Deactivation of Commercial, High-Load o-Xylene Feed VOx/TiO2 Phthalic Anhydride Catalyst by Unusual Over-Reduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- PA yield = mass-based phthalic anhydride yield [wt %]
- CO = CO content in reactor off-gas [Vol %]
- CO2 = CO2 content in reactor off-gas [Vol %]
- load = o-xylene loading in feed stream [g/Nm³]
- purity = o-xylene feed purity [wt %]
- oX = o-xylene slip in reactor off-gas [wt %]
- MA = maleic anhydride content in reactor off-gas [wt %].
- over-reduction of the active surface vanadium oxide species,
- coke formation by adsorbed reaction intermediates,
- deposition of catalyst poisons, like alkali salts,
- changing selectivity due to loss of promoters from the surface,
- fouling by deposits like dust plugging pores,
- sintering and accompanied loss of surface area,
- transformation of the TiO2 anatase phase into the catalytically inactive but thermodynamically more stable rutile phase.
2. Results
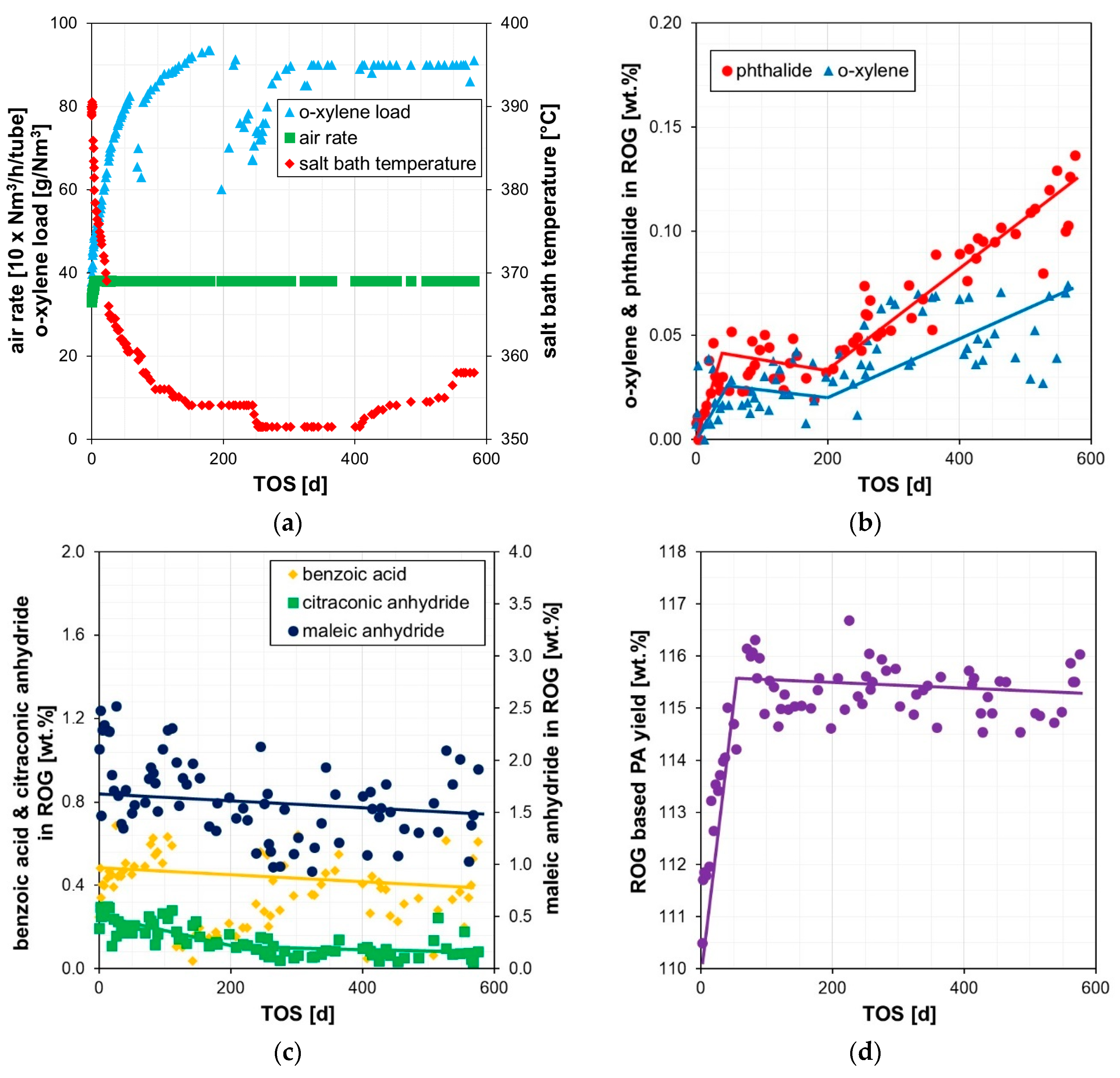
2.1. Development of Process Parameters, Reactor Performance and Catalyst Bed Temperatures
2.2. Analysis of Used Catalyst Samples
2.2.1. Crystalline Phase Composition
2.2.2. Specific Surface Area
2.2.3. Chemical Composition
3. Discussion
- abnormal, irreversible catalyst deactivation in most of the catalyst bed due to an enhanced degree of catalyst reduction,
- abnormal loss in the specific surface area/catalyst activity of the polishing layer catalyst bed due to enhanced formation of high-molecular carbon deposits plugging pores,
- both abnormal catalyst surface processes were induced by an irregular reactor shutdown during which the air purge was insufficient to remove the organic species from the reactor and the catalyst surface.
4. Materials and Methods
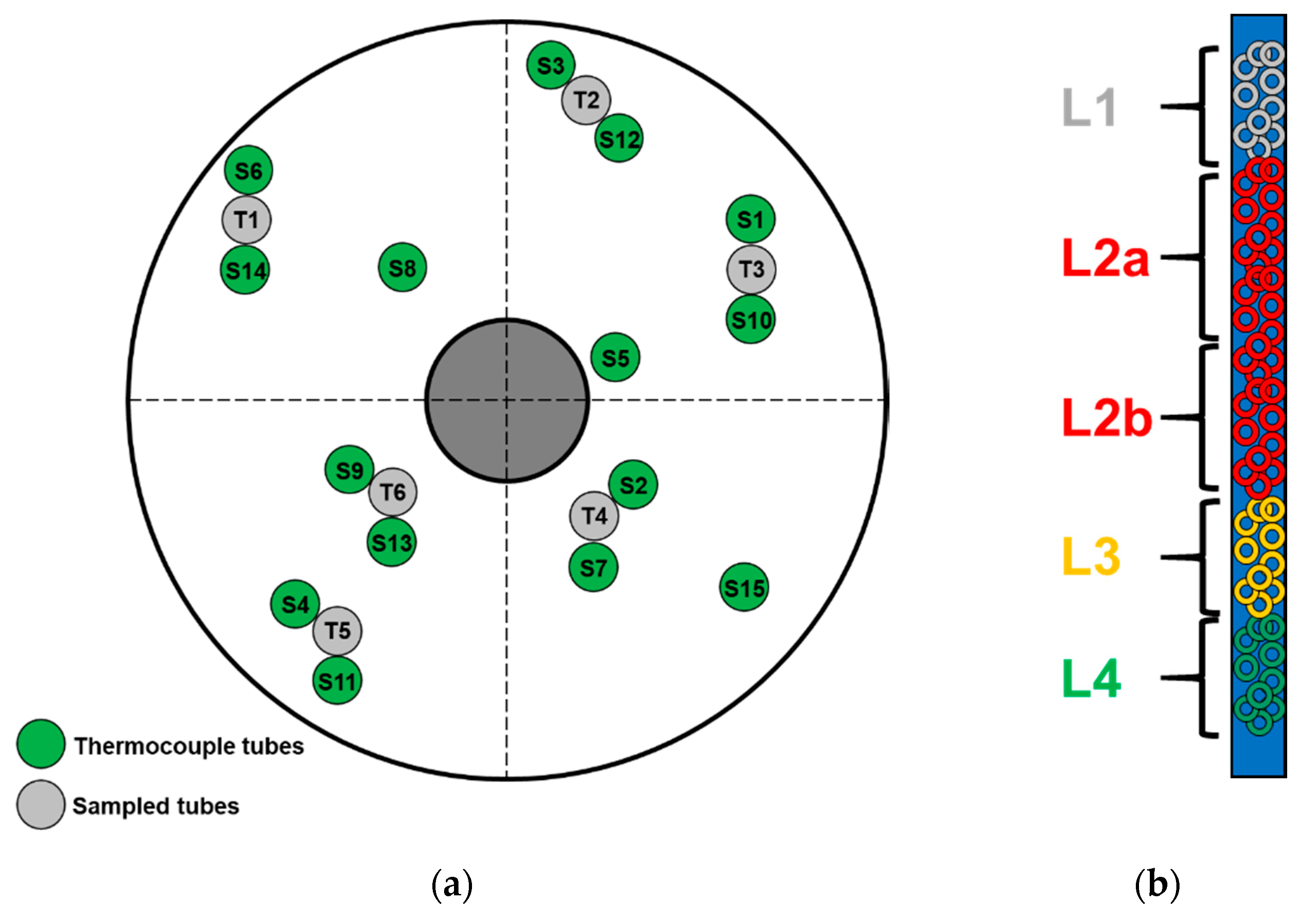
4.1. Sampling of Used Catalyst
- crystalline bulk phase compositions by X-ray diffraction measurements combined with Rietveld refinements,
