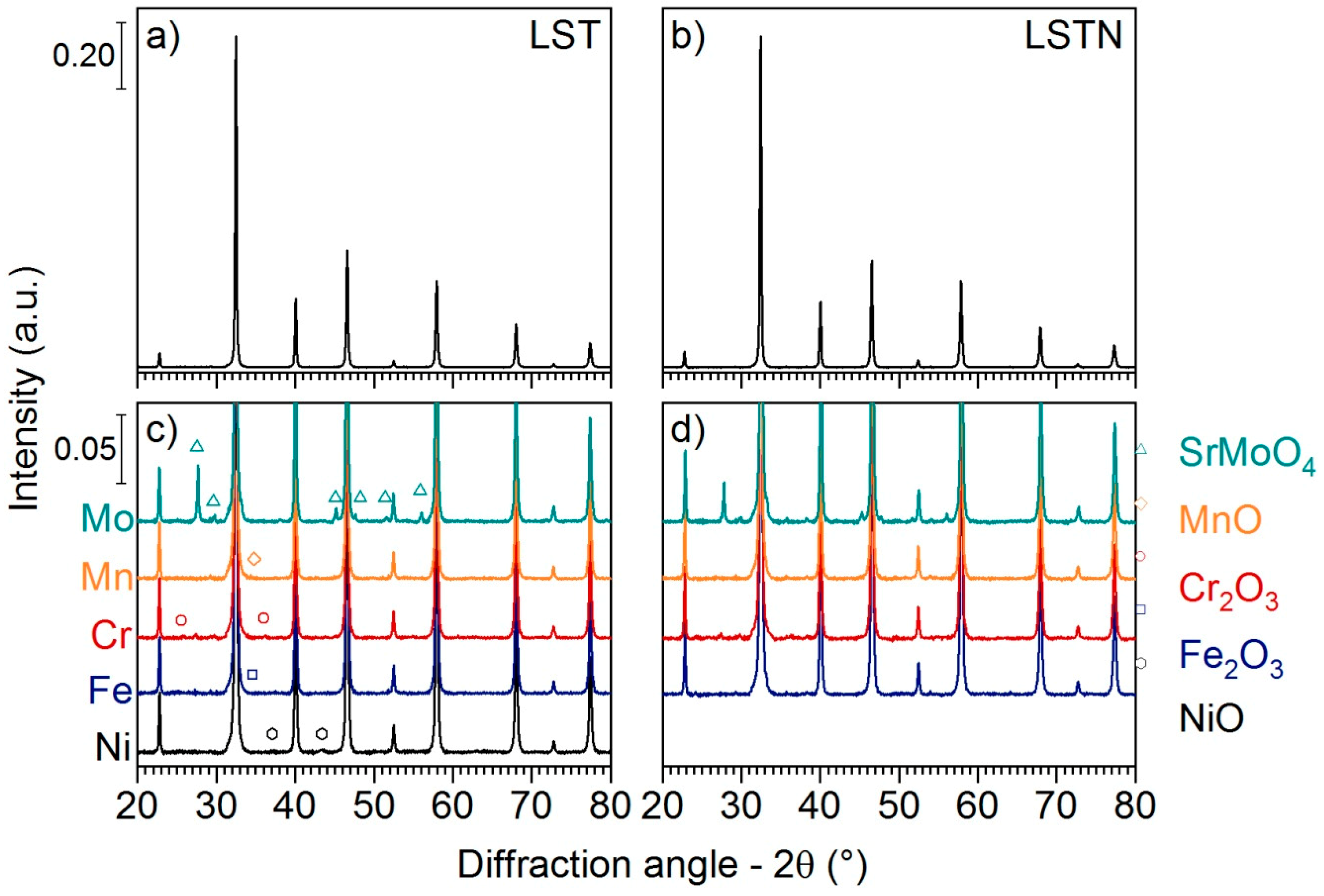
Figure 1 shows X-ray diffraction (XRD) patterns of the materials after impregnation of La
0.3Sr
0.55TiO
3±δ (LST) and La
0.3Sr
0.55Ti
0.95Ni
0.05O
3±δ (LSTN) with the metal precursors and subsequent calcination. XRD patterns are also given for LST (
Figure 1a) and LSTN (
Figure 1b). The reflections of the corresponding single metal oxides were observed in the XRD patterns of impregnated LST (LST-5Me, Me = Cr, Mn, Fe and Ni;
Figure 1c), as well as impregnated LSTN (LSTN-5Me, Me = Cr, Mn and Fe;
Figure 1d). Impregnation with the Mo precursor (LST-5Mo and LSTN-5Mo) resulted in the presence of reflections of a SrMoO
4 phase, which is indicative of the segregation of small quantities of Sr from both LST and LSTN lattices during impregnation and calcination.
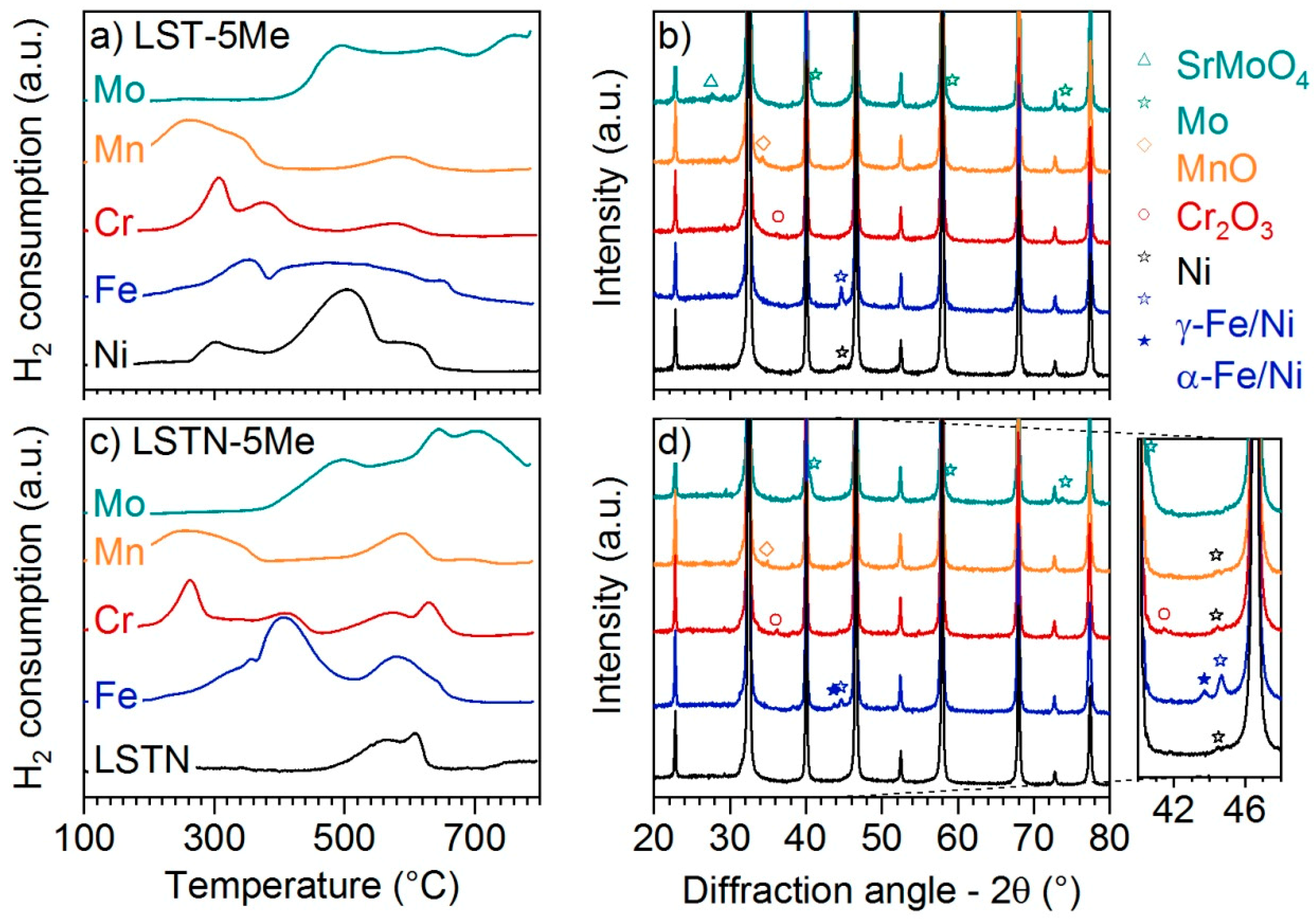
TPR experiments revealed the reduction of MnO, Cr
2O
3, Fe
2O
3 and NiO on LST up to 800 °C (
Figure 2a). SrMoO
4 was only partially reduced at these temperatures as is evident from the fact that H
2 was still consumed at 800 °C. Similar behavior was also observed on impregnated LSTN materials (
Figure 2c), on which also contributions of the LSTN support can be observed between ca. 450 °C and 650 °C. XRD measurements of the reduced materials confirmed the reduction of NiO, Fe
2O
3 and partial reduction of SrMoO
4 on LST-5Ni, LST-5Fe and LST-5Mo, respectively (
Figure 2b). No reflections of Cr and Mn metals were observed on reduced LST-5Cr and LST-5Mn, respectively. Instead, reflections of the single oxides were observed, similar to the calcined materials. This was likely due to the ex situ nature of the XRD experiments and the strong tendency of dispersed Cr and Mn particles to form oxides.
