Structure of Imidazolium-N-phthalolylglycinate Salt Hydrate: Combined Experimental and Quantum Chemical Calculations Studies


Abstract
:1. Introduction
2. Result and Discussion
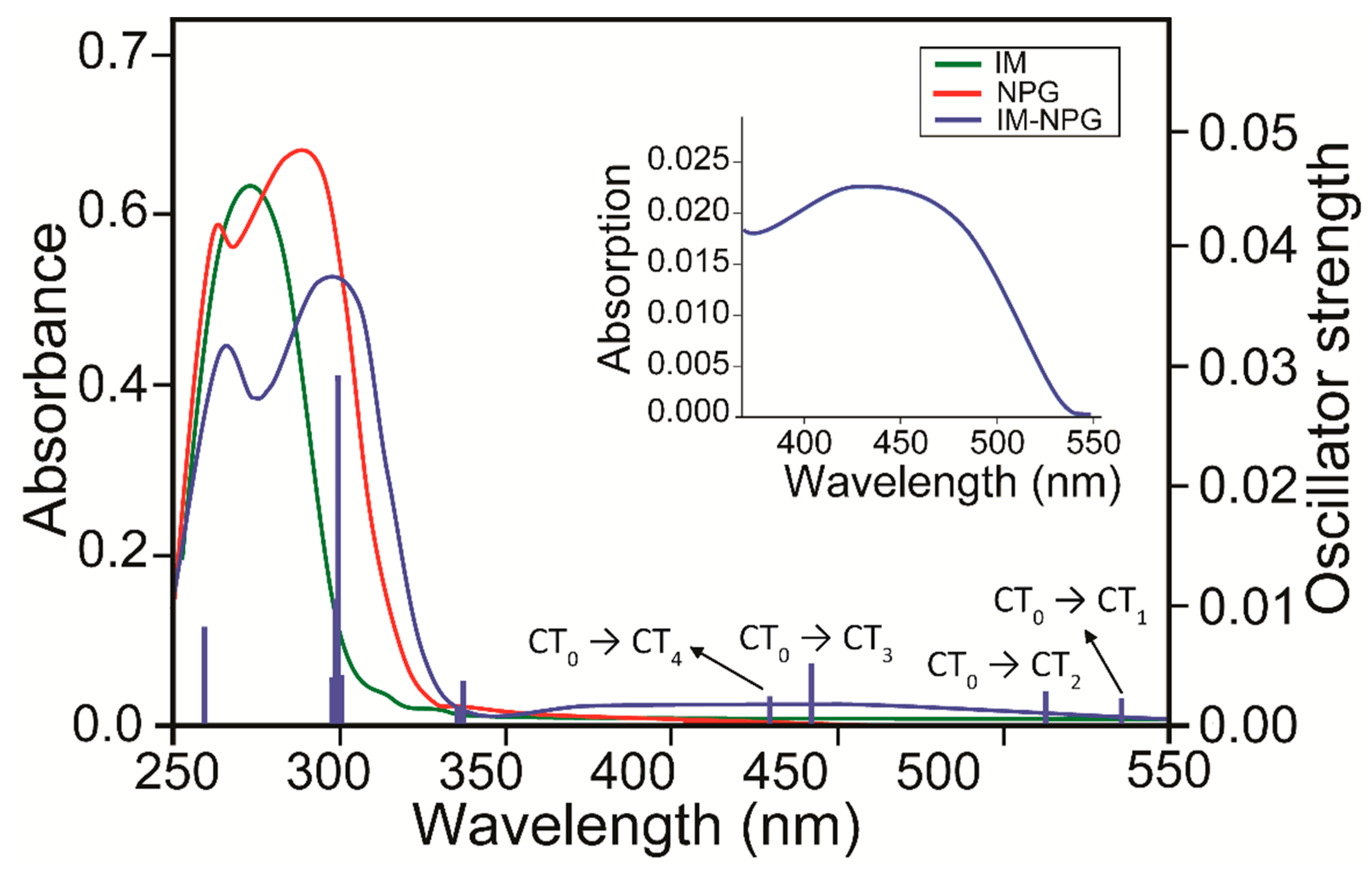
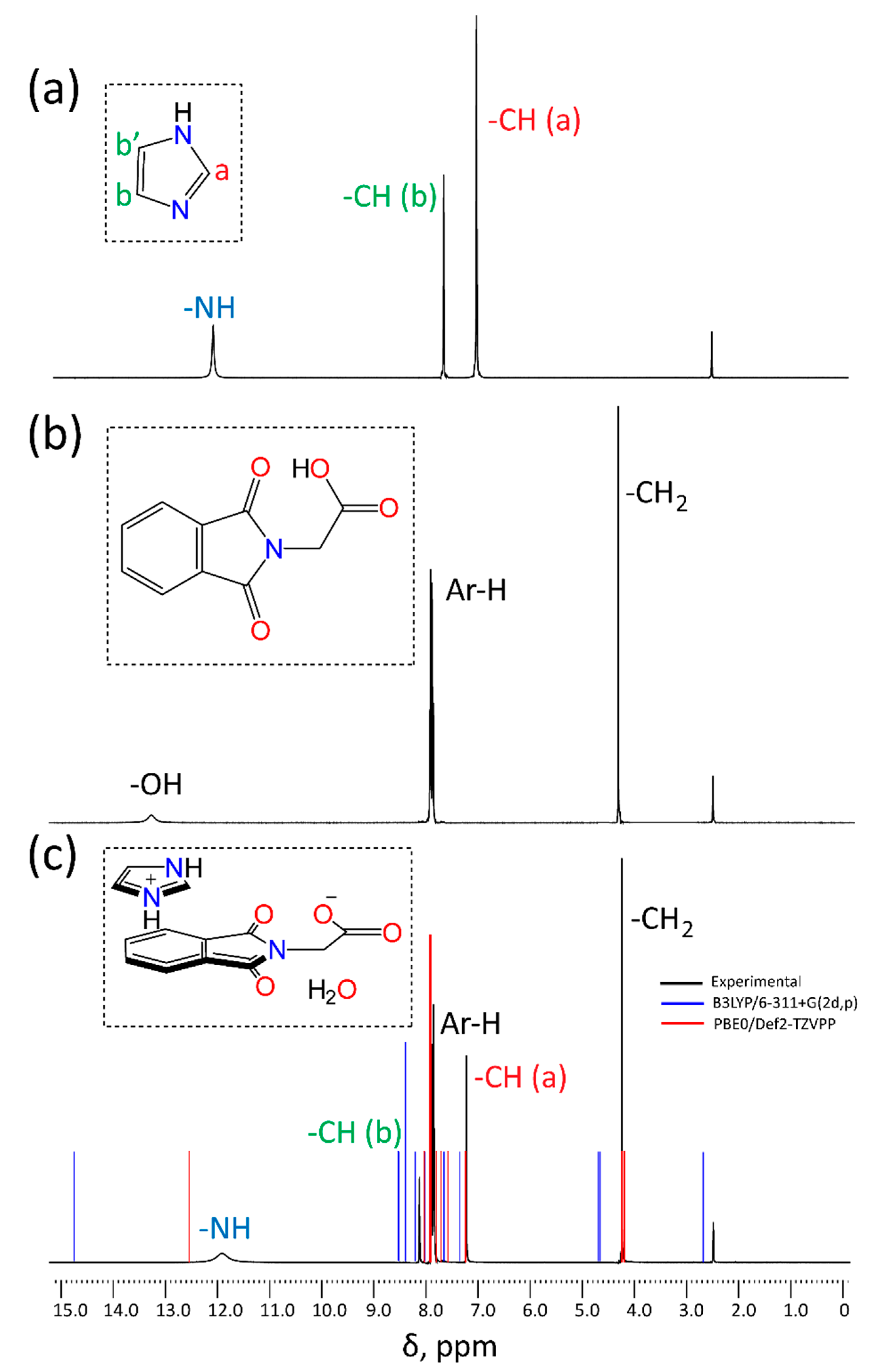
2.1. Spectroscopic Analysis
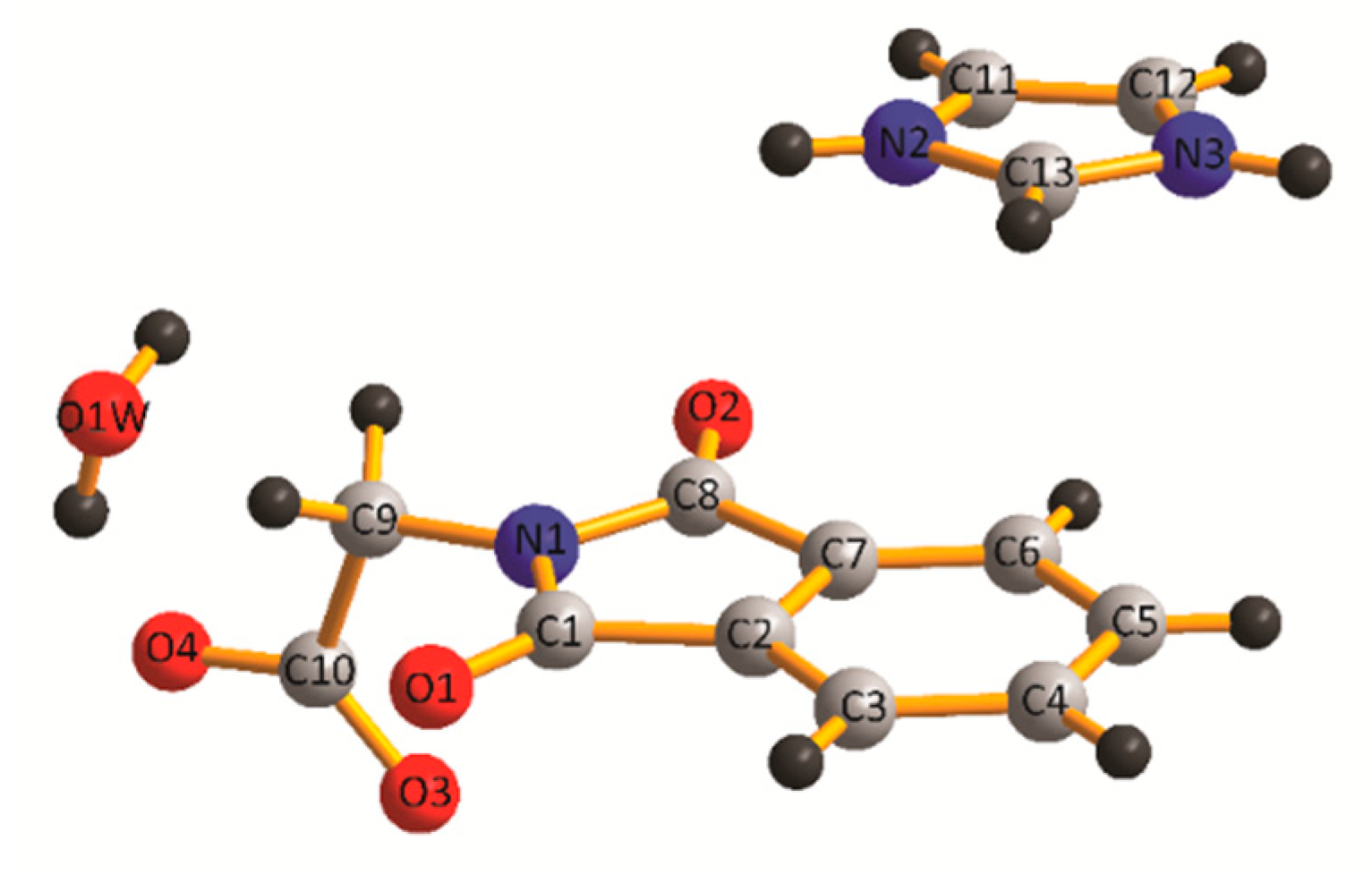
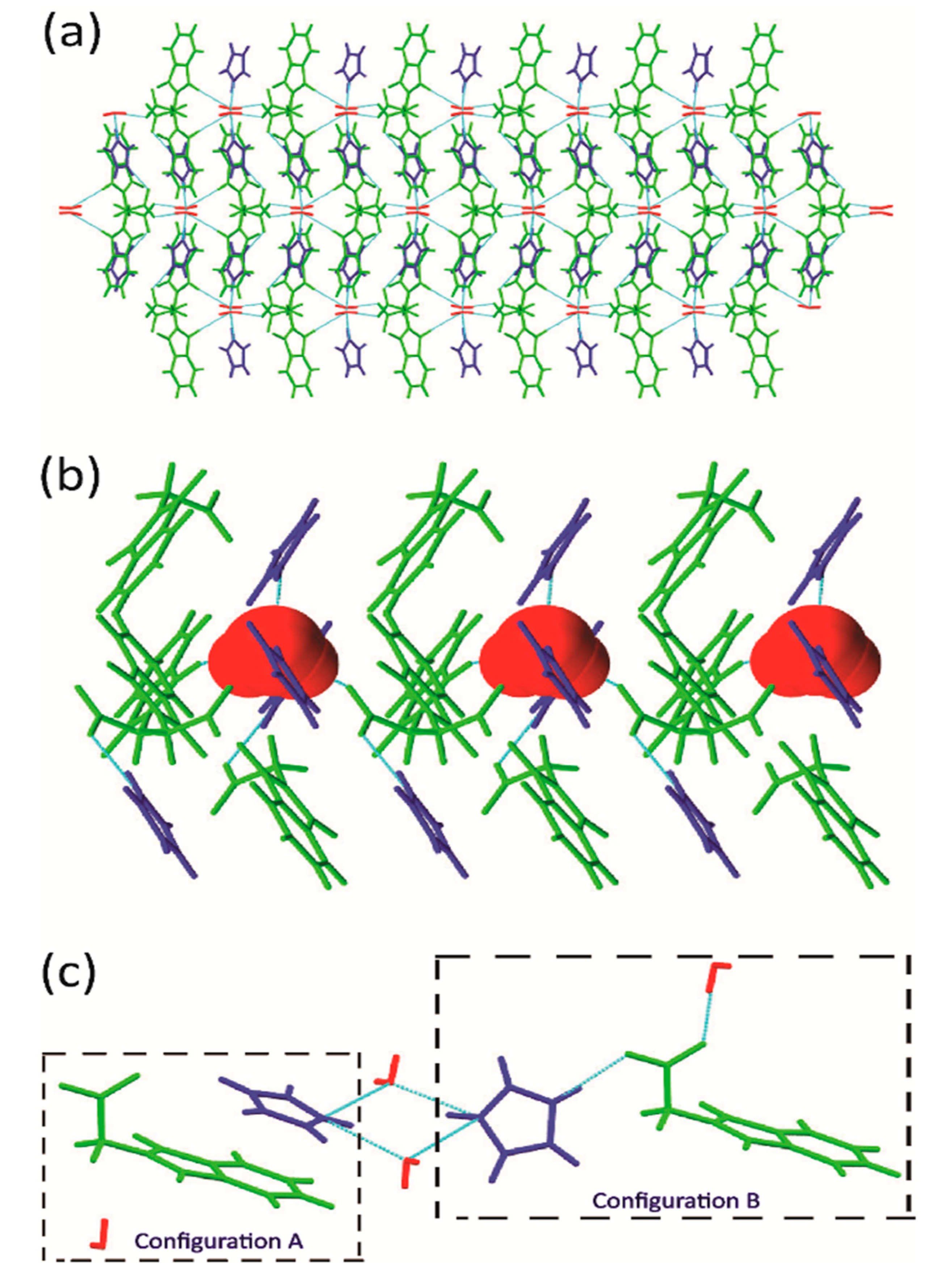
2.2. Crystal Structure of IM+–NPG−–HYD (1:1:1)
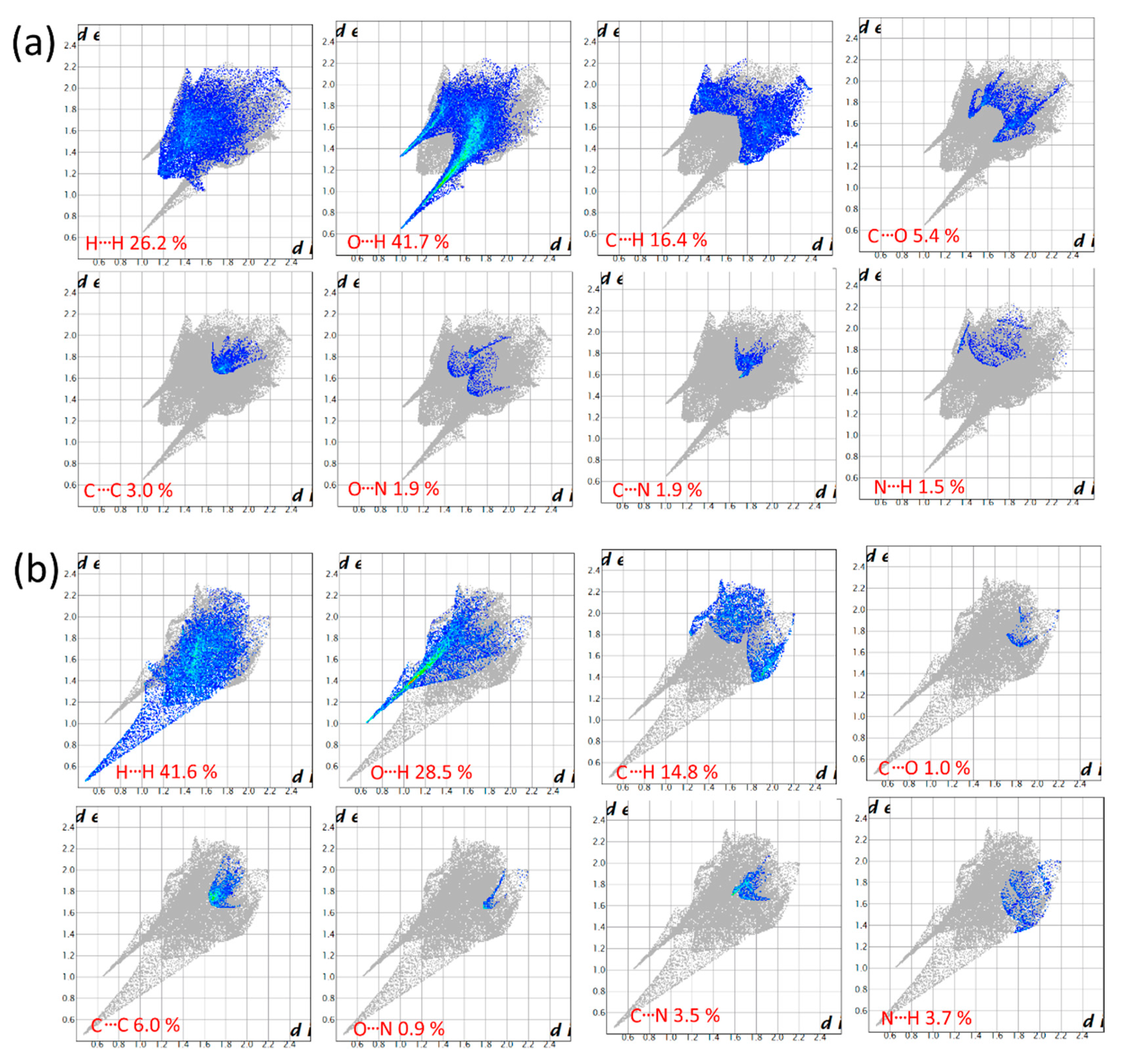
2.3. Hirshfeld Surface Analysis
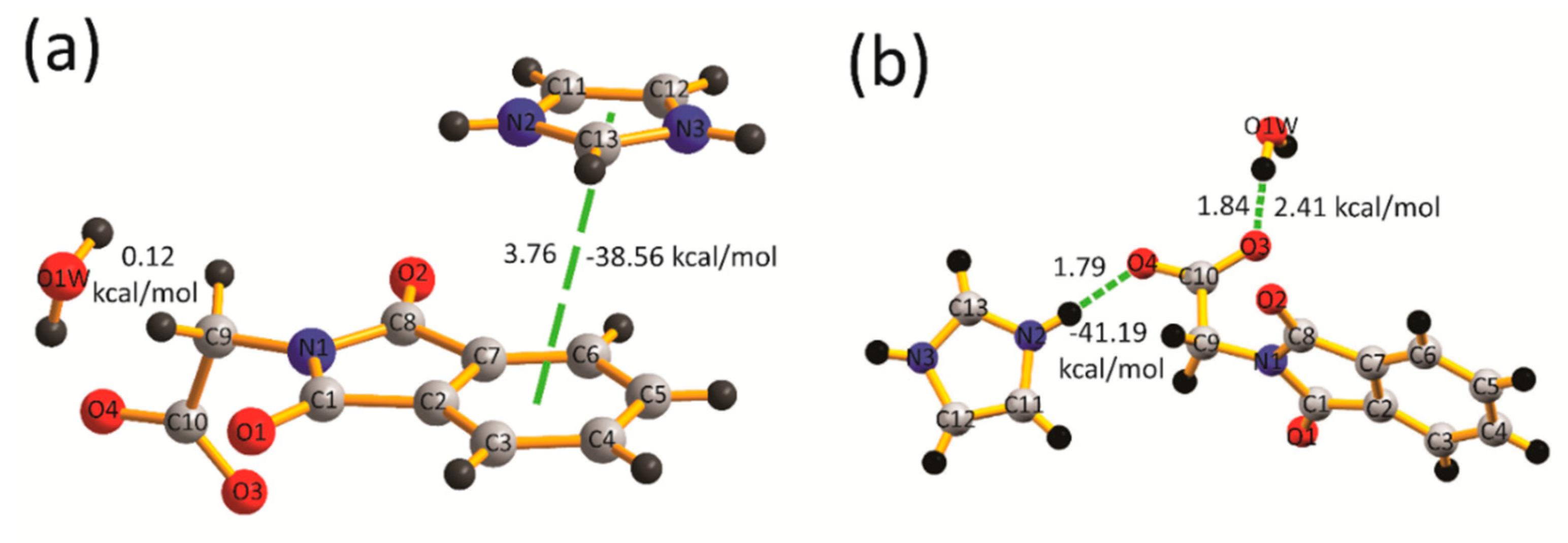
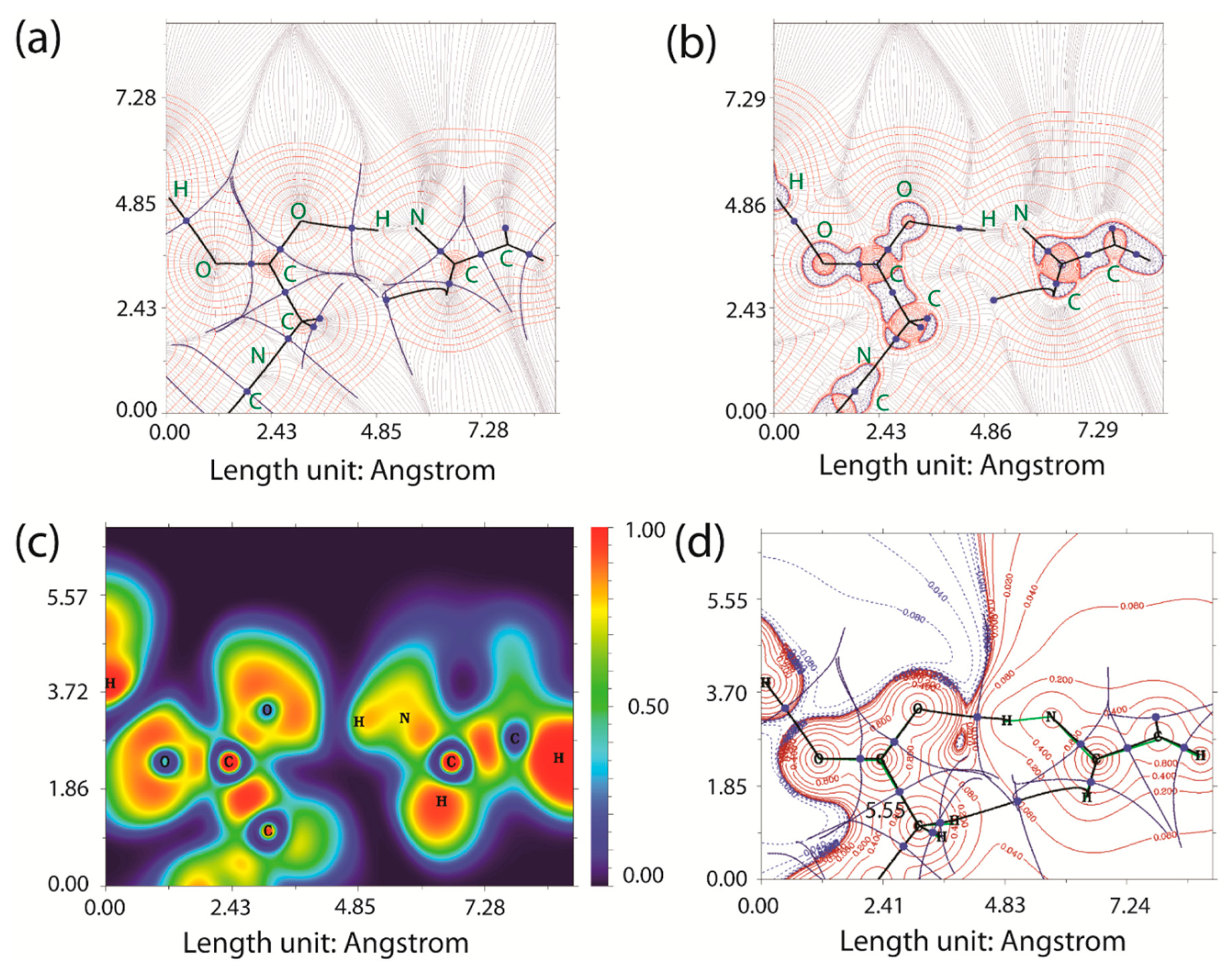
2.4. Quantum-Theory-of-Atoms-in-Molecules Analysis
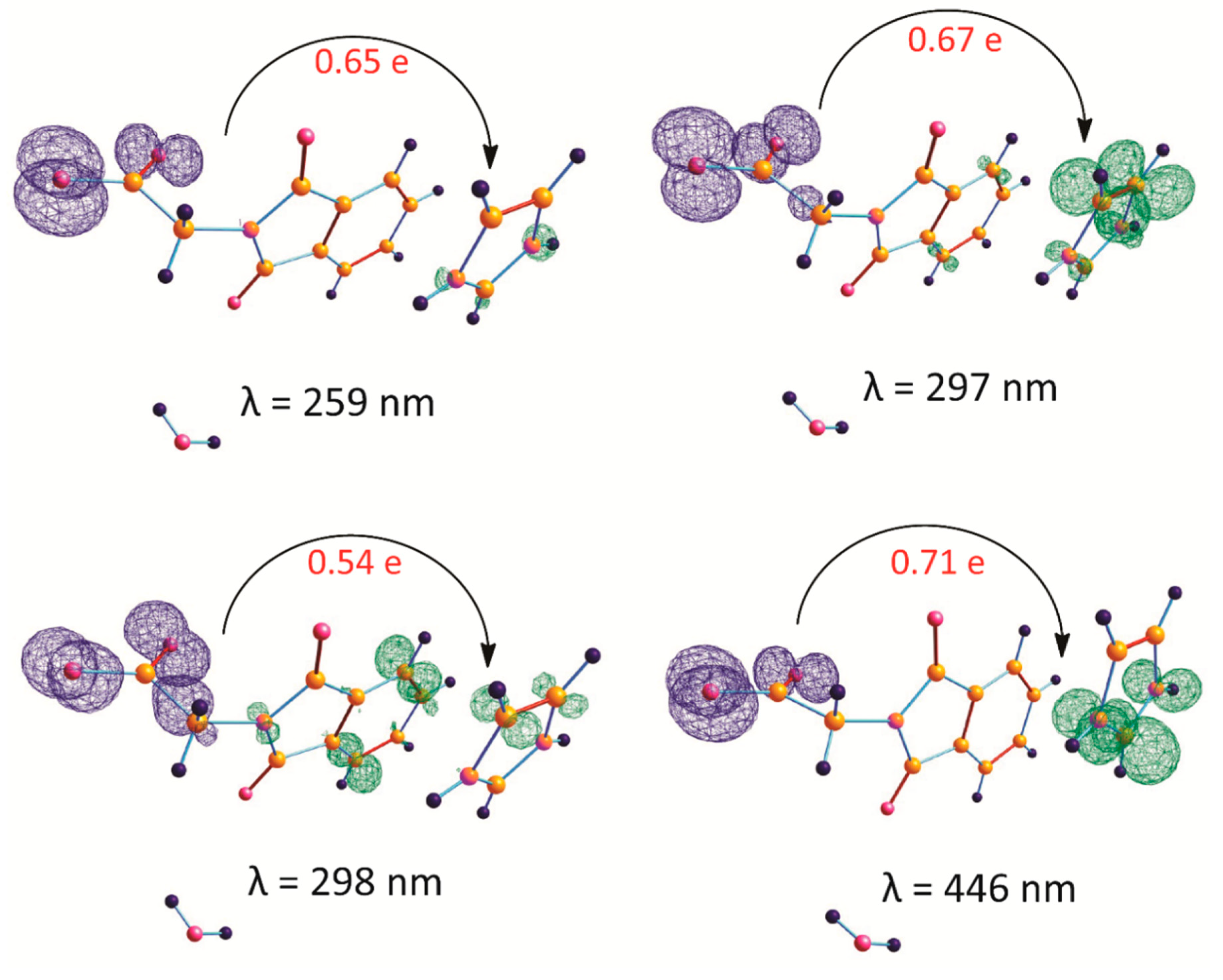
2.5. CT in IM+–NPG−–HYD
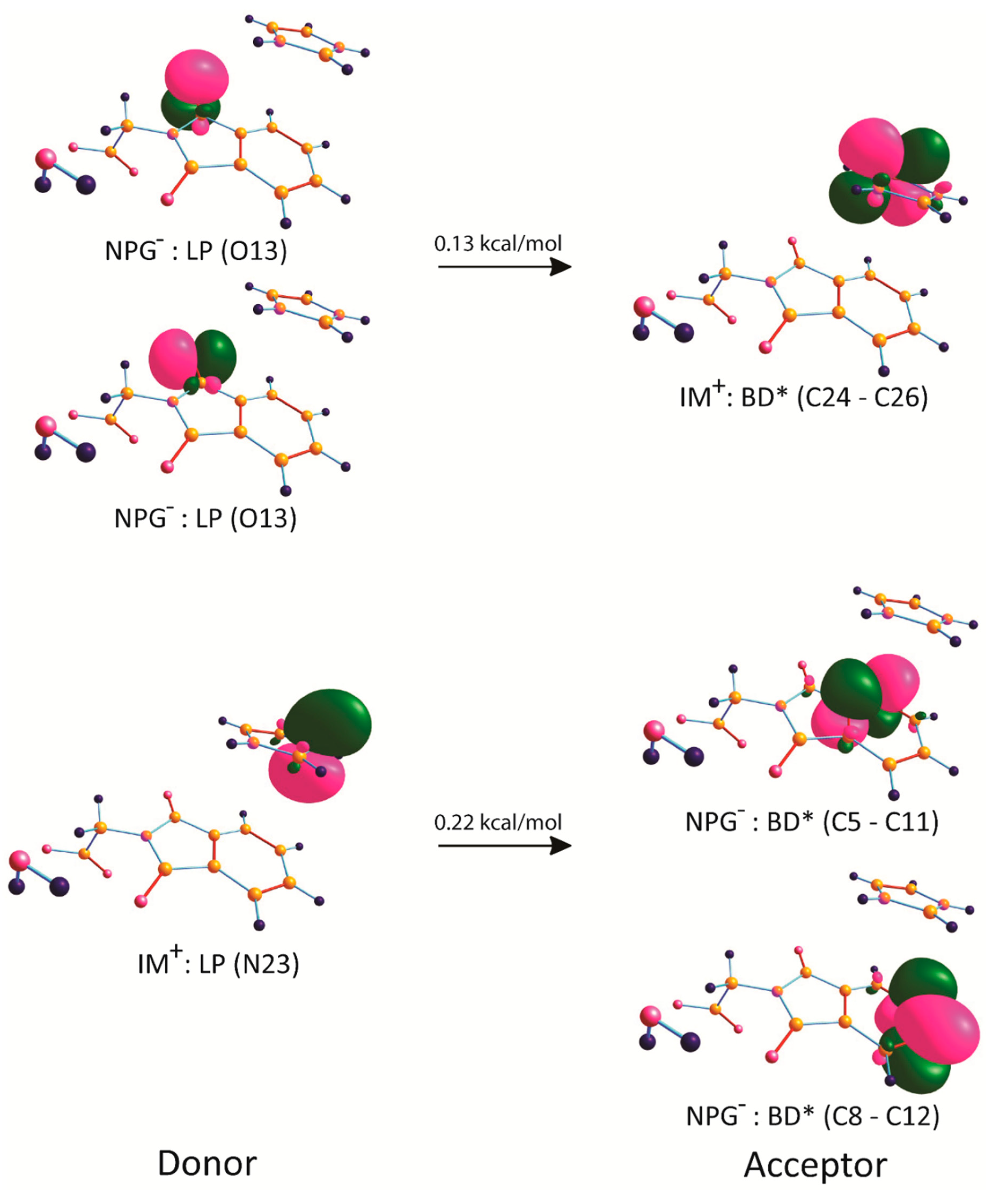
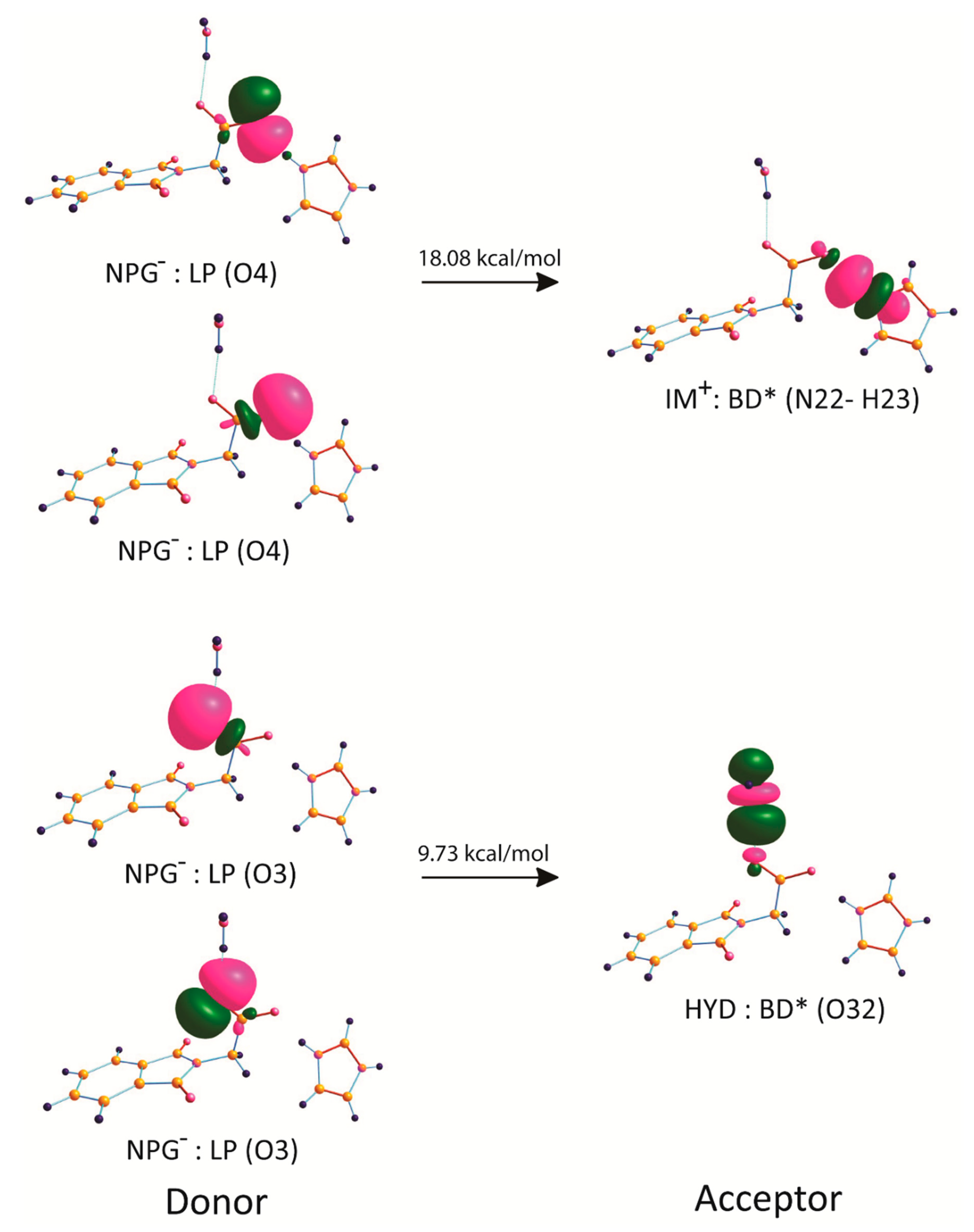
2.6. Natural Bond Orbital Analysis
3. Conclusions
4. Experimental Section
4.1. General Methods and Instrumentation
4.2. Synthesis of IM+–NPG−–HYD (1:1:1)
4.3. Single-Crystal X-ray Crystallography
5. Computational Details
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Whitesides, G.M.; Mathias, J.P.; Seto, C.T. Molecular self-assembly and nanochemistry: A chemical strategy for the synthesis of nanostructures. Science 1991, 254, 1312–1319. [Google Scholar] [CrossRef]
- Lehn, J.M. From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry. Chem. Soc. Rev. 2007, 36, 151–160. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parshall, G.W. Crystal Engineering: The Design of Organic. Solids. Mater. Sci. Monogr. 1989, 54. [Google Scholar]
- Biradha, K. Crystal engineering: From weak hydrogen bonds to co-ordination bonds. CrystEngComm 2003, 5, 374–384. [Google Scholar] [CrossRef]
- Xie, Z.; Yang, B.; Li, F.; Cheng, G.; Liu, L.; Yang, G.; Xu, H.; Ye, L.; Hanif, M.; Liu, S.; et al. Cross Dipole Stacking in the Crystal of Distyrylbenzene Derivative: The Approach toward High Solid-State Luminescence Efficiency. J. Am. Chem. Soc. 2005, 127, 14152–14153. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yang, X.; Yan, D.J. Vapor-phase π–π molecular recognition: A fast and solvent-free strategy towards the formation of co-crystalline hollow microtube with 1D optical waveguide and up-conversion emission. J. Mater. Chem. C 2017, 5, 1632–1637. [Google Scholar] [CrossRef]