- specific surface area measurements by nitrogen adsorption,
- chemical bulk contents of V, Sb, P, Na, K and Fe by atomic absorption spectroscopy,
- chemical bulk contents of C by combustion analysis.
4.2. Physico-Chemical Analysis of Used Catalyst
Author Contributions
Funding
Conflicts of Interest
References
- Suter, H. Phthalsäureanhydrid und seine Verwendung, 1st ed.; Dr. Dietrich Steinkopff Verlag: Darmstadt, Germany, 1972; pp. 1–6. [Google Scholar]
- The Global Phthalic Anhydride Market. 2018. Available online: https://www.researchandmarkets.com/reports/4515077/global-phthalic-anhydride-market-segmented-by (accessed on 26 November 2018).
- Marx, R.; Wölk, H.-J.; Mestl, G.; Turek, T. Reaction scheme of o-xylene oxidation on vanadia catalyst. Appl. Catal. 2011, 398, 37–43. [Google Scholar] [CrossRef]
- Mestl, G.; Gückel, C.; Estenfelder, M.; Käding, B. Method for Applying a Wash Coat Suspension to a Carrier Strucutre. Patent EP 2160241, 31 May 2007. [Google Scholar]
- Richter, O.; Mestl, G. Selective Oxidation of o-Xylene to Phthalic Anhydride: Still Room for Improvement in an Established Catalyst System; Jahrestreffen Reaktionstechnik: Würzburg, Germany, 22–27 May 2017. [Google Scholar]
- Gückel, C.; Dialer, H.; Estenfelder, M.; Pitschi, W. Use of a Multi-Layer Catalyst for Producing Phthalic Anhydride. Patent WO 2006092304, 2 March 2005. [Google Scholar]
- Gückel, C.; Dialer, H.; Estenfelder, M.; Pitschi, W. Method for Producing a Multi-Layer Catalyst for Obtaining Phthalic Anhydride. Patent WO 2006092305, 2 March 2005. [Google Scholar]
- Richter, O.; Mestl, G.; Lesser, D.; Marx, R.; Fromm, N.; Schulz, F.; Schinke, P.; Pitschi, W. Catalyst Arrangment with Optimized Void Fraction for the Production of Phthalic Acid Anhydride. Patent WO 2015162227, 24 April 2004. [Google Scholar]