All reflections were also encountered on the reduced LSTN-type materials. However, reduced LSTN-5Fe exhibited reflections that indicated the presence of two Fe allotropes (α-Fe and γ-Fe,
Figure 2d), which is likely a consequence of Fe/Ni alloy formation during reduction at 800 °C as it was not observed on LST-5Fe. Subsequent rapid cooling to room temperature after reduction resulted in phase separation. This is supported by phase diagrams of the Fe-Ni system, which predict partial phase decomposition of the homogeneous γ-Fe/Ni alloy phase and various phase transformations during cooling [
20]. Since cooling rates were high in these experiments (ca. 20 °C·min
−1) the presence of metallic phases, which are not at equilibrium is highly probable [
21]. Ni (111) reflections could be observed in LSTN, LSTN-5Cr and LSTN-5Mn, whereas the absence of the same in LSTN-5Fe and LSTN-5Mo can be regarded as an indication of Ni/Me alloy formation in the latter cases.
2.2. Catalytic Activity
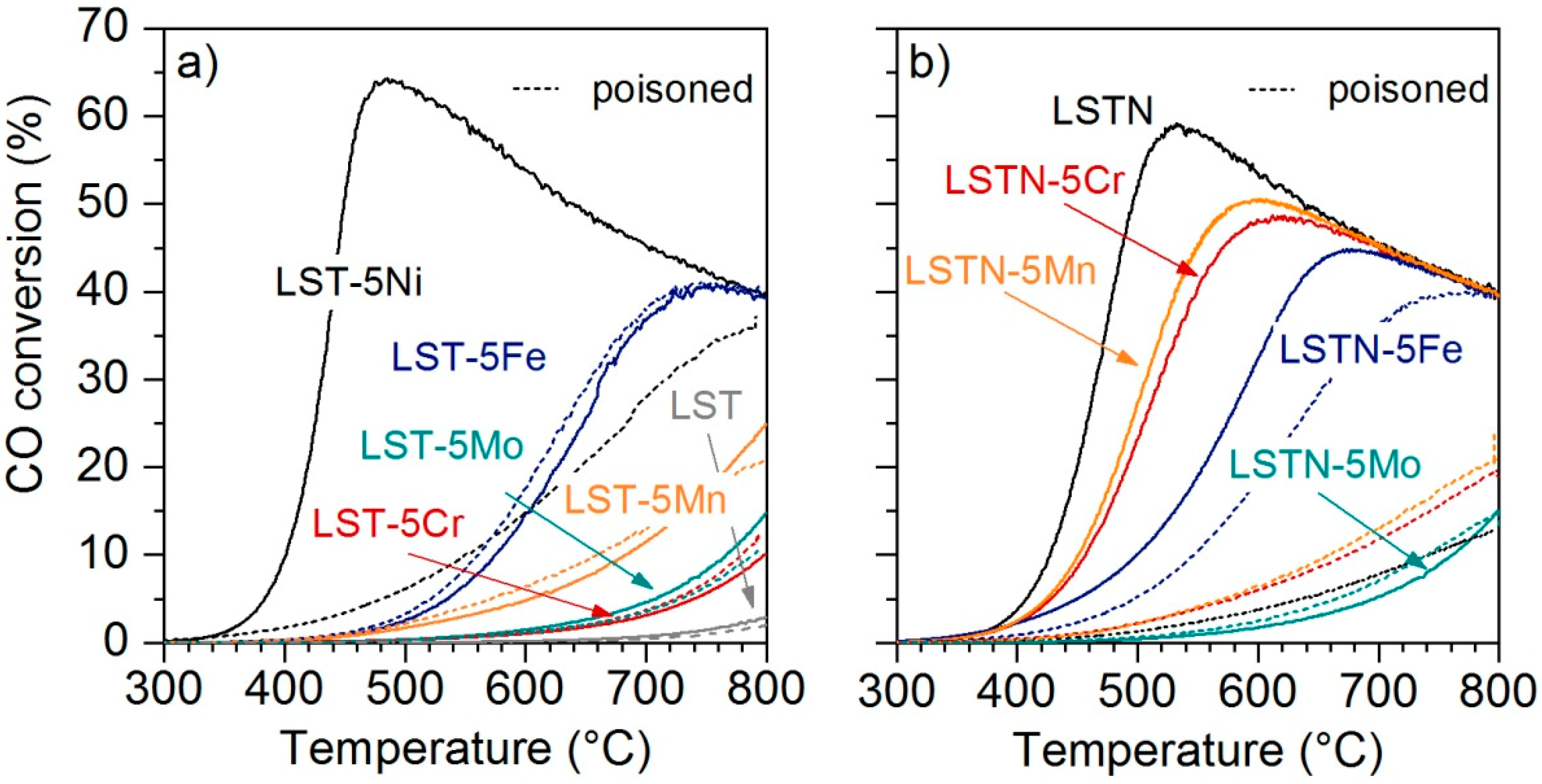
The activity of the pre-reduced catalysts towards the water gas shift (WGS) reaction was assessed by measuring CO conversion between 300 °C and 800 °C and the results are shown in
Figure 3. Activities of the catalysts produced using Ni-free LST as a support are displayed in
Figure 3a. Nickel was the most active among all metals and LST-5Ni exhibited CO conversion above 60% at 460 °C. The next best activities were shown by LST-5Fe followed by LST-5Mn and finally LST-5Mo and LST-5Cr, which showed only limited activities at reactions temperatures below 800 °C. The lack of activity of Cr and Mo is not surprising. Molybdenum was added with the specific intent to introduce an element with negligible WGS activity, but with significant sensitivity to sulfur to be used as a sacrificial agent. Chromium is mainly used in Fe/Cr-based WGS catalysts for its capability to stabilize the active Fe phase [
22]. Since LST did not show any significant WGS activity (
Figure 3a) it can be assumed that the catalytic activity of LST-Ni, LST-Fe and LST-Mn was due to the impregnated metal. The CO conversion of each catalyst after poisoning is shown by the dashed lines in
Figure 3a. Only LST-5Ni suffered severely during catalyst poisoning using 50 ppm H
2S under reaction conditions (800 °C, 1 h). The catalytic activity of LST-5Fe, LST-5Mn, LST-5Cr and LST-5Mo on the other hand, remained rather constant.
