Intramolecular sp2-sp3 Disequalization of Chemically Identical Sulfonamide Nitrogen Atoms: Single Crystal X-Ray Diffraction Characterization, Hirshfeld Surface Analysis and DFT Calculations of N-Substituted Hexahydro-1,3,5-Triazines

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Details
2.2. Crystallographic Details
2.3. Hirshfeld Surface Calculations
2.4. Theoretical Methods
3. Results
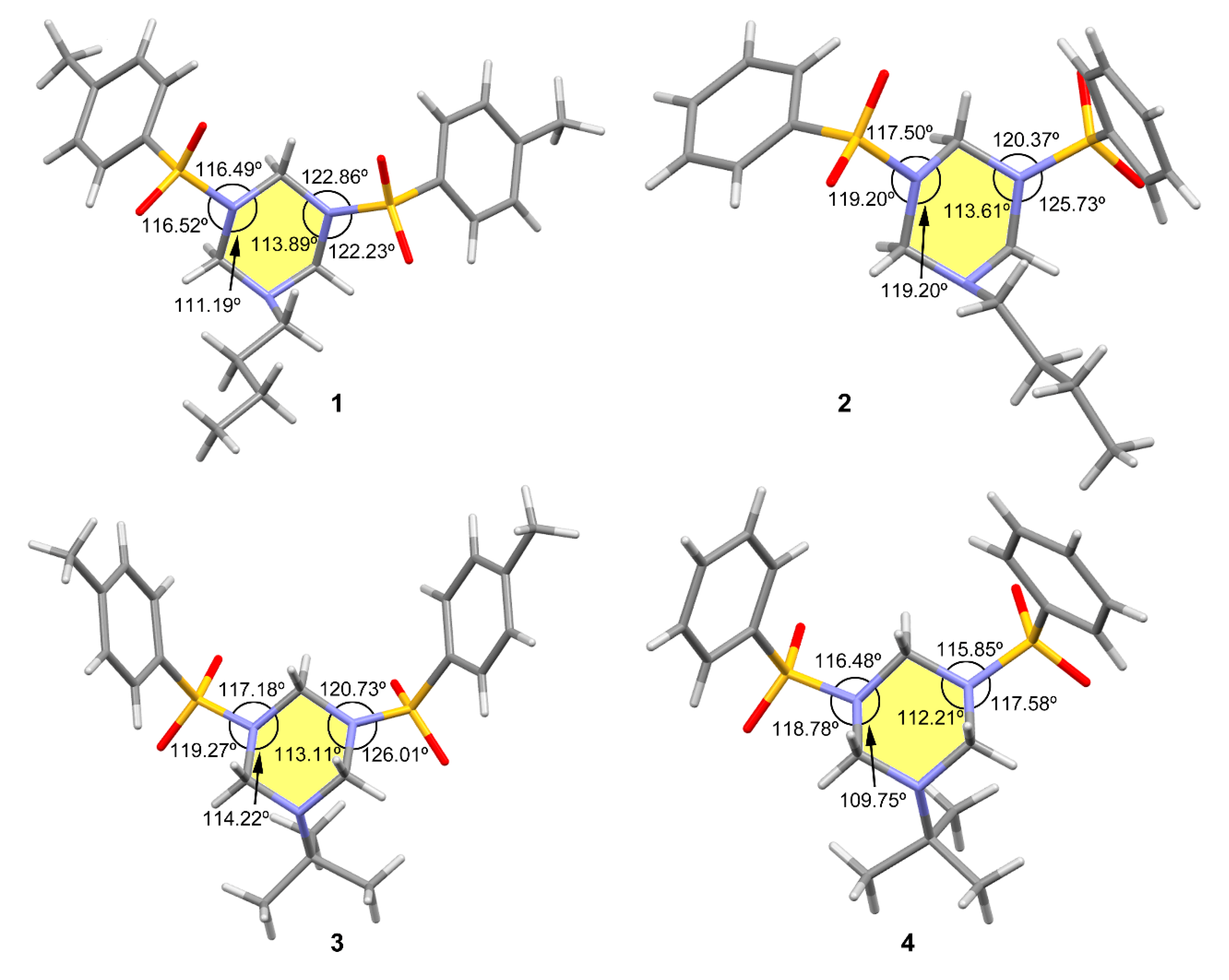
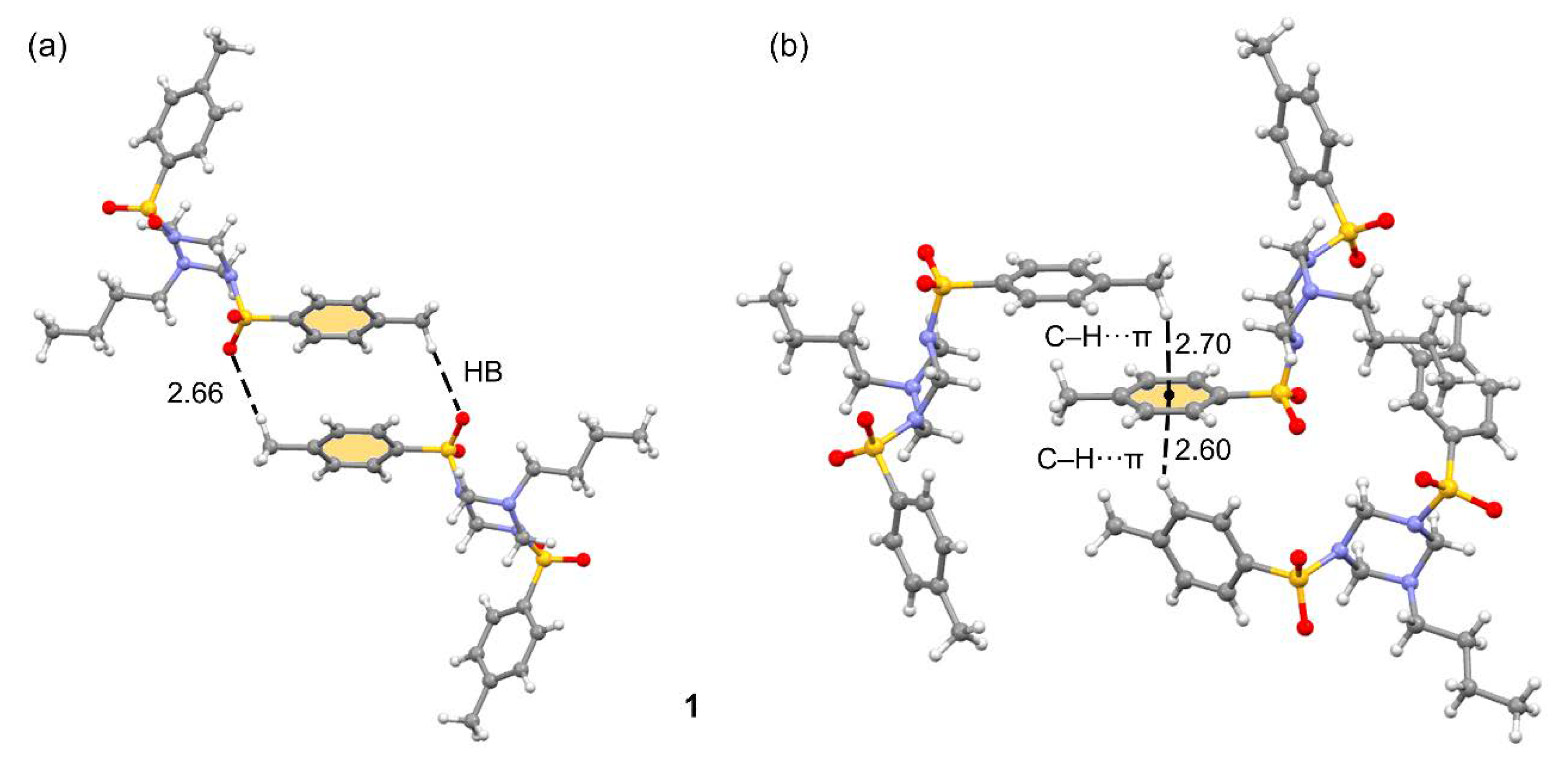
