1. Introduction
Molecular hydrogen gas detected by Cassini spacecraft indicates water-rock interaction in active hydrothermal systems within Saturn’s icy moon Enceladus [
1]. Composition and temperature of the hydrothermal system in Enceladus were estimated using the composition and the size of fine silica dust particles discovered by Cassini [
2,
3]. The dust particles emerging from the south pole of Enceladus continuously supply the Saturn’s E ring [
4]. In addition, Cassini detected a number of gas species such as carbon dioxide, ammonia, and methane in the erupting plume. Methane might be produced in hydrothermal systems by methanogens [
5] or by abiogenic reductive reactions of carbon dioxide.
Substantial amount of molecular hydrogen gas is observed to generate during serpentinization of ultramafic rocks in hydrothermal vents on the Earth [
6,
7] and in laboratories. The hydrogen molecules are involved in a sequence of reductive reactions of single carbon compounds and convert dissolved carbon dioxide into methane by mineral catalyzed hydrothermal reactions [
8,
9,
10]. Isotopic labeling of dissolved carbon dioxide is used to identify the source of the produced methane in the hydrothermal conditions [
11,
12,
13]. It was shown that high temperature water is required for the synthesis of methane from dissolved carbon dioxide.
High temperature water has different properties from ambient liquid water: low density and low dielectric constant [
14,
15,
16]. Specially water with the temperature and the pressure above the critical point (374 °C, 218 atm) is called supercritical water. Supercritical water is an ideal medium to destruct organic compounds and is studied extensively in science and technology. Oxidative reactions in supercritical water were studied using single carbon compounds such as methane, methanol, and carbon monoxide [
17,
18,
19].
Brock and Savage [
17] assumed that the gas phase reactions can be applied to reactions in supercritical water with the temperature range of 500–600 °C. They adopted the ideal gas approximation for reactants and constructed a detailed chemical kinetics model based on combustion kinetics. The model consists of 22 species and 148 reversible elementary free-radical reactions. The numbers of elementary reactions and species can be reduced to correctly predict the concentrations because the kinetics of the oxidation is controlled by a small number of elementary reactions [
17,
20]. They successfully reproduce the experimentally determined oxidation rate of methane at the high temperature above the critical point. This indicates the validity of the ideal gas approximation to describe reactions in supercritical water. It was shown that HO
2 and OH radials play crucial roles in the supercritical water oxidation. The calculated activation energy for the oxidation rate of methane becomes 36 kcal/mol, which is smaller than the experimental value of 44 kcal/mol [
19]. The predicted rate constant becomes a little higher than that determined by experimental studies when the temperature decreases to the critical temperature.
Chemical reactions in supercritical water are dominantly governed by free-radical reactions. However, free-radical reactions lose the important role in chemical reactions in hot water with the decreasing temperature [
15]. Free radicals rapidly interact with other atoms or molecules and turn into a stable molecule. Instead the acid- and base-catalyzed reactions start to act effectively with an increase in the ion product. The ion product of water is dependent on the temperature and has the maximum value of
around 300 °C [
14]. The ion product at 300 °C is nearly three orders of magnitude larger than that of ambient liquid water, suggesting a critical role of hydroniums and hydroxides in chemical reactions in hot water. Uddin et al. [
21] examined the acid–base catalytic effects on the hydration of formaldehyde using the quantum mechanical/molecular mechanics-molecular dynamics. They found that hydration of formaldehyde is efficiently catalyzed by hydronium and hydroxide.
In the present study, we perform a number of quantum chemical simulations to investigate the formation of methane through the acid–base catalyzed reduction of methanol in hot water. We apply the Gaussian 4 method of the Gaussian 16 software [
22] to locate and optimize the geometries of a reactant compound, a transition state, and a product compound. A reactant compound includes a hydronium or a hydroxide in addition to a methanol, a hydrogen molecule and some water molecules. We will show that the energy barrier for the reduction is significantly reduced by the acid–base catalytic effects. We calculate the acid–base catalyzed reduction rate of methanol and compare it with that determined by laboratory experiments.
2. Computational Method
We adopted a simple model for the reduction of methanol in hot water. A methanol interacts with a hydrogen molecule in hot water, forming a methane and a water molecule, expressed as
Water molecules as well as a hydronium or a hydroxide can get involved in the reduction as catalyst. In our previous studies, we revealed the important role of water molecules during oxidative and reductive reactions of a single carbon compound [
23,
24,
25,
26,
27]. Water molecules help to transfer a proton of a reactant molecule to proceed the reactions in aqueous solution. The energy barrier of the reaction is significantly reduced by the catalytic effect of water molecules. We included water molecules as well as a hydronium or a hydroxide in the present quantum chemical simulations to follow the motion of a proton during the reduction process.
A tentative geometry of a molecule was generated using the MOLDEN software [
28]. We employ the Gaussian 16 software [
22] to optimize the geometry and calculate the energy barrier for the reduction. The geometry was optimized using the B3LYP [
29,
30,
31] functional with the 6-311+G(3df, 2p) basis set [
32,
33,
34,
35]. Then, we applied the Gaussian 4 method [
36] to obtain the accurate electronic energy of the molecule. We used the MOLDEN software to display the geometries of the molecules.