Catalytic activities of impregnated LSTN-type catalysts are displayed in
Figure 3b. LSTN exhibited the highest activity towards the WGS reaction, followed by LSTN-5Mn and LSTN-5Cr. LSTN-5Fe also exhibited higher activity than its Ni-free counterpart LST-5Fe, whereas the low activity of LSTN-5Mo remained unchanged by the presence of Ni in the sample. The catalytic results provide an indication for Ni alloying with the impregnated metals Cr, Fe and Mn. The fact that all LSTN-5Me (Me = Cr, Fe, Mn, Mo) catalysts exhibited lower catalytic activity than LSTN, while all metals showed at least some activity towards WGS on Ni-free LST, provides evidence for close interaction between segregated Ni and Me in LSTN-5Me or coverage of the active Ni phase with the less active Me. This interaction was found to be less beneficial for WGS activity under sulfur-free conditions. However, catalytic tests after sulfur poisoning showed a stabilizing effect of the metals on WGS activity. Especially LSTN-5Fe was able to maintain comparably high levels of catalytic activity after poisoning by H
2S. It should be noted that the activity of LSTN-5Fe after poisoning did not exceed the one of LST-5Fe, which could indicate that Ni is still poisoned. Nevertheless, the improved activity of LSTN-5Fe in sulfur-free conditions compared to LST-5Fe demonstrates a potential benefit of a bimetallic catalyst.
2.3. Sulfur Uptake
Differences between the various materials were also observed in their behavior during H
2S adsorption.
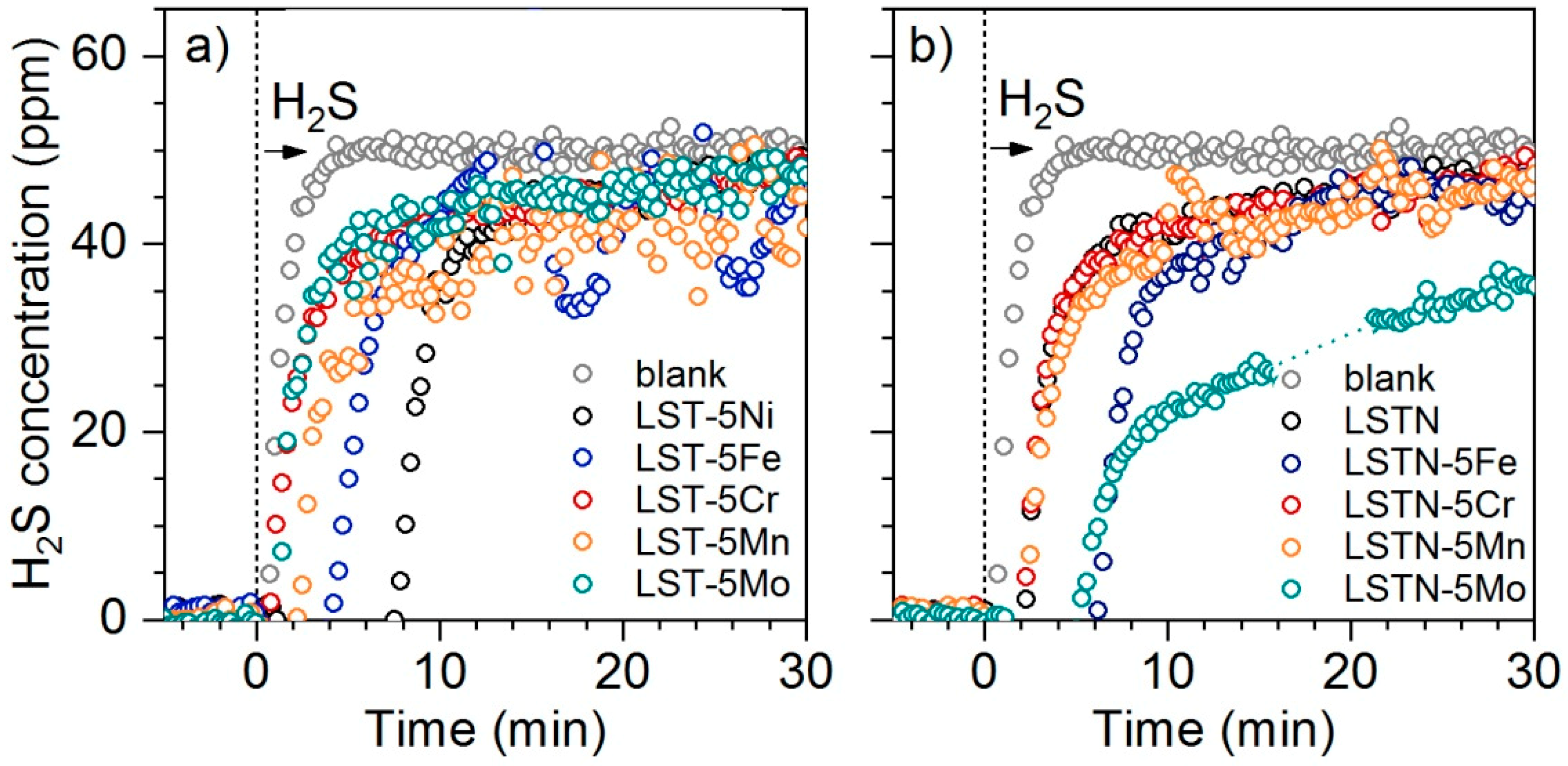
Figure 4a shows H
2S breakthrough curves for LST-5Me catalysts during sulfur loading. H
2S (50 ppm) was introduced to the reaction gas stream after 5 min equilibration time. Dosing H
2S over a blank quartz reactor resulted in negligible retention time and 50 ppm were attained after ca. 5 min. Sulfur adsorption can be observed by the increased retention times when H
2S is dosed over the catalyst bed. The longest retention time of around 8 min was recorded for LST-5Ni. The significant sulfur uptake of this sample (210 ppm by weight) is in line with its strong deactivation in terms of the catalytic activity after sulfur loading (
Figure 3a). A retention time of around 4 min and 2 min was observed for LST-5Fe and LST-5Mn, respectively. H
2S breakthrough of LST-5Mo and LST-5Cr was close to the blank experiment indicating low sulfur adsorption properties of these metals. This indicates that the ability of Mo as a sulfur scavenger was limited on these samples compared to the more conventionally applied single oxides MoO
2 and MoO
3, possibly due to the SrMoO
4 phase formed during synthesis (
Figure 1).
Interestingly, LSTN-5Mo showed significant sulfur uptake exceeding the combined sulfur storage capabilities of LSTN and LST-5Mo. This can be regarded as an indication for close interaction between segregated Ni and Mo, which appeared to significantly change the sulfur uptake properties of the material. Retention times of LSTN-5Fe, LSTN-5Cr and LSTN-5Mo were approximately the sum of those of LSTN and their LST-5Me counterparts and can be therefore explained by the increased metal content on the sample surface. Sulfur uptake of LSTN was much lower than that of LST-5Ni (ca. 40 ppm compared to 210 ppm), which can be justified using the similar argumentation that when Ni is deposited on LST by impregnation, the Ni metal surface is higher compared to pre-reduced LSTN, due to partial Ni reduction in the latter. Although no obvious advantage of metal impregnation on LSTN could be observed from these H
2S adsorption experiments because none of the metals reduced the H
2S uptake of the sample, the increased sulfur tolerance in terms of the catalytic activity towards WGS exhibited by LSTN-5Fe (
Figure 3b) could be an advantageous property. Therefore, the question arises whether it is possible to exploit the reversible segregation of metals from an LST-type host to produce both redox stable, as well as sulfur tolerant Ni/Fe catalysts.