- Richter, O.; Mestl, G.; Schulz, F.; Pitschi, W.; Fromm, N.; Schinke, P. Catalytic Converter Arrangement with Optimized Surface for Producing Phthalic Anhydride. Patent WO 2015162230, 24 April 2014. [Google Scholar]
- Nikolov, V.A.; Anastasov, A.I. Pretreatment of a Vanadia-Titania Catalyst for Partial Oxidation of o-Xylene under Industrial Conditions. Ind. Eng. Res. 1992, 31, 80–88. [Google Scholar] [CrossRef]
- Anastasov, A.I. Deactivation of industrial V2O5-TiO2 catalyst for oxidation of o-xylene into phthalic anhydride. Chem. Eng. Proc. 2003, 42, 449–460. [Google Scholar] [CrossRef]
- Saleh, R.Y.; Wachs, I.E.; Chan, S.S.; Chersich, C.C. The interaction of V2O5 with TiO2 (anatase): Catalyst evolution with calcination temperature and o-xylene oxidation. J. Catal. 1986, 98, 102–114. [Google Scholar] [CrossRef]
- Dias, C.R.; Portela, M.F.; Bond, G.C. Synthesis of Phthalic Anhydride: Catalysts, Kinetics, and Reactor Modeling. Catal. Rev. 1997, 39, 169–207. [Google Scholar] [CrossRef]
- Nikolov, V.A.; Klissurski, D.G.; Anastasov, A. I Phthalic Anhydride from o-Xylene Catalysis: Science and Engineering. Catal. Rev. 1991, 33, 319–374. [Google Scholar] [CrossRef]
- Gasior, M.; Haber, J.; Machej, T. Evolution of V2O5-TiO2 catalysts in the course of the catalytic reaction. Appl. Catal. 1987, 33, 1–14. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogenous Catalyst Deactivation and Regeneration: A Review. Catalyst 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Pernicone, N. Methods for Laboratory-Scale Evaluation of Catalyst Life in Industrial Plants. Appl. Catal. 1985, 15, 17–31. [Google Scholar] [CrossRef]
- Georgieva, A.T.; Anastasov, A.I.; Nikolov, V.A. Deactivation Properties of a High-Productive Vanadia-Titania Catalyst for Oxidation of o-Xylene to Phthalic Anhydride. Braz. J. Chem. Eng. 2008, 25, 351–364. [Google Scholar]
- Krajewski, W.; Galantowicz, M. Effect of catalyst deactivation on the process of oxidation of o-xylene to phthalic anhydride in an industrial multitubular reactor. Stud. Surf. Sci. Catal. 1999, 126, 447–452. [Google Scholar]