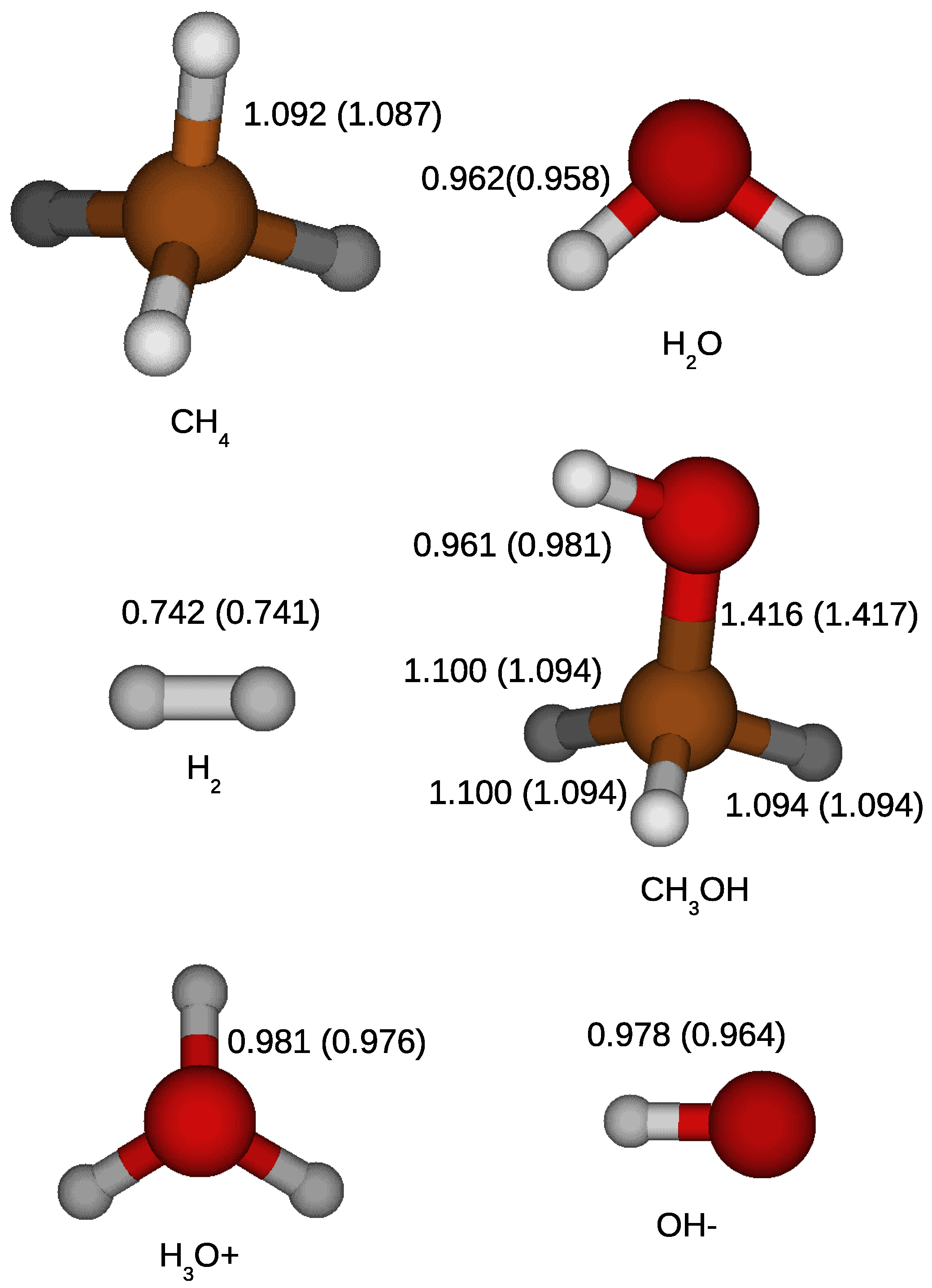
Figure 1 shows the calculated geometries of methane, water, methanol, hydrogen molecule, hydronium, and hydroxide with the calculated bond lengths as well as those determined by laboratory experiments. The geometries of the molecules were represented well by the calculations.
We used a trial geometry to locate a transition state with the synchronous transit guided quasi Newton method. The transition state was confirmed by finding a single imaginary frequency of vibration. Once we obtained a transition state, we performed the intrinsic reaction coordinate (IRC) calculation and follow the minimum energy pathway to reach a reactant compound and a product compound. The reactant compound and the product compound were further optimized with the Gaussian 4 method to calculate the electronic energies and the vibrational frequencies of atoms in the compounds. We estimate the energy barrier, , for the reduction of a methanol from the difference of the electronic energies corrected by the zero point energy between a reactant compound and a transition state.
We applied the transition state theory [
43] to calculate the reduction rate of methanol using the following equation:
where
is the tunneling correction factor to enhance the reduction rate by the tunneling effect of a proton,
,
T, and
h are the Boltzmann constant, the temperature, and the Planck constant, respectively, and
and
are the partition functions of a reactant compound and a transition state. We approximated the potential barrier by the unsymmetrical Eckart potential and estimated the tunneling correction factor following our previous study [
23]. The partition function is expressed as the product of the partition functions due to translation, rotational motion, and vibrational motion.
3. Numerical Results and Discussion
Our discussion begins with the reductive reaction of a methanol in the gas phase. A hydrogen molecule directly attacks a hydroxyl of a methanol in the gas phase reaction.
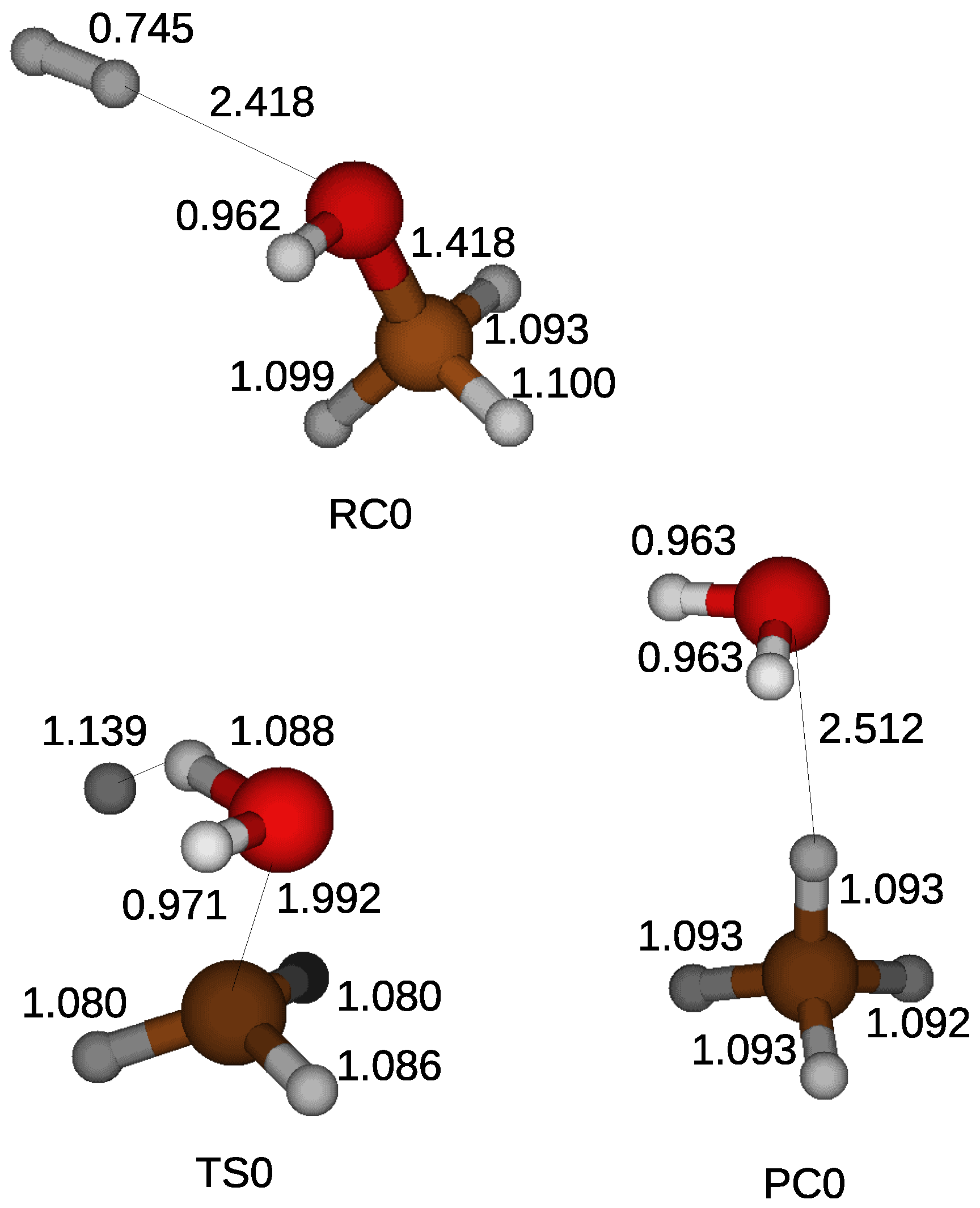
Figure 2 shows a reactant compound, a transition state, and a product compound, calculated with the Gaussian 4 method. A hydrogen molecule is weakly bonded with a hydroxyl of a methanol by a hydrogen bond in a reactant compound. The CO bond of the methanol is broken when a hydrogen molecule approaches the methanol. The approaching hydrogen molecule loses one of the hydrogen atoms, which is combined with the hydroxyl of the methanol. The transition state is formed by forming a water molecule and leaving a CH
3 behind. The CH
3 later holds the hydrogen atom and becomes a methane. The energy barrier for the reduction is calculated from the difference of the electronic energies corrected by the zero point energy between a reactant compound and a transition state.