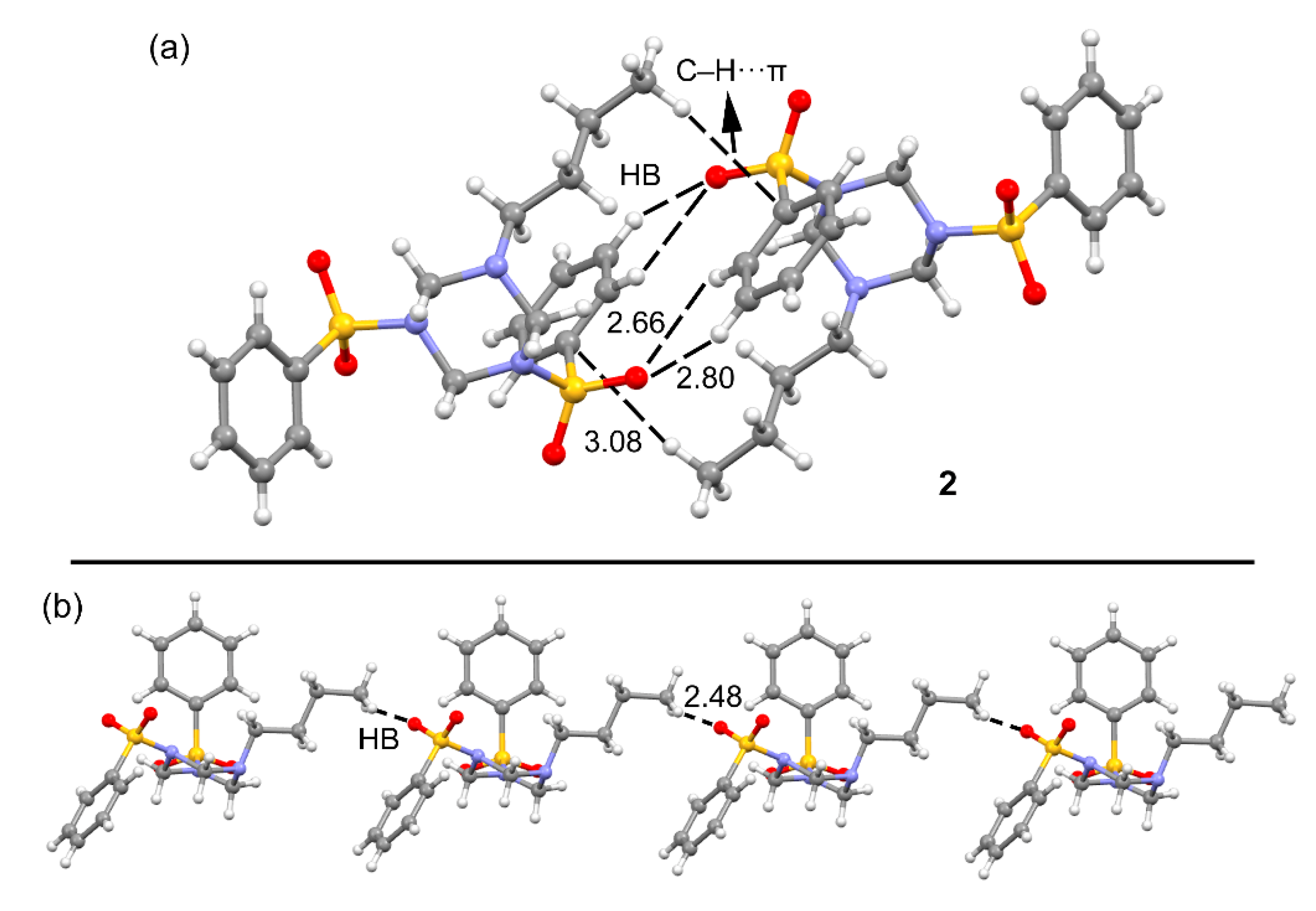
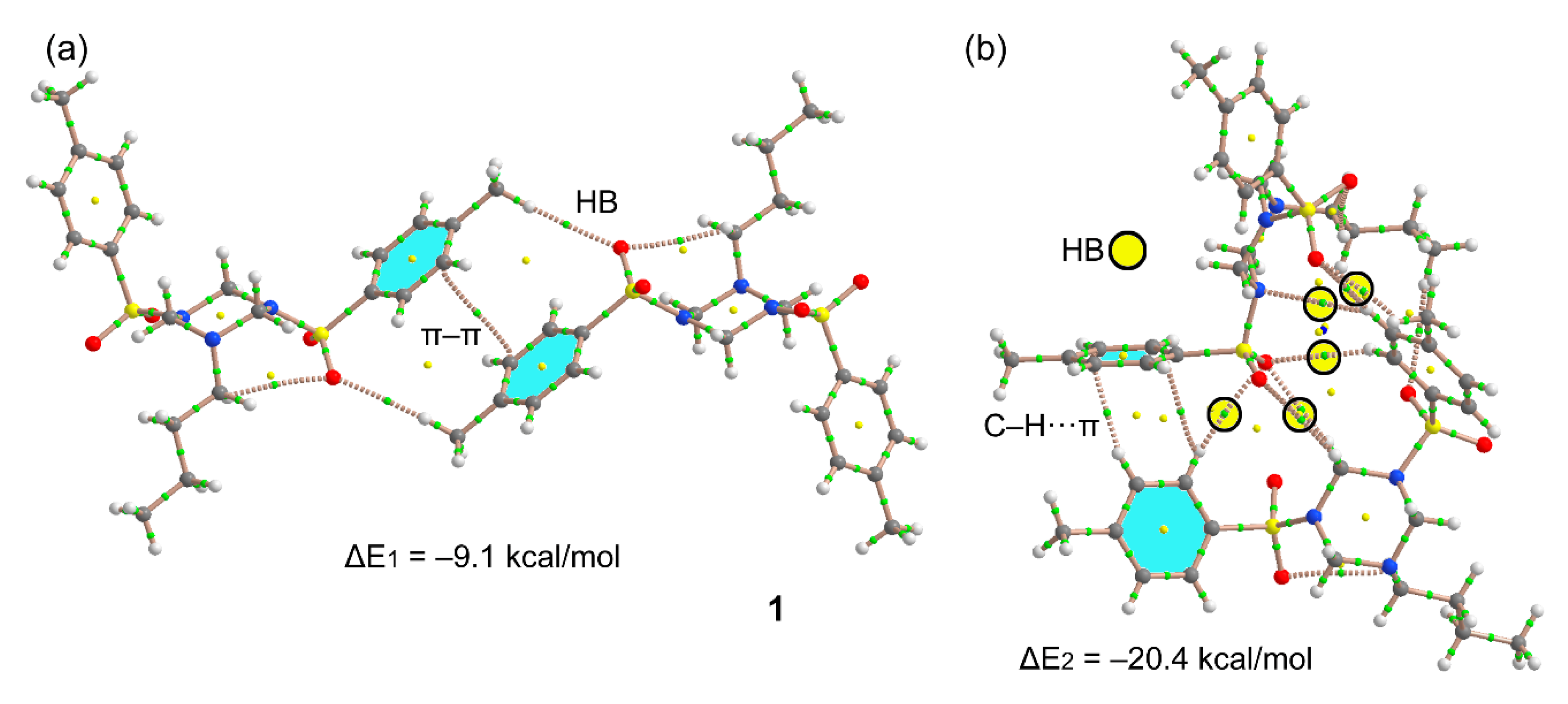
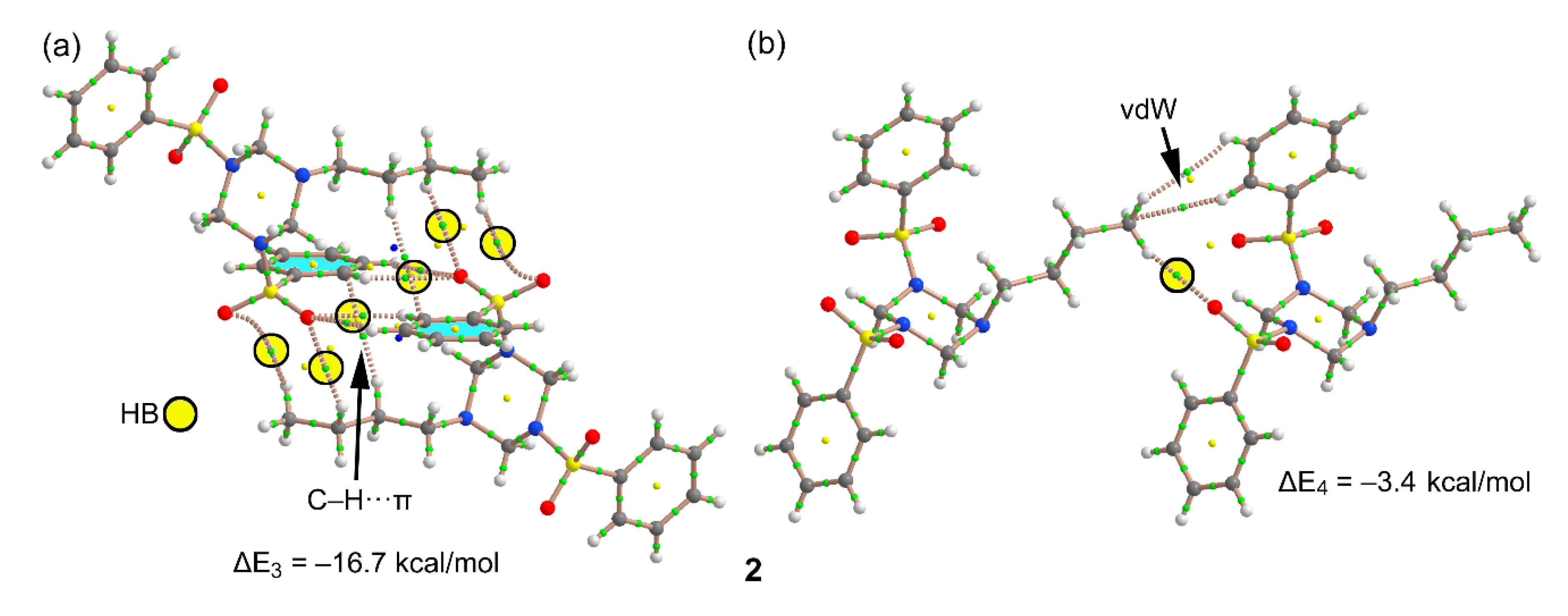