2.4. La0.3Sr0.55Ti1-x-yFexNiyO3±δ (x = 0, 0.025, 0.05; y = 0, 0.05)
Catalytic activity and sulfur uptake data indicated that Fe might be a suitable candidate to improve sulfur tolerance of LSTN while maintaining the self-regeneration property. In order to explore the potential segregation of both Fe and Ni from the perovskite-type host, both Ni and Fe were introduced at the perovskite B-site of LST to obtain compositions of the type La
0.3Sr
0.55Ti
1-x-yFe
xNi
yO
3±δ (x = 0, 0.025, 0.05; y = 0, 0.05) the denotations of which are summarized in
Table 1.
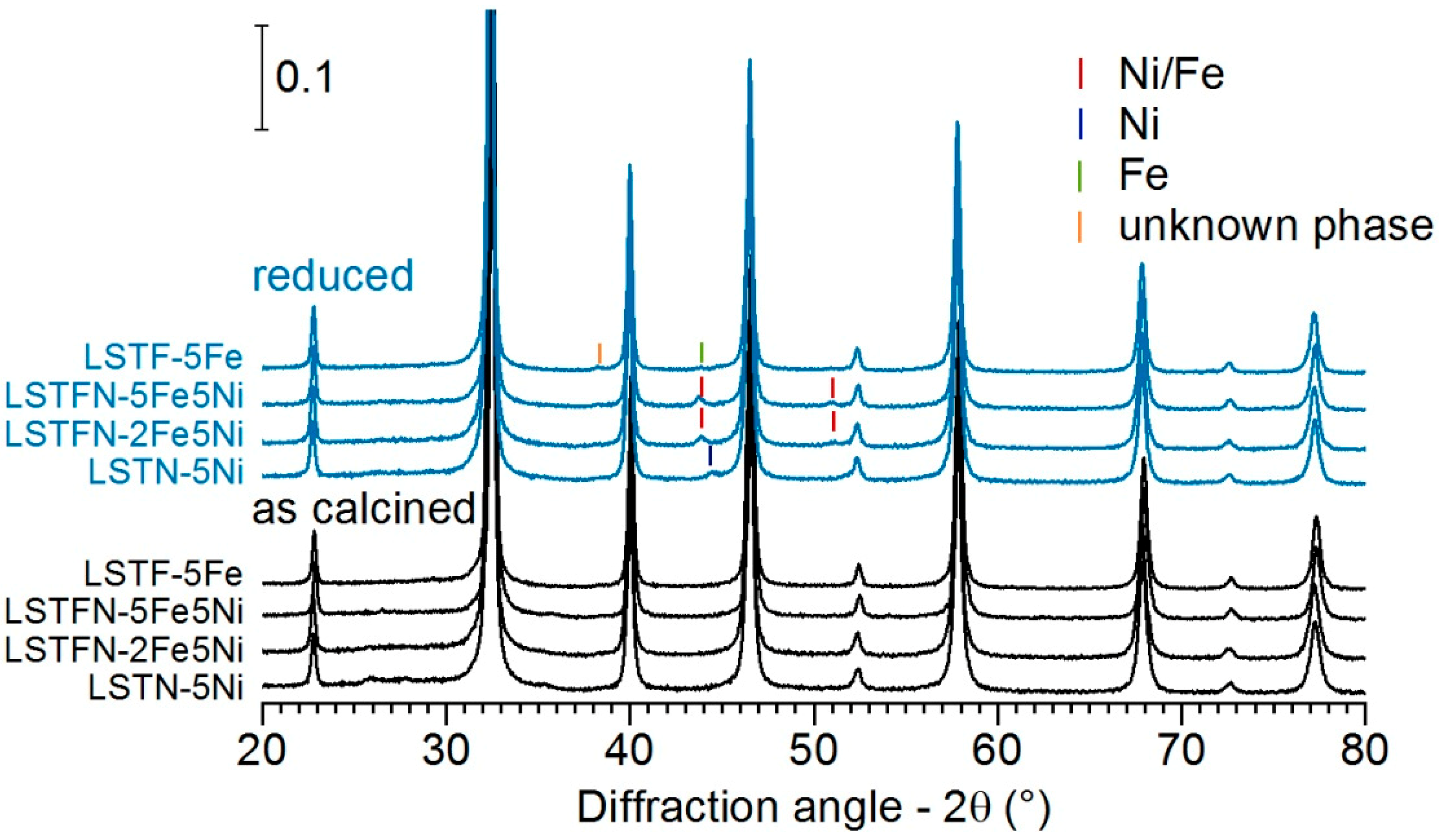
Figure 5 displays a summary of XRD patterns collected on calcined materials, as well as after 15 h reduction at 800 °C (10 vol.% H
2/Ar).
All calcined samples exhibited only reflections, which could be attributed to the perovskite host. After reduction, weak reflections belonging to metallic phases were observed. Reduced La0.3Sr0.55Ti0.95Ni0.05O3±δ (LSTN-5Ni) showed a reflection centered at 44.40°, which corresponds to the Ni (111) reflection. Reduced La0.3Sr0.55Ti0.925Fe0.025Ni0.05O3±δ (LSTFN-2Fe5Ni) and La0.3Sr0.55Ti0.9Fe0.05Ni0.05O3±δ (LSTFN-2Fe5Ni) exhibited a weak reflection at 43.77°, which is higher than one would expect for the (111) reflection of metallic Fe (43.6° 2θ), but certainly at lower angles than the Ni (111) reflection. This can be regarded as evidence for Ni/Fe alloy particle formation upon reduction of LSTFN-type materials. La0.3Sr0.55Ti0.925Fe0.025Ni0.05O3±δ (LSTF-5Fe) did not show significant reflections of metallic Fe after reduction. However, a new reflection appeared at 38.27°, which could not be assigned to any metallic phase. While metallic Ti is expected to display a reflection at around 38.4°, it is highly unlikely that it formed under these pretreatment conditions, due to the inherent stability of Ti4+. The missing reflection at 35.2° excludes this possibility conclusively. However, the observed reflection could be explained by the formation of a new perovskite-type phase of lower symmetry. Orthorhombic perovskites (such as A-site stoichiometric LaSrFeO3±δ) possess a reflection at ca. 38° corresponding to the (113) lattice planes. All other reflections might be either hidden below the dominant original perovskite phase or too weak to be observed.