- Yan, D.P.; Delori, A.; Lloyd, G.O.; Friscic, T.; Day, G.M.; Jones, W.; Lu, J.; Wei, M.; Evans, D.G.; Duan, X. A cocrystal strategy to tune the luminescent properties of stilbene-type organic solid-state materials. Angew. Chem. Int. Ed. 2011, 50, 12483–12486. [Google Scholar] [CrossRef]
- Chung, J.W.; You, Y.; Huh, H.S.; An, B.-K.; Yoon, S.-J.; Kim, S.H.; Lee, S.W.; Park, S.Y.J. Shear-and UV-induced fluorescence switching in stilbenic π-dimer crystals powered by reversible [2 + 2] cycloaddition. Am. Chem. Soc. 2009, 131, 8163–8172. [Google Scholar] [CrossRef]
- Qin, Y.; Zhang, J.; Zheng, X.; Geng, H.; Zhao, G.; Xu, W.; Hu, W.; Shuai, Z.; Zhu, D. Charge-Transfer Complex Crystal Based on Extended-π-Conjugated Acceptor and Sulfur-Bridged Annulene: Charge-Transfer Interaction and Remarkable High Ambipolar Transport Characteristics. Adv. Mater. 2014, 26, 4093–4099. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, J.H.; Ma, Z.Y.; Xu, W.; Zhao, G.Y.; Geng, H.; Di, C.A.; Hu, W.P.; Shuai, Z.G.; Singh, K.; et al. Fullerene/sulfur-bridged annulene cocrystals: Two-dimensional segregated heterojunctions with ambipolar transport properties and photoresponsivity. J. Am. Chem. Soc. 2013, 135, 558–561. [Google Scholar] [CrossRef]
- Zhang, J.; Geng, H.; Virk, T.S.; Zhao, Y.; Tan, J.H.; Di, C.A.; Xu, W.; Singh, K.; Hu, W.P.; Shuai, Z.G.; et al. Sulfur-Bridged Annulene-TCNQ Co-Crystal: A Self-Assembled ‘‘Molecular Level Heterojunction’’ with Air Stable Ambipolar Charge Transport Behavior. Adv. Mater. 2012, 24, 2603–2607. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Ahn, S.; Kim, J.B.; Schenck, C.; Hiszpanski, A.M.; Oh, S.; Schiros, T.; Loo, Y.-L.; Nuckolls, C. Using self-organization to control morphology in molecular photovoltaics. J. Am. Chem. Soc. 2013, 135, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Wuest, J.D. Co-crystals give light a tune-up. Nat. Chem. 2012, 4, 74–75. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Yang, H.; Meng, Q.; Lin, H.; Wei, M. Two-Component Molecular Materials of 2,5-Diphenyloxazole Exhibiting Tunable Ultraviolet/Blue Polarized Emission, Pump-enhanced Luminescence, and Mechanochromic Response. Adv. Funct. Mater. 2014, 24, 587–594. [Google Scholar] [CrossRef]