Table 1 shows the energy barriers as well as the free energy difference between a reactant compound and a transition state. The energy barrier becomes 90.4 kcal/mol when a methanol reacts with a hydrogen molecule in the gas phase.
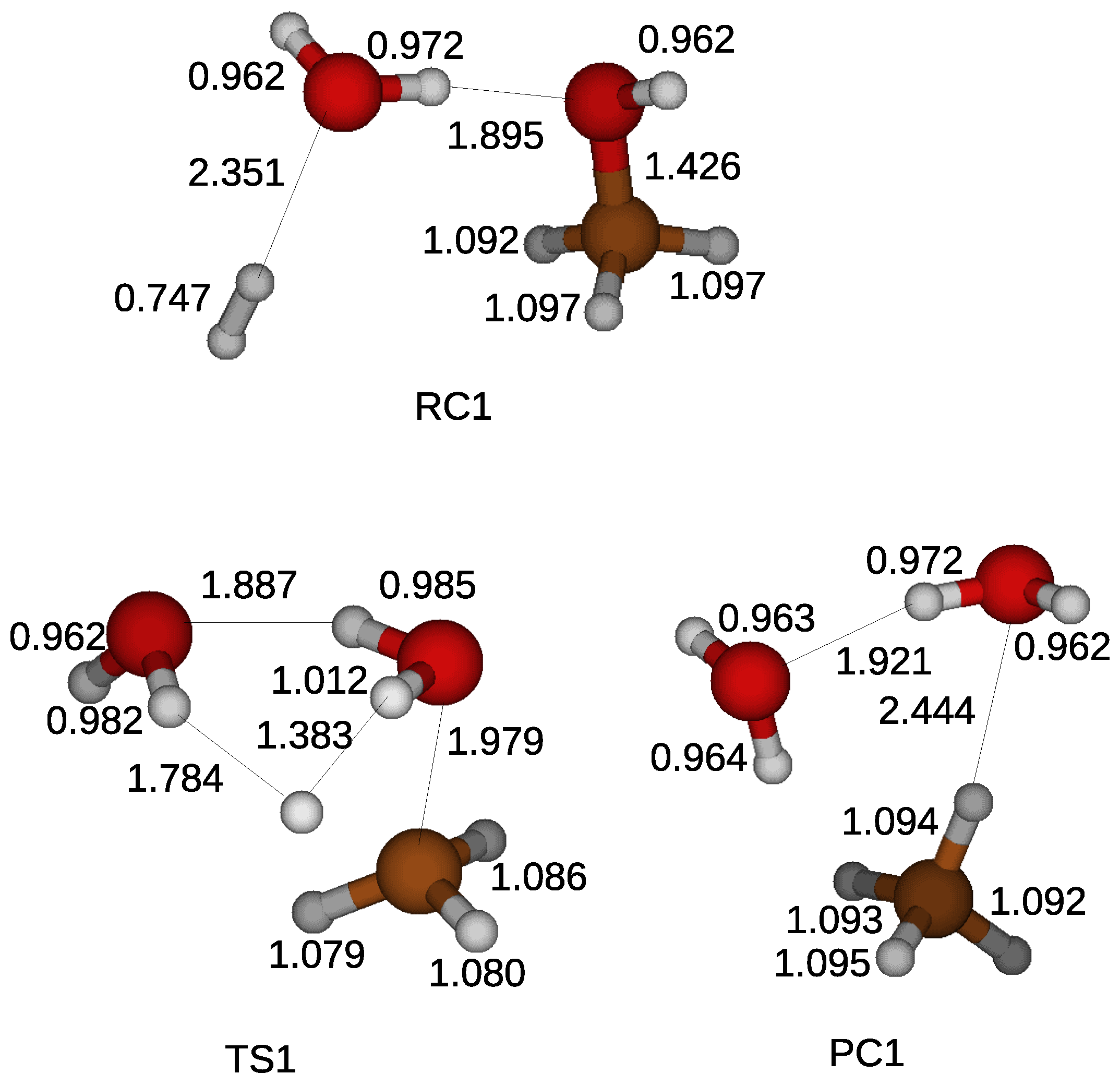
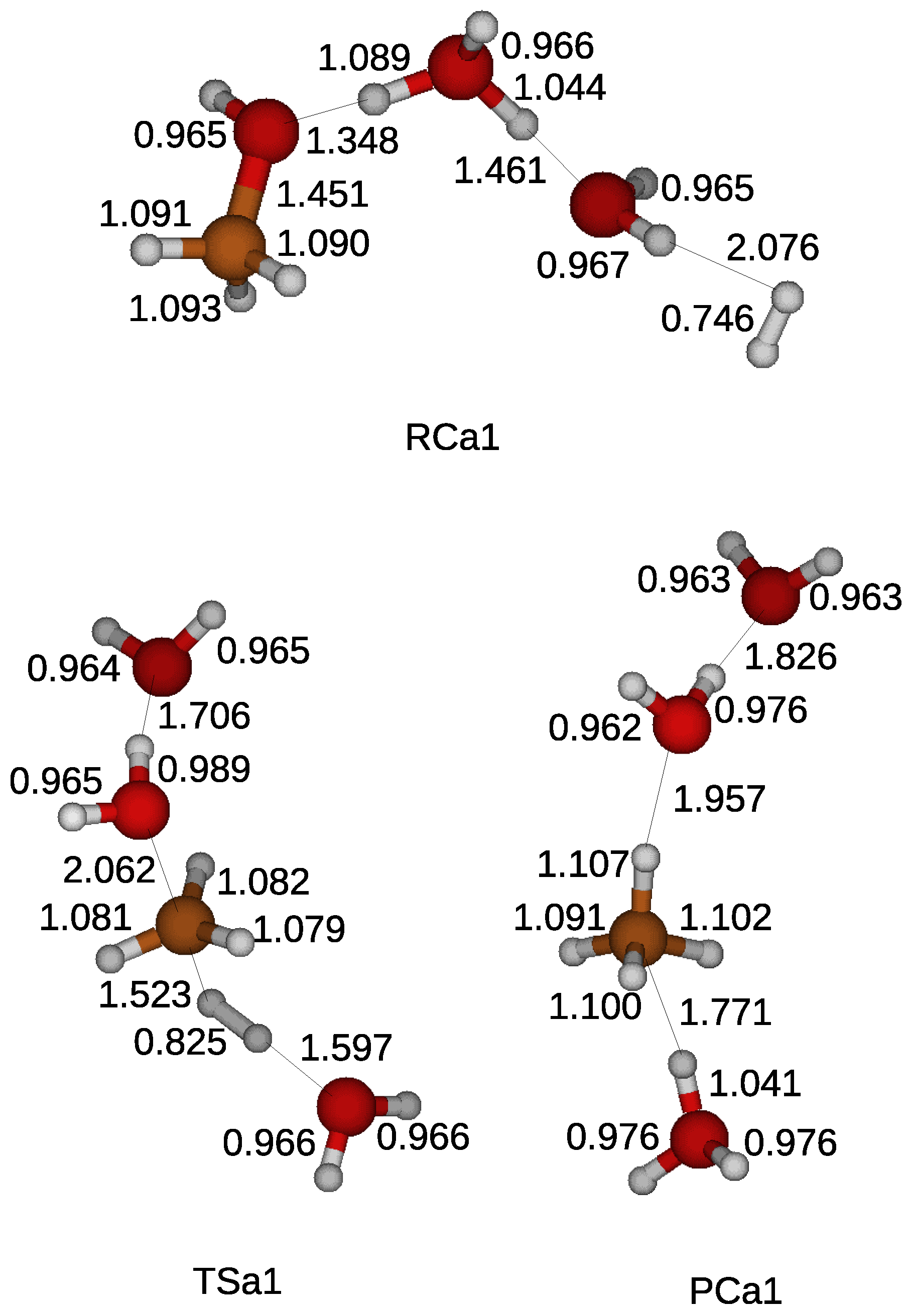
A water molecule was introduced in the quantum chemical simulation to examine the effect of water molecules on the reduction of a methanol. A water molecule was weakly bonded with a hydroxyl of a methanol and a hydrogen molecule by hydrogen bonds in a reactant compound as shown in
Figure 3. As in the gas phase reaction, the reduction process began with the cleavage of the CO bond of the methanol. A hydrogen atom of a water molecule was shared with the hydroxyl liberated from the methanol. Two water molecules appeared when a hydrogen atom was transferred from a hydrogen molecule in a transition state. A methane molecule arose after taking the other hydrogen atom in a product compound. The calculated energy barrier of 90.8 kcal/mol was similar to that for the gas phase reduction, indicating the ineffective catalyst of a water molecule.