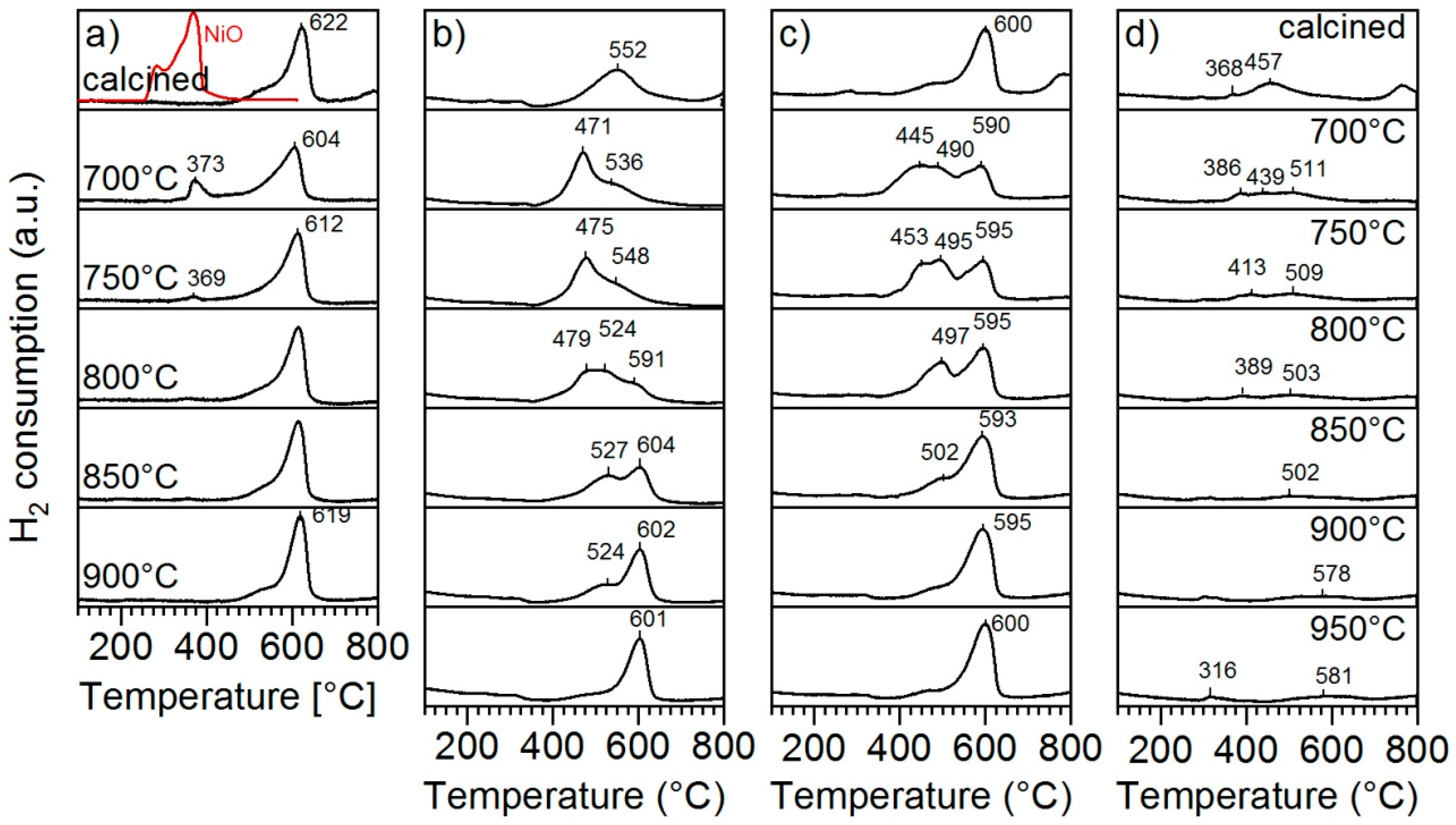
The materials were subjected to TPR and reoxidation cycles to verify the structural reversibility and to determine the temperature at which segregated metals are reversibly incorporated into the host perovskite lattice. In these experiments, TPR profiles were followed by an isothermal reduction at 800 °C. The materials were then subsequently reoxidised at the indicated temperature (700 °C, 750 °C, 800 °C, 850 °C, 900 °C and 950 °C) before the next TPR profile was collected. The TPR profile is sensitive to the nature and coordination environment of reducible metal species and such experiments can, therefore, be exploited to determine the reoxidation temperature needed to reestablish the state of the reducible metal species in the initial calcined material [
2,
23].
Figure 6a displays the TPR redox cycles obtained on LSTN-5Ni. The reduction feature of NiO (ca. 370 °C) disappears after reoxidation at T
reox ≥ 800 °C, thus indicating successful and complete Ni reincorporation at this temperature [
2]. The initial TPR of calcined LSTFN-2Fe5Ni (
Figure 6b) was not as well defined as the one recorded for LSTN-5Ni. Instead of the distinct double feature, the sample exhibited a broad reduction peak between 400 °C and 650 °C. After reduction and subsequent reoxidation at low temperatures (700 °C and 750 °C) the sample exhibited a low temperature feature peaking at around 475 °C. This feature then transitioned into the previously observed double feature for reoxidation above 850 °C, which could be interpreted as the temperature at which both Fe and Ni are reincorporated into the perovskite host.
The sample with higher Fe content (LSTFN-5Fe5Ni;
Figure 6c) exhibited the reduction feature, which was attributed previously to a two-step reduction process of Ni [
24]. However, peak reduction temperatures were shifted to lower temperatures by 22 °C compared to the ones recorded for LSTN-5Ni in
Figure 6a. Reoxidation at lower temperatures caused the formation of a new reduction feature between 350 °C and 550 °C also on this sample, which can be attributed to the reoxidation of Ni/Fe oxides at the perovskite surface. Reestablishment of the initial reduction profile was achieved after reoxidation at 850 °C. Interestingly, LSTF-5Fe (
Figure 6d) did not exhibit as an extensive reduction as the other samples; the reduction features were broad and attenuated with increasing reoxidation temperature. In this case, TPR-redox cycling seemed unsuitable to accurately trace the reoxidation temperature necessary for Fe reincorporation and demonstrates the limited reducibility of Fe in LSTF-5Fe in absence of Ni.
Even though TPR provides important insight in the reducibility of the materials, only an element specific method, such as X-ray absorption spectroscopy (XAS) can be used to ultimately differentiate between individual contributions of two or more reducible species. Therefore, XAS was applied to investigate the effect of the simultaneous presence of both Fe and Ni in LSTFN-type samples on the reduction and reoxidation of the individual metals.
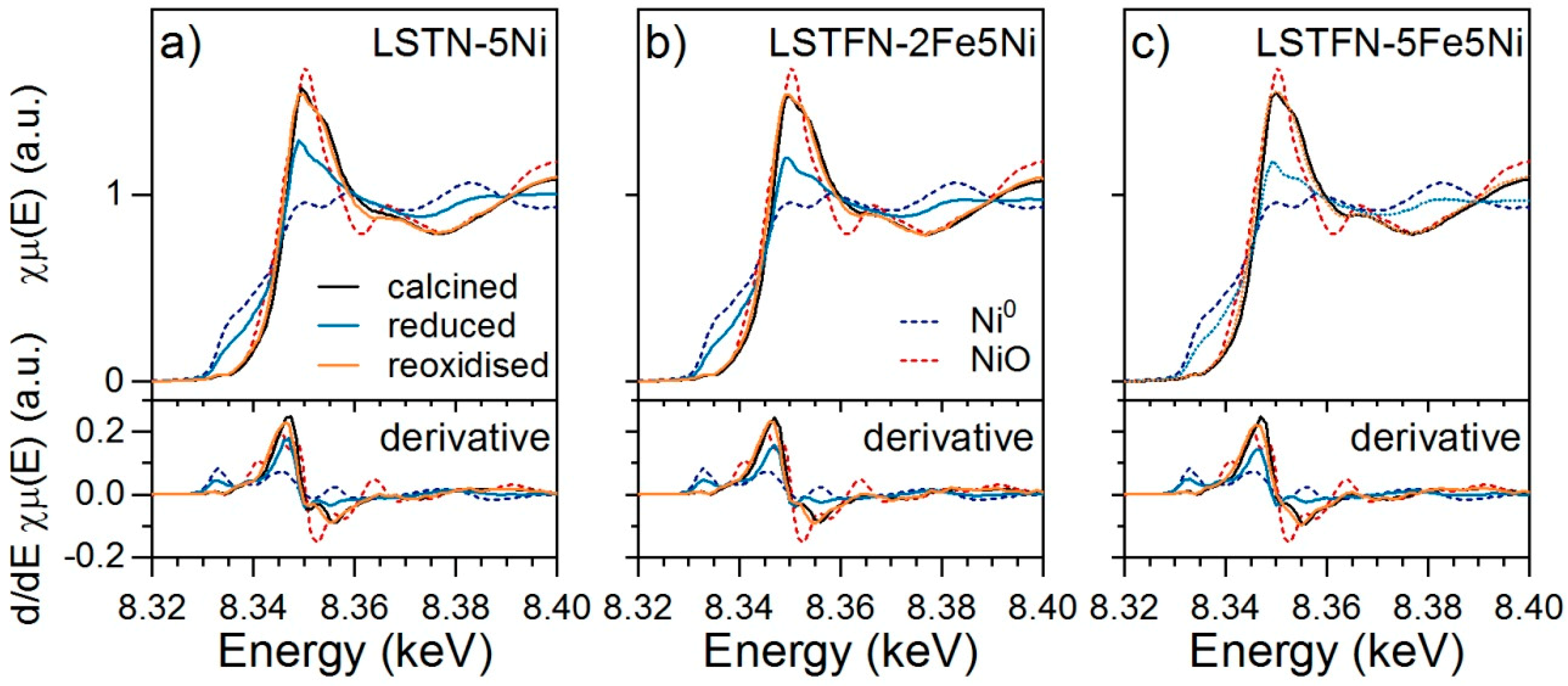
Figure 7 displays the Ni K-edge (8.333 keV) X-ray absorption near edge structure (XANES) spectra of the Ni-containing samples LSTN-5Ni (
Figure 7a), LSTFN-2Fe5Ni (
Figure 7b) and LSTFN-5Fe5Ni (
Figure 7c) in their calcined state, as well as after reduction (10 vol.% H
2/Ar, 800 °C, 15 h) and reoxidation (20 vol.% O
2/N
2, 800 °C, 2 h).