- Tsutsumi, J.Y.; Yamada, T.; Matsui, H.; Haas, S.; Hasegawa, T. Competition between charge-transfer exciton dissociation and direct photocarrier generation in molecular donor-acceptor compounds. Phys. Rev. Lett. 2010, 105, 226601. [Google Scholar] [CrossRef] [PubMed]
- Vincent, V.M.; Wright, J.D. Competition between charge-transfer exciton dissociation and direct photocarrier generation in molecular donor-acceptor compounds. J. Chem. Soc. Faraday Trans. 1974, 70, 58–71. [Google Scholar] [CrossRef]
- Horiuchi, S.; Ishii, F.; Kumai, R.; Okimoto, Y.; Tachibana, H.; Nagaosa, N.; Tokura, Y. Ferroelectricity near room temperature in co-crystals of nonpolar organic molecules. Nat. Mater. 2005, 4, 163–166. [Google Scholar] [CrossRef]
- Tayi, A.S.; Shveyd, A.K.; Sue, A.C.H.; Szarko, J.M.; Rolczynski, B.S.; Cao, D.; Kennedy, T.J.; Sarjeant, A.A.; Stern, C.L.; Paxton, W.F.; et al. Room-temperature ferroelectricity in supramolecular networks of charge-transfer complexes. Nature 2012, 488, 485–489. [Google Scholar] [CrossRef]
- Bosshard, C.; Wong, M.S.; Pan, F.; Gunter, P.; Gramlich, V. Self-assembly of an acentric co-crystal of a highly hyperpolarizable merocyanine dye with optimized alignment for nonlinear optics. Adv. Mater. 1997, 9, 554–557. [Google Scholar] [CrossRef]
- Pan, F.; Wong, M.S.; Gramlich, V.; Bosshard, C.; Gunter, P.J. A Novel and Perfectly Aligned Highly Electro—Optic Organic Cocrystal of a Merocyanine Dye and 2,4-Dihydroxybenzaldehyde. J. Am. Chem. Soc. 1996, 118, 6315–6316. [Google Scholar] [CrossRef]
- Morimoto, M.; Irie, M. A diarylethene cocrystal that converts light into mechanical work. J. Am. Chem. Soc. 2010, 132, 14172–14178. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical preparation of co-crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef] [PubMed]