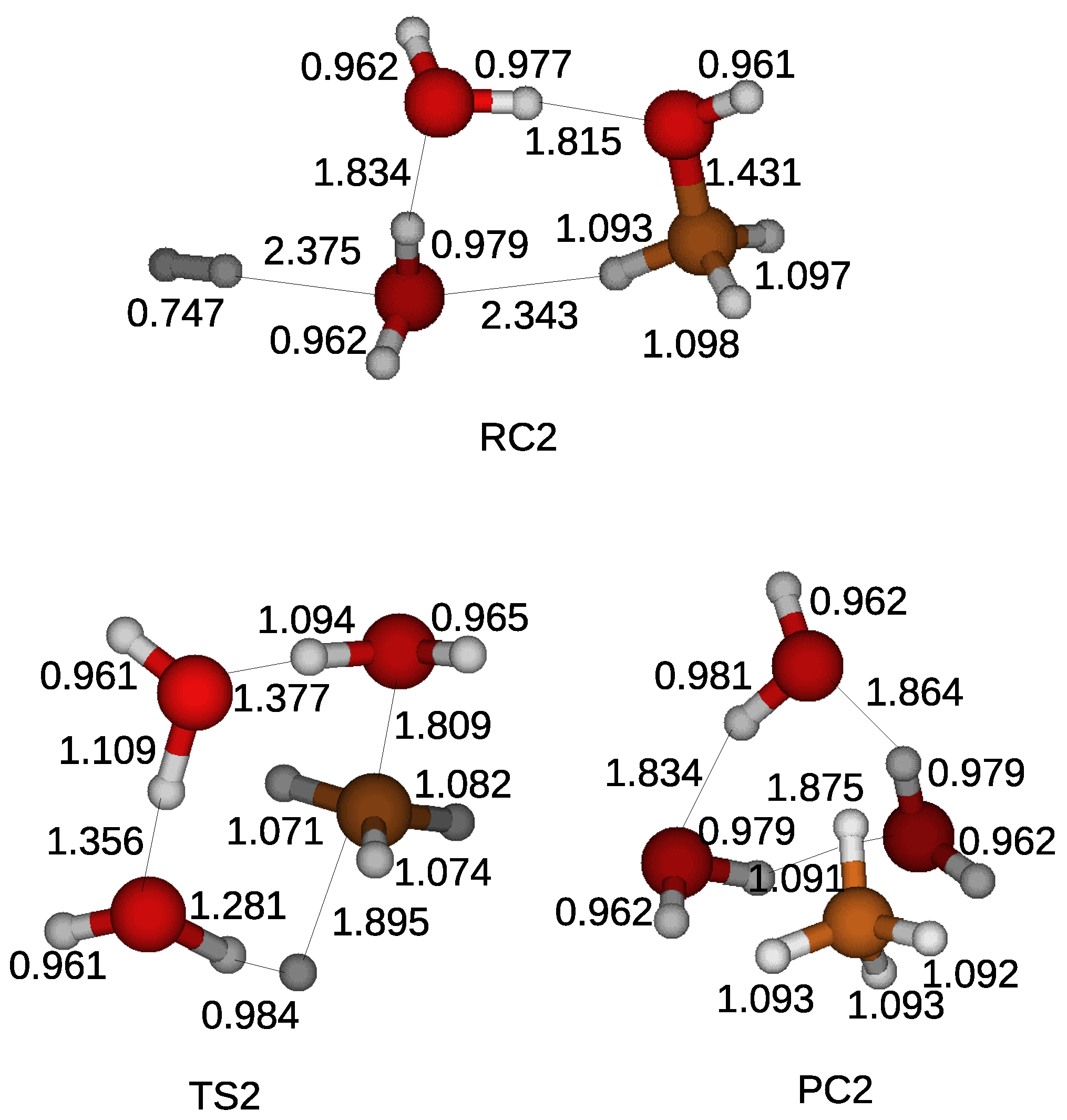
One more water molecule is included in the simulation because our previous studies showed the cooperative work of water molecules to encourage the effective delivery of a hydrogen atom in a compound [
23]. It was shown that the energy barrier efficiently decreases with an increase in the number of water molecules in the simulation. There were some possible locations to place a hydrogen molecules and two water molecules in the simulation. We discovered that the low energy barrier for the reduction of methanol was obtained when a hydrogen molecule approaches a carbon atom of a methanol from the opposite side with respect to the hydroxyl of the methanol as shown in the transition state of
Figure 4. An approaching hydrogen molecule induced the break of the CO bond of the methanol and it being divided into two hydrogen atoms. A water molecule near a hydrogen molecule caught one of the hydrogen atoms and transport another extra hydrogen atom toward the other water molecule. The CH
3 gained a hydrogen atom and turns into a methane. The energy barrier was reduced by 13 kcal/mol, implying the catalytic role of water molecules.