The Ni K-edge spectra of the calcined materials displayed an intense whiteline with a high energy shoulder that can be taken as characteristic of Ni adopting the coordination environment of Ti [
2,
25,
26]. The edge energy was also higher than in the case of Ni
2+ in NiO. After reduction, the spectra corresponded to a linear combination of the spectra of Ni adopting the coordination environment of the B-site after calcination and Ni
0. Although TPR analysis showed reversibility only after reoxidation at 850 °C (
Figure 6), the shape of the XANES spectra and thus the state of Ni were completely reversible in this redox cycle, which was carried out at lower temperatures. It was also observed that the contribution of the Ni
0 reference to the spectra of the reduced samples increased with increasing Fe concentration. This was confirmed by linear combination fit (LCF) of the spectra indicating that the amount of Ni
0 increased from 52% in LSTN-5Ni to 62% in LSTFN-2Fe5Ni and to 67% in LSTFN-5Fe5Ni. LCF results are summarized in
Figure S1 and the corresponding fit results are shown in
Figure S2.
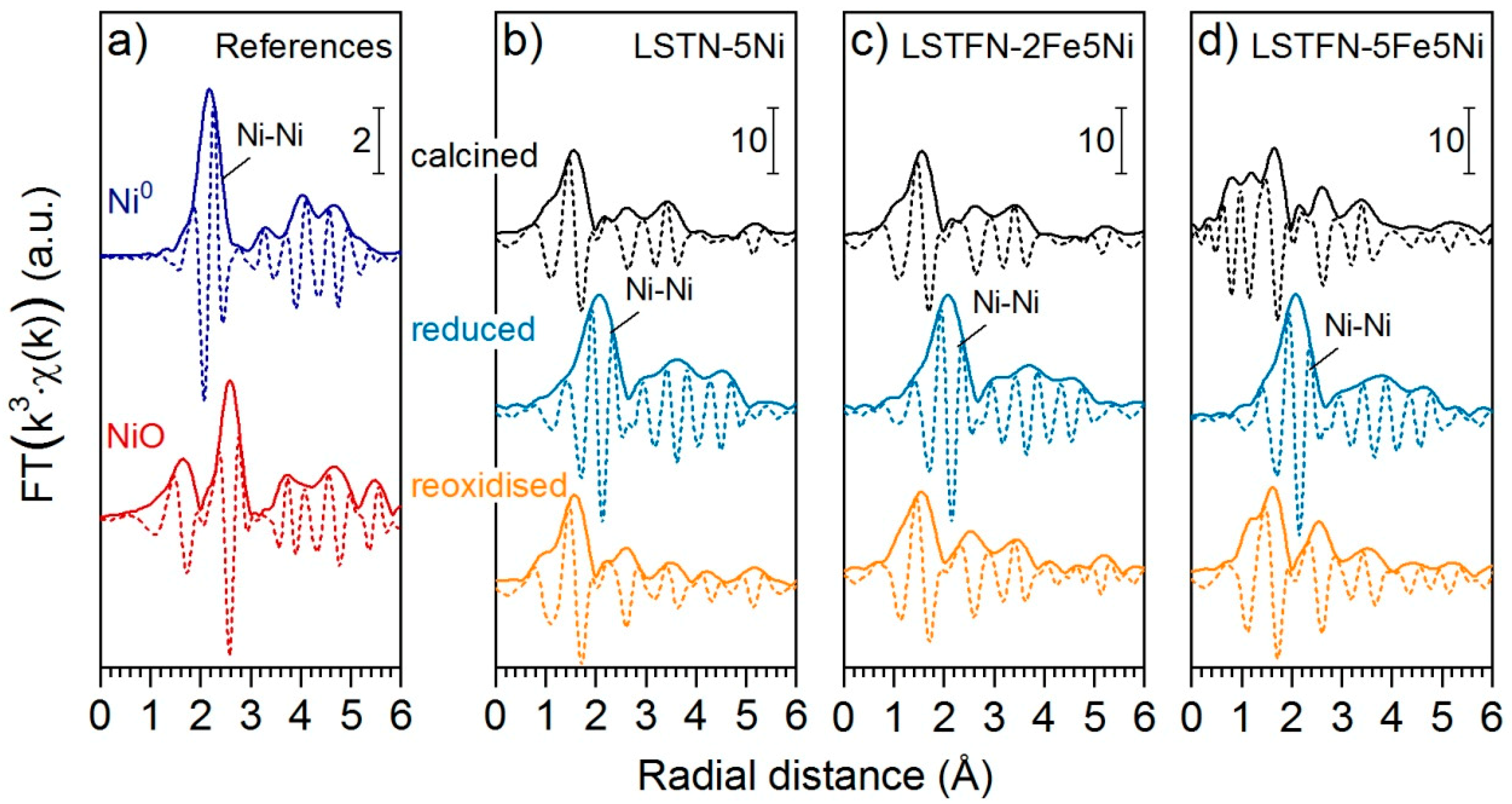
Evidence that Ni was not only reduced, but also segregated and formed metallic particles is provided by the extended X-ray absorption fine structure of the Ni K-edge. The k
3-weighted data is shown in
Figure S3 for all samples, as well as Ni references. The radial distances of coordination shells become obvious through Fourier transformation of this data, as shown in
Figure 8. After reduction, the feature attributed to a Ni-Ni coordination shell appeared at 2.15 Å. This feature was present for all samples so that metal particle formation can be assumed likewise.