- Shimozawa, M.; Hashimoto, K.; Ueda, A.; Suzuki, Y.; Sugii, K.; Yamada, S.; Imai, Y.; Kobayashi, R.; Itoh, K.; Iguchi, S.; et al. Quantum-disordered state of magnetic and electric dipoles in an organic Mott system. Nat. Commun. 2017, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ueda, A.; Yamada, S.; Isono, T.; Kamo, H.; Nakao, A.; Kumai, R.; Nakao, H.; Murakami, Y.; Yamamoto, K.; Nishio, Y.; et al. Hydrogen-bond-dynamics-based switching of conductivity and magnetism: A phase transition caused by deuterium and electron transfer in a hydrogen-bonded purely organic conductor crystal. J. Am. Chem. Soc. 2014, 136, 12184–12192. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Hatakeyama, A.; Enomoto, M.; Kumai, R.; Murakami, Y.; Mori, H. Modulation of a Molecular π-Electron System in a Purely Organic Conductor that Shows Hydrogen-Bond-Dynamics-Based Switching of Conductivity and Magnetism. Chem. Eur. J. 2015, 21, 15020–15028. [Google Scholar] [CrossRef] [PubMed]
- Collini, E. Cooperative effects to enhance two-photon absorption efficiency: Intra-versus inter-molecular approach. Phys. Chem. Chem. Phys. 2012, 14, 3725–3736. [Google Scholar] [CrossRef]
- Ono, T.; Sugimoto, M.; Hisaeda, Y. Multicomponent Molecular Puzzles for Photofunction Design: Emission Color Variation in Lewis Acid–Base Pair Crystals Coupled with Guest-to-Host Charge Transfer Excitation. J. Am. Chem. Soc. 2015, 137, 9519–9522. [Google Scholar] [CrossRef]
- Narayanan, A.; Cao, D.; Frazer, L.; Tayi, A.S.; Blackburn, A.K.; Sue, A.C.-H.; Ketterson, J.B.; Stoddart, J.F.; Stupp, S.I. Ferroelectric Polarization and Second Harmonic Generation in Supramolecular Cocrystals with Two Axes of Charge-Transfer. J. Am. Chem. Soc. 2017, 139, 9186–9191. [Google Scholar] [CrossRef]
- Zhu, W.; Zheng, R.; Fu, X.; Fu, H.; Shi, Q.; Zhen, Y.; Dong, H.; Hu, W. Revealing the Charge-Transfer Interactions in Self-Assembled Organic Cocrystals: Two-Dimensional Photonic Applications. Angew. Chem. Int. Ed. 2015, 54, 6785–6789. [Google Scholar] [CrossRef]
- Mulliken, R.S. Molecular compounds and their spectra. II. J. Am. Chem. Soc. 1952, 74, 811–824. [Google Scholar] [CrossRef]