It would be valuable if we could compare our calculated reduction rate with that determined by experimental studies. Unfortunately we failed to find any kinetics study for the reduction of methanol by hydrogen molecule in hot water because it might take long to perform an experiment in a laboratory. We used experimental results of Seewald et al. [
44] and estimated the reduction rate of methanol approximately as follows. The concentration of reactant compound decreased due to the formation of methane and the rate is represented as
where
is the concentration of reactant compound consisted of methanol and hydrogen molecule at time of
t and
k is a proportional constant. Since methane increased through the conversion of the reactant compound, the concentration of methane at time of
t is given by
where
and
are the concentrations of methane and the reactant compound at
, respectively. We determined three unknown constants:
,
, and
k using the experimental data of Seewald et al., in which the concentration of methane in aqueous solution started increasing from 0 mmol/kg and becomes 0.006 and 0.012 mmol/kg at 48 and 216 h, respectively, when the water temperature is 300 °C. The concentration of methane at
t hours is expressed as
From this equation we obtain the experimentally determined reduction rate of methanol as
The transition state theory is applied to calculate the reduction rates of methanol in the gas phase as well as that when one or two water molecules are introduced in the simulation. The reduction rates of methanol increase with the increasing temperature. The highest reduction rate of methanol was obtained when two water molecules were involved in the reduction due to the lowest energy barrier shown in the
Table 1.
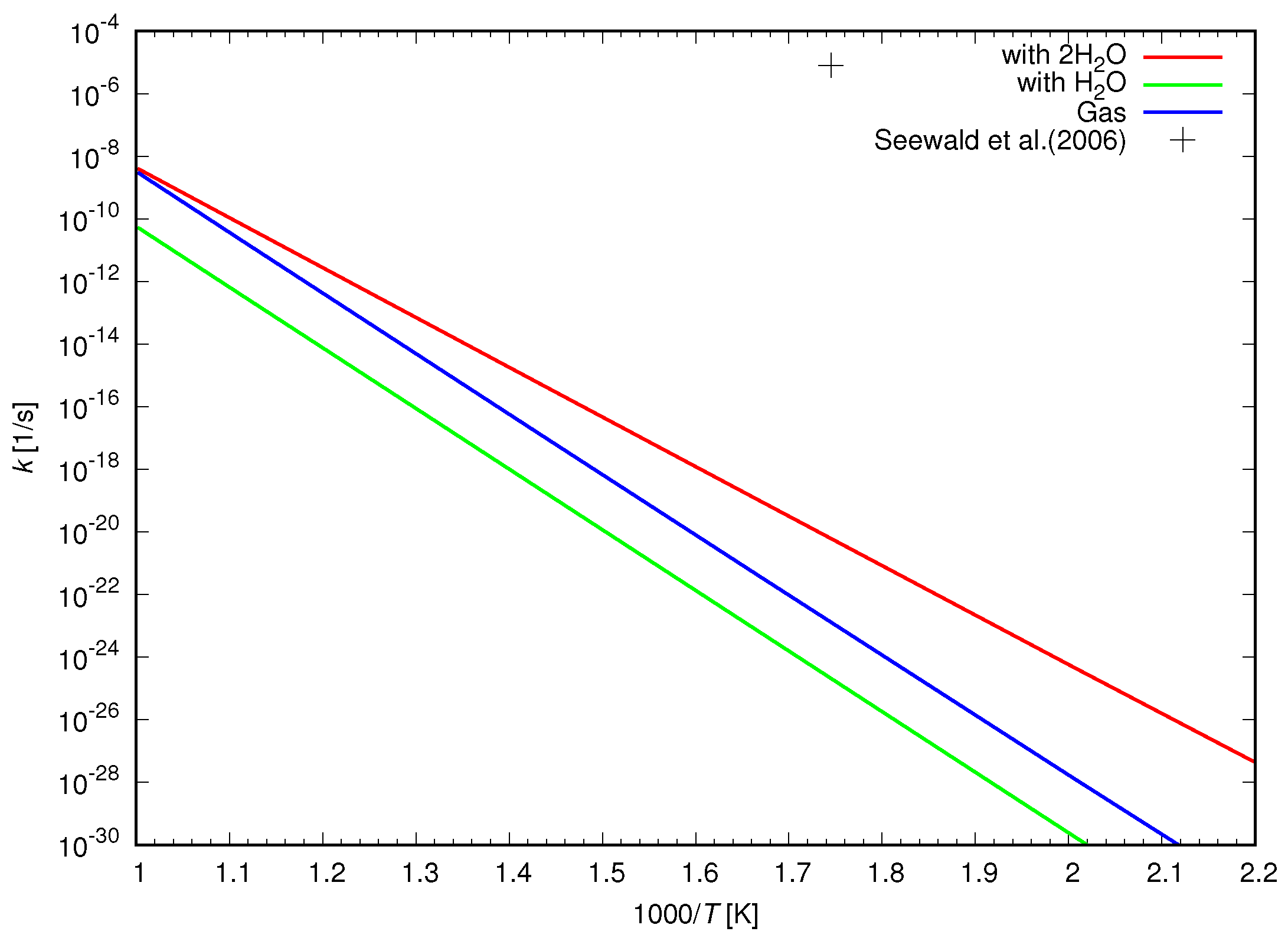
Figure 5 shows the calculated reduction rate as well as that given by Equation (
6). The calculated reduction rate was 15 orders of magnitude lower than that determined by the experimental study.