- Castillo-Araiza, C.O.; López-Isunza, F. The role of catalyst activity on the steady state and transient behavior of an industrial-scale fixed bed catalytic reactor for the partial oxidation of o-xylene on V2O5/TiO2 catalysts. Chem. Eng. J. 2011, 176, 26–32. [Google Scholar] [CrossRef]
- Calderbank, P.H. Kinetics and Yields in the Catalytic Oxidation of o-Xylene to Phthalic Anhydride with V2O5 Catalysts. Adv. Chem. Ser. 1974, 133, 646–653. [Google Scholar]
- Dias, C.R.; Portela, M.F.; Bond, G.C. Oxidation of o-Xylene to Phthalic Anhydride over V2O5/TiO2 Catalyst I. Influence of Catalyst Composition, Preparation Method and Operating Conditions on Conversion and Product Selectivities. J. Catal. 1995, 157, 344–352. [Google Scholar] [CrossRef]
- Dias, C.R.; Portela, M.F.; Bond, G.C. Oxidation of o-Xylene to Phthalic Anhydride over V2O5/TiO2 Catalyst II. Transient Catalytic Behaviour. J. Catal. 1995, 157, 353–358. [Google Scholar] [CrossRef]
- Dias, C.R.; Portela, M.F.; Bond, G.C. Oxidation of o-Xylene to Phthalic Anhydride over V2O5/TiO2 Catalyst III. Study of Organic Residie Formed on the Catalyst Surface. J. Catal. 1996, 162, 284–294. [Google Scholar] [CrossRef]
- Dias, C.R.; Portela, M.F.; Bond, G.C. Oxidation of o-Xylene to Phthalic Anhydride over V2O5/TiO2 Catalyst Part 4. Mathematical Modelling Study and Analysis of the Reaction Network. J. Catal. 1996, 164, 276–287. [Google Scholar]
- Dias, C.R.; Portela, M.F.; Bond, G.C. Deactivation of V2O5/TiO2 catalyst in the oxidation of o-xylene to phthalic anhydride. Stud. Surf. Sci. Catal. 1994, 88, 475–482. [Google Scholar]
- Mongkhonsi, T.; Kershenbaum, L. The effect of deactivation of a V2O5/TiO2 (anatase) industrial catalyst on reactor behavior during the partial oxidation of o-xylene to phthalic anhydride. Appl. Catal. 1998, 170, 33–48. [Google Scholar] [CrossRef]