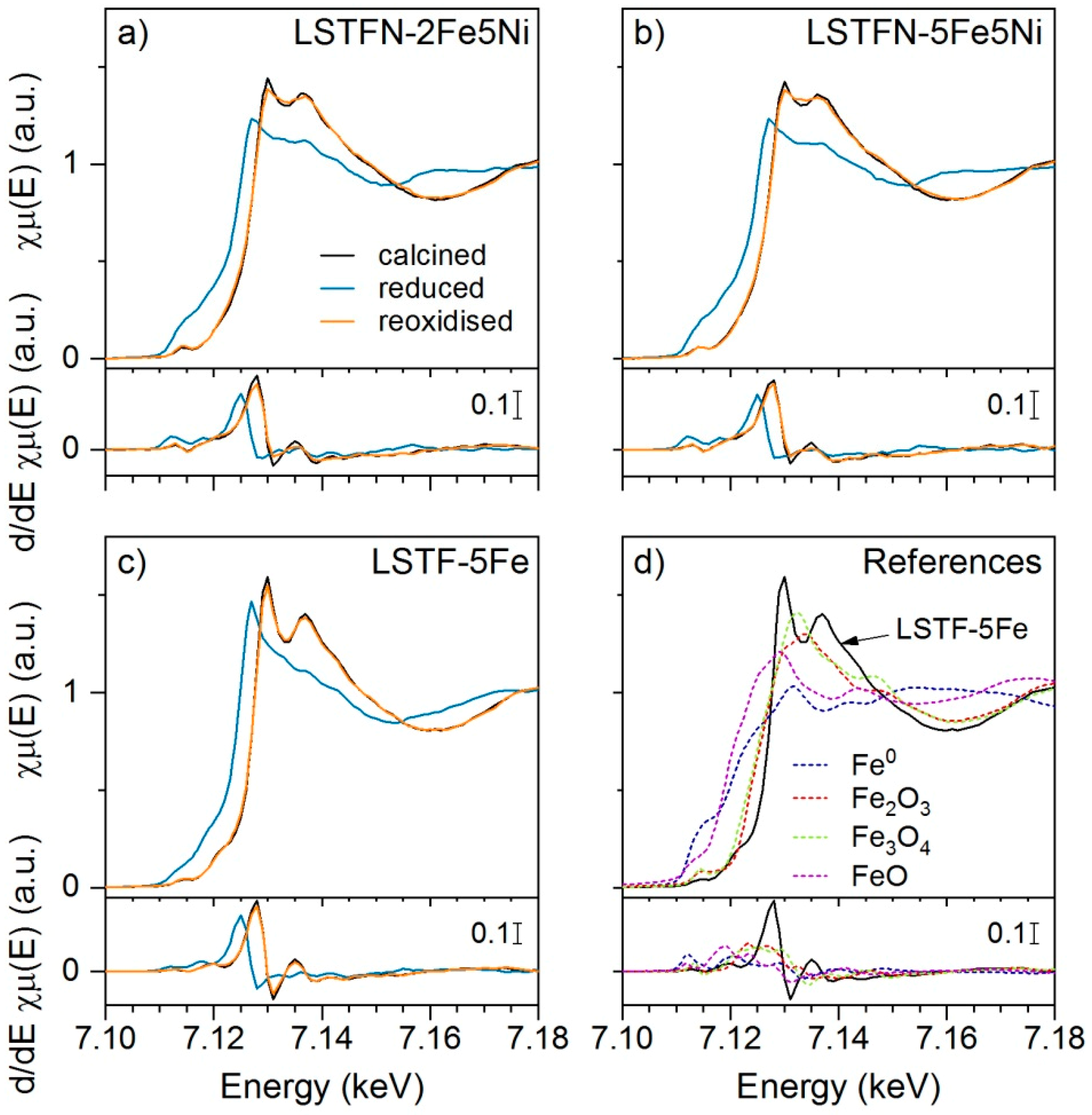
Fe K-edge (7.112 keV) XANES data was obtained on the Fe-containing samples LSTFN-2Fe5Ni, LSTFN-5Fe5Ni and LSTF-5Fe and spectra of calcined, reduced and reoxidized samples are displayed in
Figure 9 along with spectra of Fe
0, FeO, Fe
3O
4 and Fe
2O
3 reference compounds. Significant changes in the shape of the XANES could be observed also for Fe K-edge absorption spectra over the redox cycle. Clear Fe reduction could be observed by a decrease in whiteline intensity, as well as a shift in absorption edge energy (E
0). This shift is best determined through the position of the first maximum in the derivative of the absorption curves, which changes from 7.128 keV for calcined materials to 7.125 keV for reduced materials, corresponding to a decreased ionization energy, which is typically observed for reduced states. However, in the spectra of the reduced samples, the whiteline did not correspond to a simple linear combination of the reference spectrum of Fe
0 and the spectrum of calcined LSTFN- or LSTF-type materials, thus suggesting other states of Fe.
No suitable fits could be obtained through LCF analysis using all displayed Fe reference spectra. This may be linked to the presence of other Fe-containing phases as was already suggested from the XRD patterns in
Figure 1. Furthermore, the XANES of LSTFN-2Fe5Ni and LSTFN-5Fe5Ni was not completely restored in the reoxidized materials as can be seen in the region of the local minimum at around 7.135 keV. LCF of the spectra of the reoxidized materials indicated the presence of Fe
3O
4 (ca. 17%) and thus incomplete reincorporation of Fe under the applied reoxidation conditions.
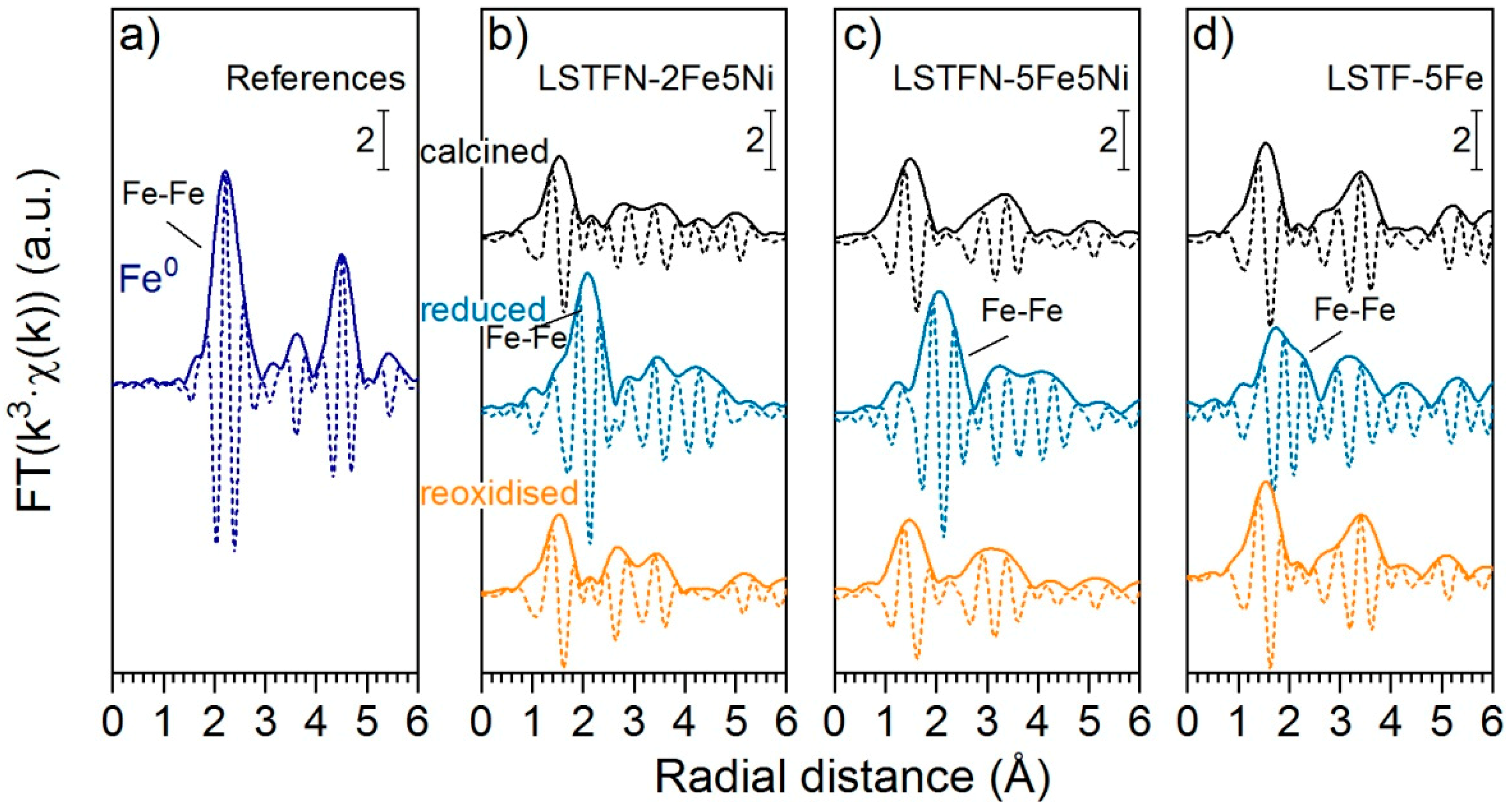
The k
3-weighted Fe K-edge (7.112 keV) EXAFS data of LSTFN-2Fe5Ni, LSTFN-5Fe5Ni and LSTF-5Fe is shown in
Figure S4, whereas the Fourier transformed data is shown in
Figure 10. Compared to the Ni
0 reference in
Figure 8, Fe
0 in the Fe foil displayed the first coordination shell at a slightly longer radial distance (2.23 Å) and similar to the Ni K-edge data contributions of this feature, could be found in the spectra of the reduced samples. This indicates that besides Ni Fe was also partially reduced to Fe
0 and was present in the form of metal particles. Interestingly, the contribution of this feature to the spectra of the reduced samples decreased with decreasing Ni/Fe ratios along the series LSTFN-2Fe5Ni > LSTFN-5Fe5Ni > LSTF-5Fe, which suggests that larger Ni content favors Fe reduction. Hence, the positive influence of one metal on the reducibility and segregation of the other metal could be observed.