- Thomas, L.H.; Jones, A.O.F.; Kallay, A.A.; McIntyre, G.J.; Wilson, C.C. Engineering short, strong, charge-assisted hydrogen bonds in benzoic acid dimers through cocrystallization with proton sponge. Cryst. Growth Des. 2016, 16, 2112–2122. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.D. Design of crystalline molecular networks with charge-assisted hydrogen bonds. Chem. Commun. 2005, 47, 5838–5842. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Grepioni, F. Intermolecular interactions in nonorganic crystal engineering. Acc. Chem. Res. 2000, 33, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Aaker€oy, C.B.; Beatty, A.M.; Lorimer, K.R. Charge-assisted hydrogen bonds and halogen-halogen interactions in organic salts: Benzylammonium benzoates and pentafluorobenzoates. Struct. Chem. 1999, 10, 229–242. [Google Scholar] [CrossRef]
- Braga, D.; Draper, S.M.; Champeil, E.; Grepioni, F. Inorganic–organometallic crystal synthesis. The role of charge-assisted C–H…O and C–H…Cl hydrogen bonds in crystalline [(η5-C5H5) 2Co][H2PO4]· 3H2O and [(η6-C6H5Me) 2Cr][Cl]. J. Organomet. Chem. 1999, 573, 73–77. [Google Scholar] [CrossRef]
- Bankiewicz, B.; Matczak, P.; Palusiak, M. Electron density characteristics in bond critical point (QTAIM) versus interaction energy components (SAPT): The case of charge-assisted hydrogen bonding. J. Phys. Chem. A 2012, 116, 452–459. [Google Scholar] [CrossRef]
- Matczak, P. Intramolecular C–H···H–C contacts in diheteroaryl ketones and thioketones: A theoretical analysis. Bull. Chem. Soc. Jpn. 2016, 89, 92–102. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Silverstein, G.C. Bassler Spectrometric Identification of Organic Compounds. J. Chem. Educ. 1962, 39, 546. [Google Scholar] [CrossRef]
- Ganduri, R.; Swain, D.; Cherukuvada, S.; Row, T.N.G. Role of functionalities in structural analogues urocanic acid and l-histidine, toward the formation of anhydrous and hydrated molecular salts. Cryst. Growth Des. 2019, 19, 1845–1852. [Google Scholar] [CrossRef]
- Gunnam, A.; Suresh, K.; Nangia, A. Salts and salt cocrystals of the antibacterial drug pefloxacin. Cryst. Growth Des. 2018, 18, 2824–2835. [Google Scholar] [CrossRef]