Another catalytic effect needed to be considered because the catalytic effect of water molecules was too weak to enhance the reduction rate of methanol in hot water. As noted in the introduction, the significant increase in the ion product in hot water implies a crucial role of hydronium and hydroxide as catalysts of the reduction. At first, we considered the acid-catalyzed reaction, in which a reactant compound was comprised of a methanol, a hydrogen molecule, a water molecule, and a hydronium.
Figure 6 displays a reactant compound, a transition state, and a product compound when a hydronium catalyzes the reduction of a methanol. A hydronium connected with the hydroxyl of a methanol and a water molecule by hydrogen bonds in a reactant compound. The reduction process started when a hydrogen atom of a hydronium was delivered to a hydroxyl of a methanol. A hydrogen molecule and a water molecule moved toward the opposite side of the methanol with respect to the hydroxyl. A transition state was built when a water molecule was detached from the CH
3. The CH
3 grabbed a hydrogen atom from a hydrogen molecule and left a hydrogen atom behind. A hydronium was recovered in a product compound when a water molecule received the hydrogen atom. The lowest energy barrier of 45 kcal/mol was obtained by considering the catalytic effect of a hydronium.
We increased the number of water molecules included in the simulations as shown in
Table 1. Similar energy barriers for the reduction were found. With an increase in the number of water molecules, more water molecules were required to move to new positions in a transition state. Entropy difference between a reactant compound and a transition state increases with the increasing number of water molecules. The lowest free energy difference is obtained when a single water molecule is included in the simulation.
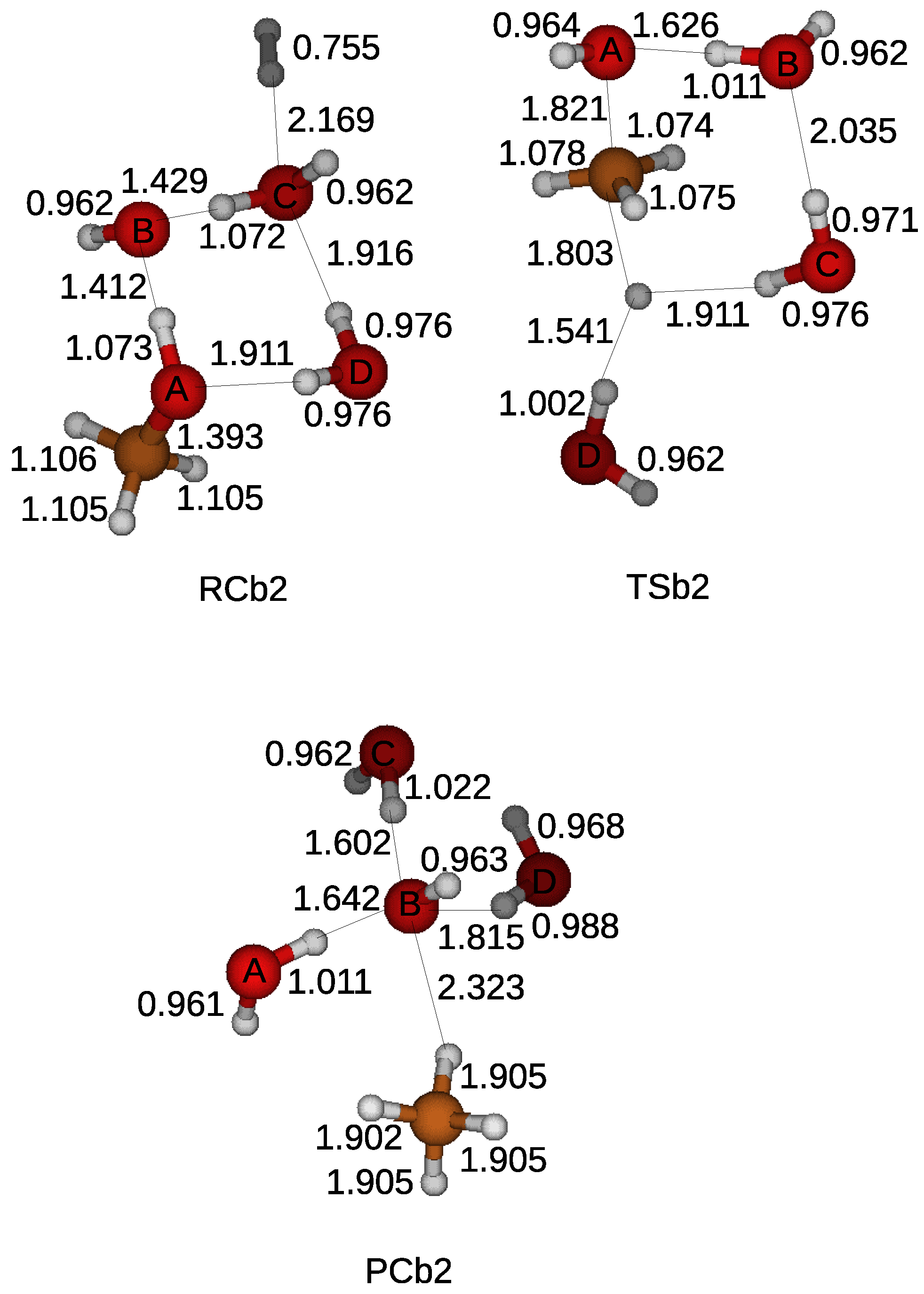
Next, the base-catalyzed reduction was examined by including a hydroxyl in the simulation.