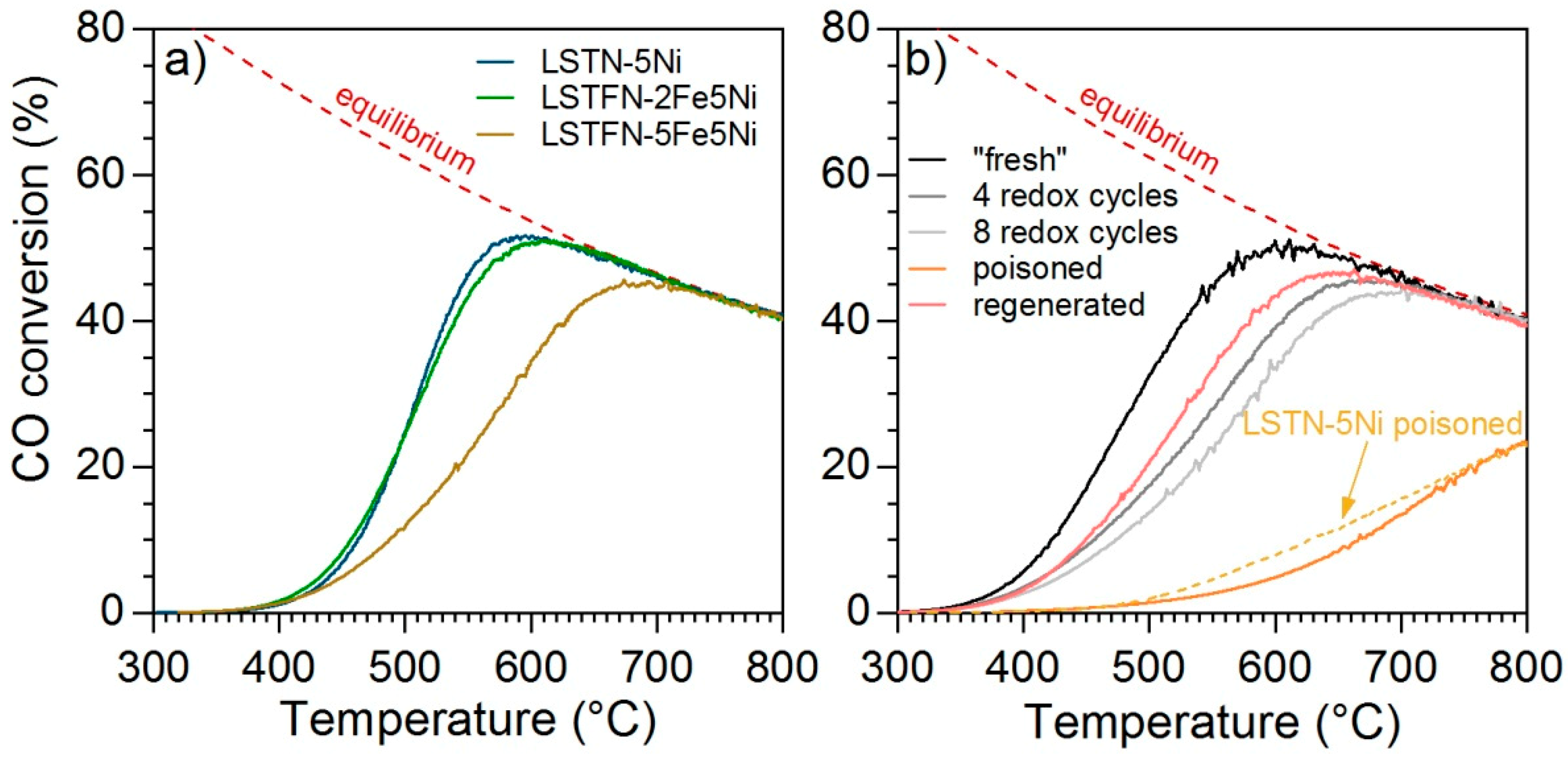
Figure 11a shows CO conversion curves for the Ni-containing samples LSTN-5Ni, LSTFN-2Fe5Ni and LSTFN-5Fe5Ni. It is apparent that the conversion curves shifted to higher temperatures with increasing Fe concentration, which corresponds to a decrease in catalytic activity. This is in contrast to the previous observation that the presence of Fe increases Ni reducibility (
Figure S1). The expected effect would be a larger amount of active Ni
0 and thus higher catalytic activity. On the other hand, the observation is in line with the decrease in WGS activity observed for LSTN-5Fe compared to LSTN in
Figure 3b and the indication of Ni/Fe alloy formation during reduction provided by this catalytic activity data, as well as XRD (
Figure 5). Since the addition of small quantities of Fe did not appear to be detrimental for catalytic activity, LSTFN-2Fe5Ni was selected for testing catalytic activity with respect to its redox stability, as well as sulfur tolerance. However, it can be seen in
Figure 11b that CO conversion decreased over the number of redox cycles, which could be a consequence of Ni/Fe particle growth, due to the incomplete Fe reincorporation over the redox cycles observed by XANES (
Figure 9). Despite the fact that Ni re-incorporated completely during reoxidation of reduced LSTFN-2Fe5Ni (
Figure 7), the remaining Fe oxide at the surface may have caused particle growth and thus the observed catalyst deactivation over the consecutive redox cycles.
The small amounts of Fe (2.5 mol.% or 0.75 wt.%) also did not provide additional stability against poisoning by sulfur, which can be realized from the CO conversion measured after catalyst poisoning by H
2S (
Figure 11b). CO conversion was as low as in poisoned LSTN. A complete redox cycle recovered catalytic activity, which was even improved compared to the catalytic activity measured before poisoning. The reason behind this phenomenon was not further investigated as any obvious improvement in performance in terms of the catalytic activity of LSTFN-2Fe5Ni in neither sulfur-free nor sulfur-containing reaction gas feeds could be observed. Nevertheless, it indicates that sulfur can also be successfully removed from LSTFN-type oxides over oxidation-reduction cycles at 800 °C, which could be potentially exploited to completely regenerate these materials if catalyst redox stability can be achieved. The conditions to achieve full reversibility of activity after poisoning should be the aim of future work.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}