- Oomens, J.; Steill, J.D. Free carboxylate stretching modes. J. Phys. Chem. 2008, 112, 3281–3283. [Google Scholar] [CrossRef] [PubMed]
- Steill, J.D.; Oomens, J. Gas-phase deprotonation of p-hydroxybenzoic acid investigated by IR spectroscopy: Solution-phase structure is retained upon ESI. J. Am. Chem. Soc. 2009, 131, 13570–13571. [Google Scholar] [CrossRef] [PubMed]
- Arenas-García, J.R.; Ruiz, D.H.; Vasquez, K.M.; Rojas, H.M.; Hopfl, H. Modification of the supramolecular hydrogen-bonding patterns of acetazolamide in the presence of different cocrystal formers: 3:1, 2:1, 1:1, and 1:2 cocrystals from screening with the structural isomers of hydroxybenzoic acids, aminobenzoic acids, hydroxybenzamides, aminobenzamides, nicotinic acids, nicotinamides, and 2, 3-dihydroxybenzoic acids. Cryst. Growth Des. 2012, 12, 811–824. [Google Scholar]
- Gao, L.; Zhang, X.-R.; Chen, Y.-F.; Liao, Z.-L.; Wang, Y.-Q.; Zou, X.-Y. A new febuxostat imidazolium salt hydrate: Synthesis, crystal structure, solubility, and dissolution study. J. Mol. Struct. 2019, 1176, 633–640. [Google Scholar] [CrossRef]
- Mandal, A.; Rissanen, K.; Mal, P. Unravelling substitution effects on charge transfer characteristics in cocrystals of pyrene based donors and 3, 5-dinitrobenzoic acid. CrystEngComm 2019, 21, 4401–4408. [Google Scholar] [CrossRef]
- Mandal, A.; Swain, P.; Nath, B.; Sau, S.; Mal, P. Unipolar to ambipolar semiconductivity switching in charge transfer cocrystals of 2, 7-di-tert-butylpyrene. CrystEngComm 2019, 21, 981–989. [Google Scholar] [CrossRef]
- Usman, R.; Khan, A.; Wang, M.; Luo, Y.; Sun, W.; Sun, H.; Du, C.; He, N. Investigation of charge-transfer interaction in mixed stack donor–acceptor cocrystals toward tunable solid-state emission characteristics. Cryst. Growth Des. 2018, 18, 6001–6008. [Google Scholar] [CrossRef]
- Zhu, W.; Zheng, R.; Zhen, Y.; Yu, Z.; Dong, H.; Fu, H.; Shi, Q.; Hu, W. Rational design of charge-transfer interactions in halogen-bonded co-crystals toward versatile solid-state optoelectronics. J. Am. Chem. Soc. 2015, 137, 11038–11046. [Google Scholar] [CrossRef]
- Bader, R. Atoms in Molecules; Oxford University Press: Oxford, UK, 1994; Available online: https://global.oup.com/academic/product/atoms-in-molecules-9780198558651?cc=sa&lang=en& (accessed on 5 February 2020).