Figure 7 shows that hydrogen bonds connect the hydroxyl with a methanol and a water molecule in a reactant compound. A water molecule with the label D cut the hydrogen bonds and moved toward the opposite side of the methanol with respect to the hydroxyl of the methanol. A hydroxyl of the label B took a hydrogen atom from a water molecule with the label C, which turned into a hydroxyl. A transition state appeared when a hydrogen molecule split into two hydrogen atoms and one of them was captured by the hydroxyl with the label C. The cleavage of the CO bond of the methanol lead to the formation of a hydroxyl with the label A and a CH
3. The hydroxyl with the label A took a hydrogen atom from a water molecule with the label B, while the CH
3 became a methane molecule by taking an isolated hydrogen atom. A product compound was comprised of a methane, a hydroxyl, and three water molecules. It was shown that a hydroxyl acts as a catalyst in the base-catalyzed reaction. The lowest energy barrier was found when two or three water molecules are included in the simulations as displayed in
Table 1. The free energy difference became lowest when two water molecules are included in the simulation due to the smaller change of entropy during the reduction. The lowest energy barrier reduced by the catalytic effect of a hydroxyl becomes 51 kcal/mol, which was a little higher than the energy barrier of the acid catalyzed reduction.
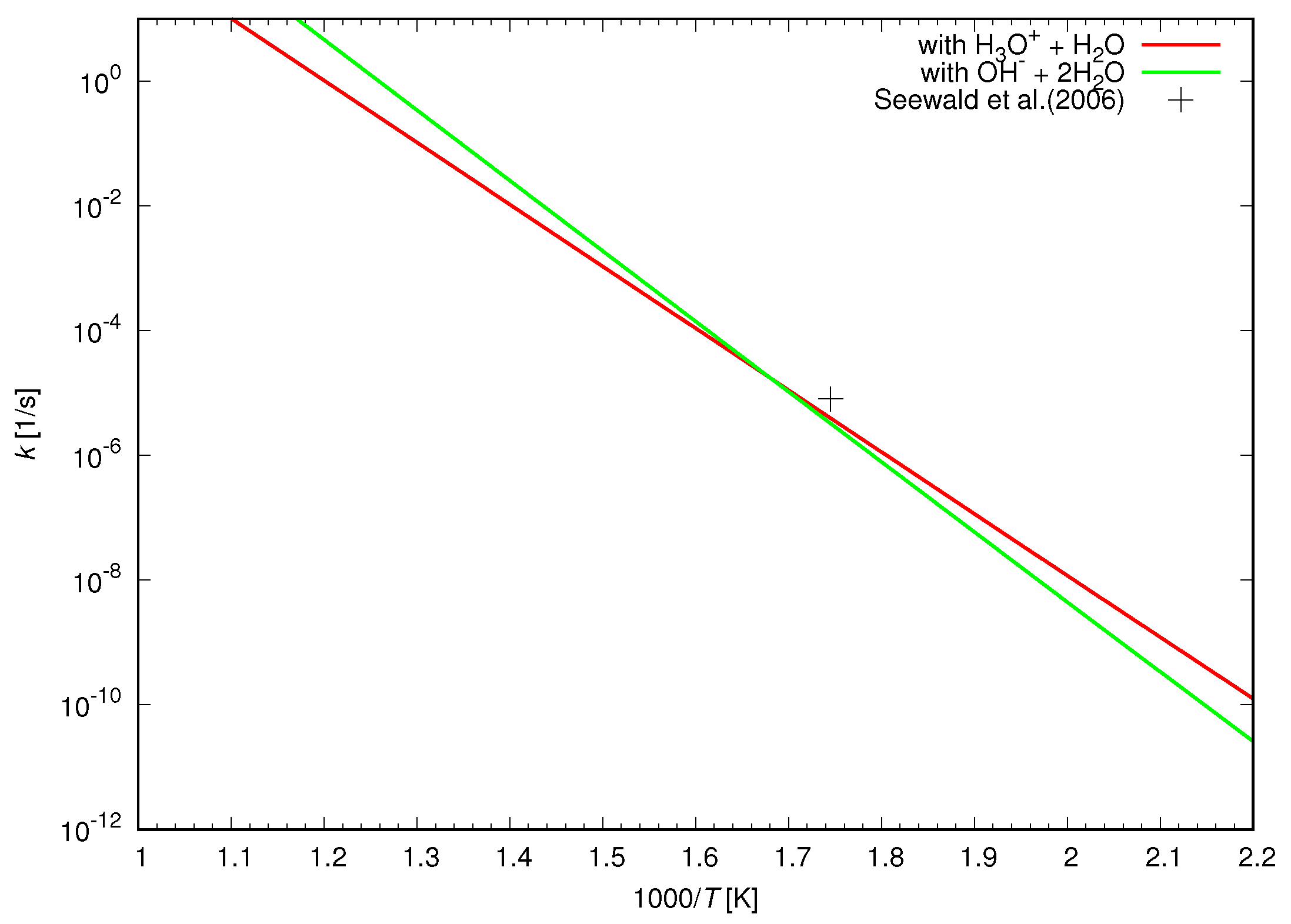
Figure 8 presents the calculated acid–base catalyzed reduction rates of methanol. A hydronium and a water molecule are introduced in the simulation of the acid catalyzed reduction as displayed in
Figure 6, while a hydroxyl and two water molecules are included in the base catalyzed reduction as shown in
Figure 7. The acid catalyzed reduction rate is similar to the base catalyzed reduction rate except a little shallower slope due to the lower energy barrier. The reduction rate of methanol is significantly enhanced by the acid–base catalytic effect and becomes 15 orders of magnitude larger than that catalyzed by water molecules alone as shown in
Figure 5. The acid–base catalyzed reduction rates successfully reproduce the reduction rate of methanol determined by the experimental study. This indicates the active role of a hydronium and a hydroxyl as a catalyst in the reduction of methanol in hot water.
We calculated the reduction rate of methanol, assuming that the rate was mainly determined by the reduction of a reactant compound. A reactant compound was formed after a methanol encountered a hydronium or a hydroxy in hot water. We estimated the mean collision time between a methanol and a hydronium, which, in the neutral condition, was nearly the same with the mean collision time between a methanol and a hydroxyl. The mean free path of a hydronium before encountering another methanol is given by
where
is the number density of hydronium and
is the cross section for collision between a methanol and a hydronium. We estimate the cross section as
by assuming that a hydronium and a methanol are approximately represented by spheres with radii of 1 Å and 2 Å. A hydronium travelled in hot water to encounter a methanol, frequently colliding with a number of water molecules. We described the motion of a hydronium in hot water as a diffusion process and estimate the mean collision time between a methanol and a hyronium as
where
is kinematic viscosity of hot water. While a methanol interacts with a hydronium, a hydrogen molecule needs to approach to form a reactant compound. The mean collision time between a methanol and a hydrogen molecule was two orders of magnitude longer than the interaction time between a methanol and a hydronium when
. Then the formation rate of a reactant compound was approximately estimated as
The formation rate of a reactant compound depends on the water temperature because the density, the ion product, and the viscosity of water are dependent on the temperature. We adopted the data given in Akiya and Savage [
14] and calculate the formation rate.