- Bond, G.C.; König, P. The Vanadium Pentoxide-Titanium Dioxide System. J. Catal. 1982, 77, 309–322. [Google Scholar] [CrossRef]
- Nikolov, V.A.; Klissurski, D.G.; Hadjiivanov, K.I. Deactivation of a V2O5-TiO2 Catalyst for the Oxidation of o-Xylene to Phthalic Anhydride. Stud. Surf. Sci. Catal. 1987, 34, 173–182. [Google Scholar]
- Wainwright, M.S.; Hoffman, T.W. The Oxidation of Ortho-xylene on Vanadium Pentoxide Catalysts. I. Transient Kinetic Measurements. Can. J. Chem. Eng. 1977, 55, 552–556. [Google Scholar] [CrossRef]
- Chapman, D.M. Behavior of titania-supported vanadia and tungsta SCR catalysts at high temperatures in reactant streams: Tungsten and vanadium oxide and hydroxide vapor pressure reduction by surficial stabiblization. Appl. Catal. 2011, 392, 143–150. [Google Scholar] [CrossRef]
- Lesser, D.; Mestl, G.; Turek, T. Transient behavior of vanadyl pyrophosphate catalyst during the partial oxidation of n-butane in industrial-sized, fixed bed reactors. Appl. Catal. 2016, 510, 1–10. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, O.; Mestl, G. Deactivation of Commercial, High-Load o-Xylene Feed VOx/TiO2 Phthalic Anhydride Catalyst by Unusual Over-Reduction. Catalysts 2019, 9, 435. https://doi.org/10.3390/catal9050435
Richter O, Mestl G. Deactivation of Commercial, High-Load o-Xylene Feed VOx/TiO2 Phthalic Anhydride Catalyst by Unusual Over-Reduction. Catalysts. 2019; 9(5):435. https://doi.org/10.3390/catal9050435
Chicago/Turabian StyleRichter, Oliver, and Gerhard Mestl. 2019. "Deactivation of Commercial, High-Load o-Xylene Feed VOx/TiO2 Phthalic Anhydride Catalyst by Unusual Over-Reduction" Catalysts 9, no. 5: 435. https://doi.org/10.3390/catal9050435
APA StyleRichter, O., & Mestl, G. (2019). Deactivation of Commercial, High-Load o-Xylene Feed VOx/TiO2 Phthalic Anhydride Catalyst by Unusual Over-Reduction. Catalysts, 9(5), 435. https://doi.org/10.3390/catal9050435