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P. Characterization of CHO hydrogen bonds on the basis of the charge density. J. Phys. Chem. A 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: DFT computations with periodic boundary conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Varghese, S.; Kim, J.H.; Yoon, S.-J.; Kwon, O.K.; An, B.-K.; Gierschner, J.; Park, S.Y. Tailor-made highly luminescent and ambipolar transporting organic mixed stacked charge-transfer crystals: An isometric donor–acceptor approach. J. Am. Chem. Soc. 2013, 135, 4757–4764. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yi, Y.; Li, Y.; Kim, E.-G.; Coropceanu, V.; Brédas, J.-L. Prediction of remarkable ambipolar charge-transport characteristics in organic mixed-stack charge-transfer crystals. J. Am. Chem. Soc. 2012, 134, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F.-W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Cromer, D.T.; Waber, J.T. International Tables for X-Ray Crystallography, Kynoch. Birm. Engl. 1952, 3, 257–269. [Google Scholar]
- Sheldrick, G.M. SAINT (Version 6.02), SADABS (Version 2.03); Bruker AXS: Madison, WI, USA, 2002. [Google Scholar]
- Sheldrick, G.M. SADABS, Program for Empirical Absorption Correction of Area Detector Data. Available online: https://www.scienceopen.com/document?vid=5cab3651-c60c-4e6d-89cc-c55396e9e2dc (accessed on 5 February 2020).
- Sheldrick, G.M. SAINT and XPREP, version 5.1; Siemens Industrial Automation Inc.: Madison, WI, USA, 1995. [Google Scholar]
- Sheldrick, G.M. SHELXTL, version 5.1; Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 1998; Volume 4, p. 18. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. Gaussian 09 Citation; Gaussian. Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Frisch, M.J.T.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.C.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, G.A.N.B.; Petersson, H.; et al. Gaussian 09 Citation. Available online: https://gaussian.com/g09citation/ (accessed on 5 February 2020).