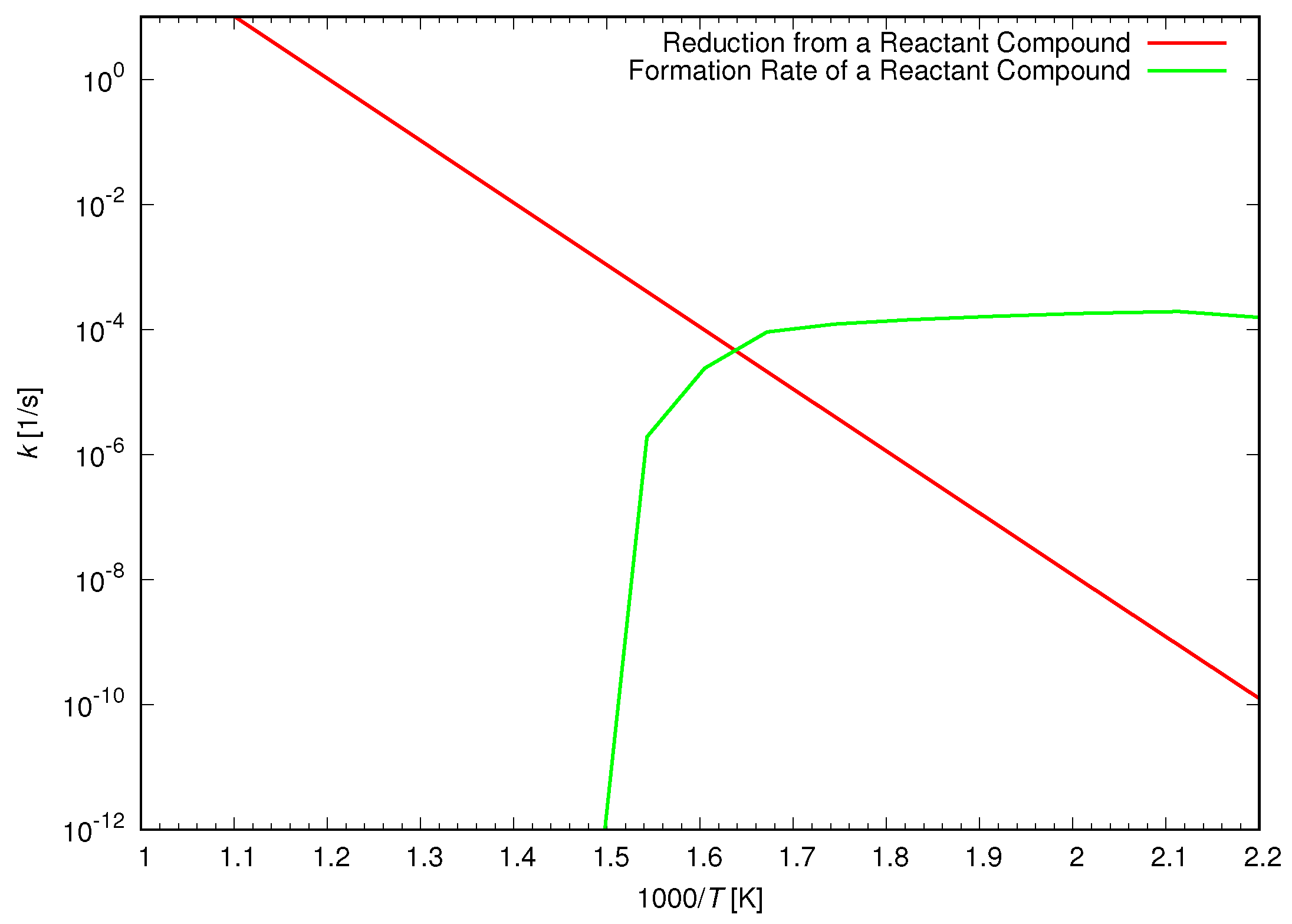
Figure 9 displays the acid catalyzed reduction rate of a reactant compound as well as the formation rate of a reactant compound. The formation rate was nearly the constant value at the low temperature, while it decreased rapidly around 374 °C of the critical temperature. The reduction rate of methanol was approximately represented by the reduction rate of a reactant compound at the low temperature because a reactant compound was formed rapidly before the reduction took place. On the other hand, at the temperature above the critical temperature, we cannot obtain the reduction rate of methanol using the present method due to such slow formation rate of a reactant compound. The reduction rate was mainly determined by the formation rate of a reactant compound at the high temperature. However, in the high temperature water of supercritical water, we needed to consider free radical reactions in the reduction of methanol.
4. Conclusions
We have performed a number of quantum chemical simulations to examine the reduction process of methanol in hot water such as hydrothermal vents. We used the Gaussian 16 software and applied the Gaussian 4 method to find optimized structures of a reactant compound, a transition state, and a product compound. A reactant compound is comprised of a methanol and a hydrogen molecule, while a methane and a water molecule form a product compound. A methanol is transformed into a methane and a water molecule by capturing a hydrogen molecule in the gas phase. Water molecules around a methanol promote the reduction process in aqueous solution by transferring a proton effectively. Two water molecules actively transfer a proton of a hydrogen molecule to a hydroxyl of the methanol to proceed the reduction. The catalytic effect of two water molecules reduces the energy barrier for the reduction by 13 kcal/mol. However, the energy barrier is still too high to reproduce the reduction rate of methanol determined by laboratory experiments.
Hot temperature water exhibits very different characteristics from ambient liquid water: a low dielectric constant, fewer hydrogen bonds, and higher ion product. Broken hydrogen bonds lead to the formation of radicals and ions in hot water. Radical reactions play crucial roles in the very high temperature water such as supercritical water. With a decrease in the temperature, the contribution of ions to the reaction is expected to increase in the hot water. We examined the acid–base catalytic effects on the reduction process of methanol. Either a hydronium or a hydroxyl is included in the simulation when a acid catalyzed or a base catalyzed reduction is examined. The energy barrier for the reduction of methanol is significantly reduced when we consider the acid–base reduction of methanol. The energy barriers for the acid and base catalyzed reduction become 45 kcal/mol and 51 kcal/mol, respectively.
We apply the transition state theory and calculate the reduction rate of methanol considering the tunneling effect of a proton. The reduction rate of methanol is significantly enhanced by the catalytic effect of water molecules, but it is still much lower than the reduction rate determined by an experiment. Huge drop of the energy barrier is found when a hydronium or a hydroxyl is introduced in the simulation as a catalyst of the reduction. The acid catalyzed reduction rate has a similar magnitude to the base catalyzed reduction rate. The calculated reduction rate of methanol agrees well with that determined by the experimental study. The present study suggests the active roles of hydroniums and hydroxyls as catalysts in the reduction of methanol in hot water.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}