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Adamo, C.; Scuseria, G.E.; Barone, V. Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model. J. Chem. Phys. 1999, 111, 2889. [Google Scholar] [CrossRef]
- Flaig, D.; Maurer, M.; Hanni, M.; Braunger, K.; Kick, L.; Thubauville, M.; Ochsenfeld, C. Benchmarking hydrogen and carbon NMR chemical shifts at HF, DFT, and MP2 levels. J. Chem. Theory Comput. 2014, 10, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm. 2009, 11, 19–32. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vibrational Mode | Experimental | Calculated |
|---|---|---|
| νsv(-OH) | 3502.27 | 3636 |
| νsv(-NH) | 3410.16 | 3502 |
| νsv(-CH) | 3173.10 | 3249 |
| νsv(-CH2) | 2934.58 | 3007 |
| νsv(-C=O) | 1774.59 | 1745 |
| νav(-O-C=O) | 1708.60 | 1710 |
| νsv(-O-C=O) | 1419.98 | 1426 |
| Bond | DFT | X-ray | Bond | DFT | X-ray | ||
|---|---|---|---|---|---|---|---|
| Conf. A | Conf. B | Conf. A | Conf. B | ||||
| O1-C1 | 1.20911 | 1.20774 | 1.207(3) | C1-C2 | 1.49332 | 1.49195 | 1.487(3) |
| O2-C8 | 1.21626 | 1.20817 | 1.213(3) | C2-C3 | 1.38389 | 1.38260 | 1.384(3) |
| O3-C10 | 1.24804 | 1.22823 | 1.243(3) | C2-C7 | 1.39069 | 1.39056 | 1.386(3) |
| O4-C10 | 1.25007 | 1.28972 | 1.264(3) | C3-C4 | 1.39878 | 1.39831 | 1.391(4) |
| N1-C1 | 1.38606 | 1.40023 | 1.392(3) | C4-C5 | 1.39823 | 1.39655 | 1.391(4) |
| N1-C8 | 1.40031 | 1.39845 | 1.381(3) | C5-C6 | 1.39897 | 1.39694 | 1.392(4) |
| N1-C9 | 1.44965 | 1.43814 | 1.448(3) | C6-C7 | 1.38346 | 1.38324 | 1.374(4) |
| N2-C11 | 1.37802 | 1.37584 | 1.373(3) | C7-C8 | 1.49368 | 1.49164 | 1.497(3) |
| N2-C13 | 1.32984 | 1.32155 | 1.325(3) | C9-C10 | 1.56229 | 1.53819 | 1.526(4) |
| N3-C12 | 1.37684 | 1.37268 | 1.370(3) | C11-C12 | 1.35969 | 1.36419 | 1.344(4) |
| N3-C13 | 1.32869 | 1.34207 | 1.322(3) | ||||
| IM+··· NPG ̶ | 3.23483 | 3.359(3) | |||||
| IM+··· NPG ̶ | 2.45662 | 2.665(3) | |||||
| D–H···A | D···A (Å) | H···A (Å) | D–H···A (deg) | Symmetry Code |
|---|---|---|---|---|
| O1W–H1···O2 | 2.9249(5) | 2.13 | 152 | |
| N2–H2···O4 | 2.6577(4) | 1.81 | 167 | ½ − x,1/2 − y,1 − z |
| O1W–H1···O3 | 2.7059(5) | 1.84 | 173 | |
| N3–H3A···O1W | 2.7525(5) | 1.98 | 150 | |
| C3–H3···O1 | 3.3427(6) | 2.47 | 157 | x,1 − y,1/2 + z |
| C9–H9A···O1 | 3.4578(6) | 2.51 | 164 | ½ − x, − ½ + y,1/2 − z |
| C11–H11···O3 | 3.4064(6) | 2.54 | 156 | x,1 − y,1/2 + z |
| C12–H12···O2 | 3.4421(6) | 2.55 | 161 | − x,y,1/2 − z |
| C13–H13···O4 | 3.1838(5) | 2.27 | 166 | x, − 1 + y,z |
| IM+–NPG−–HYD | ρ(r) | ∇2ρ(r) | V(r) | G(r) | H(r) | Einta | Eintb |
|---|---|---|---|---|---|---|---|
| Configuration A | |||||||
| N22···C5–C11 | 0.00699 | 0.02326 | −0.00311 | 0.00446 | 0.00135 | 0.9757 | 1.2006 |
| C25···C11 | 0.00654 | 0.02020 | −0.00316 | 0.00410 | 0.00094 | 0.9914 | 1.1037 |
| O2···O32 | 0.00404 | 0.01502 | −0.00241 | 0.00308 | 0.00067 | 0.7561 | 0.8291 |
| Configuration B | |||||||
| N22–H23···O4–C21 | 0.03967 | 0.12031 | –0.03865 | 0.03436 | −0.00428 | 12.1266 | 9.2497 |
| C21–O3···H34–O32 | 0.03096 | 0.11224 | –0.02988 | 0.02897 | −0.00091 | 9.3749 | 7.7987 |
| C26–H27···H19–C18 | 0.00299 | 0.01240 | –0.00149 | 0.00229 | 0.00080 | 0.4674 | 0.6164 |
| Donor NBO (i) | Acceptor NBO (j) | E (2) kcal/mol |
|---|---|---|
| Configuration A | ||
| IM+: LP (N23) | NPG-: BD* (C5–C11) | 0.07 |
| IM+: LP (N23) | NPG-: BD* (C8–C12) | 0.15 |
| NPG–: LP (O13) | IM+: BD* (C24–C26) | 0.05 |
| NPG–: LP (O13) | IM+: BD* (C24–C26) | 0.08 |
| Configuration B | ||
| NPG–: LP (O4) | IM+: BD* (N22–H23) | 4.03 |
| NPG–: LP (O4) | IM+: BD* (N22–C26) | 0.21 |
| NPG–: LP (O4) | IM+: BD* (N22–H23) | 14.05 |
| NPG–: LP (O4) | IM+: BD* (N22–C26) | 0.23 |
| NPG–: LP (O4) | IM+: BD* (N22–C30) | 0.20 |
| NPG–: LP (O4) | IM+: BD* (N22–C30) | 0.05 |
| NPG–: LP (O4) | IM+: BD* (N22–H23) | 0.11 |
| NPG–: LP (O4) | IM+: BD* (N22–C30) | 0.31 |
| NPG–: LP (O3) | HYD: BD* (O32–H34) | 3.04 |
| NPG–: LP (O3) | HYD: BD* (O32–H34) | 6.69 |
| Parameters | IM+–NPG−–HYD |
|---|---|
| Empirical formula | C13H13N3O5 |
| Formula weight | 291.27 |
| Temperature (K) | 100(2) |
| Crystal system | Monoclinic |
| Space group | C2/c |
| a (Å) | 20.982(3) |
| b (Å) | 7.8082(10) |
| c (Å) | 17.943(3) |
| α (°) | 90 |
| β (°) | 119.808(7) |
| γ (°) | 90 |
| Volume (Å3) | 2550.7(7) |
| Z | 8 |
| ρcalc (g/cm3) | 1.5168 |
| μ (mm–1) | 0.119 |
| F(000) | 1216.7 |
| Crystal size (mm3) | 0.33 × 0.21 × 0.14 |
| Radiation | Mo Kα (λ = 0.71073) |
| 2θ range for data collection (°) | 5.68 to 50.1 |
| Index ranges | –28 ≤ h ≤ 27, –10 ≤ k ≤ 10, –23 ≤ l ≤ 23 |
| Reflections collected | 18701 |
| Independent reflections | 2265 (Rint = 0.0943, Rsigma = 0.0639) |
| Data/restraints/parameters | 2265/0/193 |
| Goodness-of-fit on F2 | 1.093 |
| Final R indexes [I>=2σ (I)] | R1 = 0.0526, wR2 = 0.1094 |
| Final R indexes (all data) | R1 = 0.0669, wR2 = 0.1168 |
| Largest diff. peak/hole/e Å–3 | 0.37/–0.41 |
| CCDC | 1915942 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usman, M.; Khan, R.A.; Jaafar, M.H.; Alsalme, A.; Tabassum, S. Structure of Imidazolium-N-phthalolylglycinate Salt Hydrate: Combined Experimental and Quantum Chemical Calculations Studies. Crystals 2020, 10, 91. https://doi.org/10.3390/cryst10020091
Usman M, Khan RA, Jaafar MH, Alsalme A, Tabassum S. Structure of Imidazolium-N-phthalolylglycinate Salt Hydrate: Combined Experimental and Quantum Chemical Calculations Studies. Crystals. 2020; 10(2):91. https://doi.org/10.3390/cryst10020091
Chicago/Turabian StyleUsman, Mohammad, Rais Ahmad Khan, Mohammad H. Jaafar, Ali Alsalme, and Sartaj Tabassum. 2020. "Structure of Imidazolium-N-phthalolylglycinate Salt Hydrate: Combined Experimental and Quantum Chemical Calculations Studies" Crystals 10, no. 2: 91. https://doi.org/10.3390/cryst10020091
APA StyleUsman, M., Khan, R. A., Jaafar, M. H., Alsalme, A., & Tabassum, S. (2020). Structure of Imidazolium-N-phthalolylglycinate Salt Hydrate: Combined Experimental and Quantum Chemical Calculations Studies. Crystals, 10(2), 91. https://doi.org/10.3390/cryst10020091