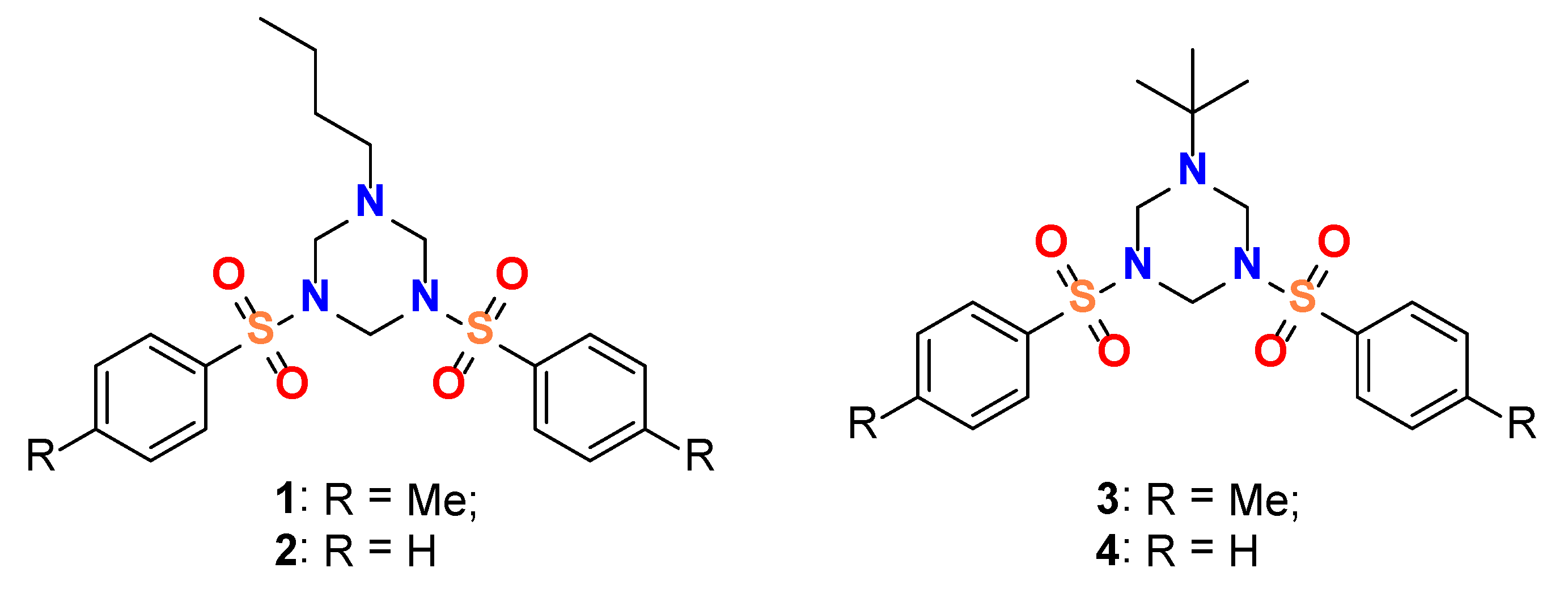
3.1. Structural Description
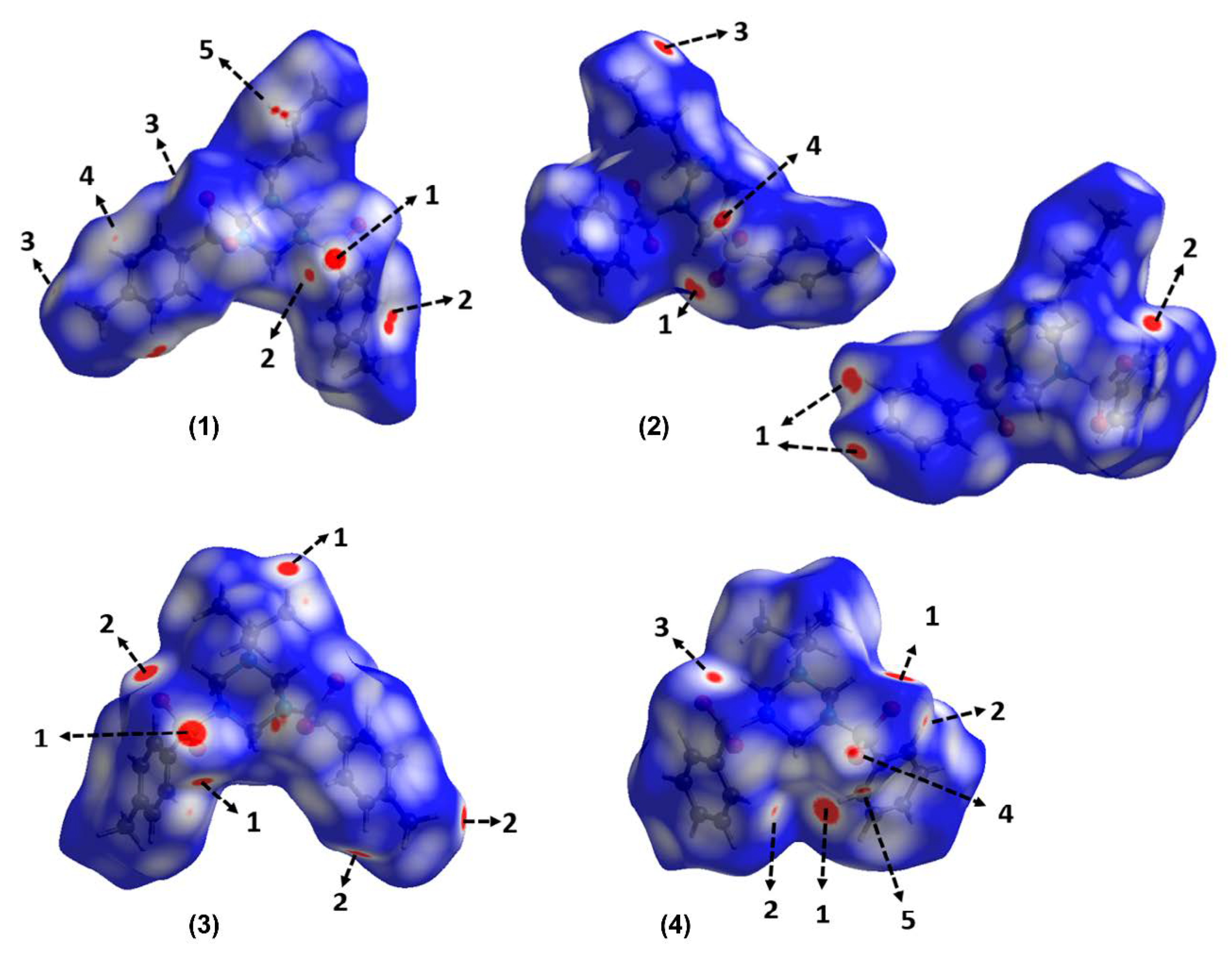
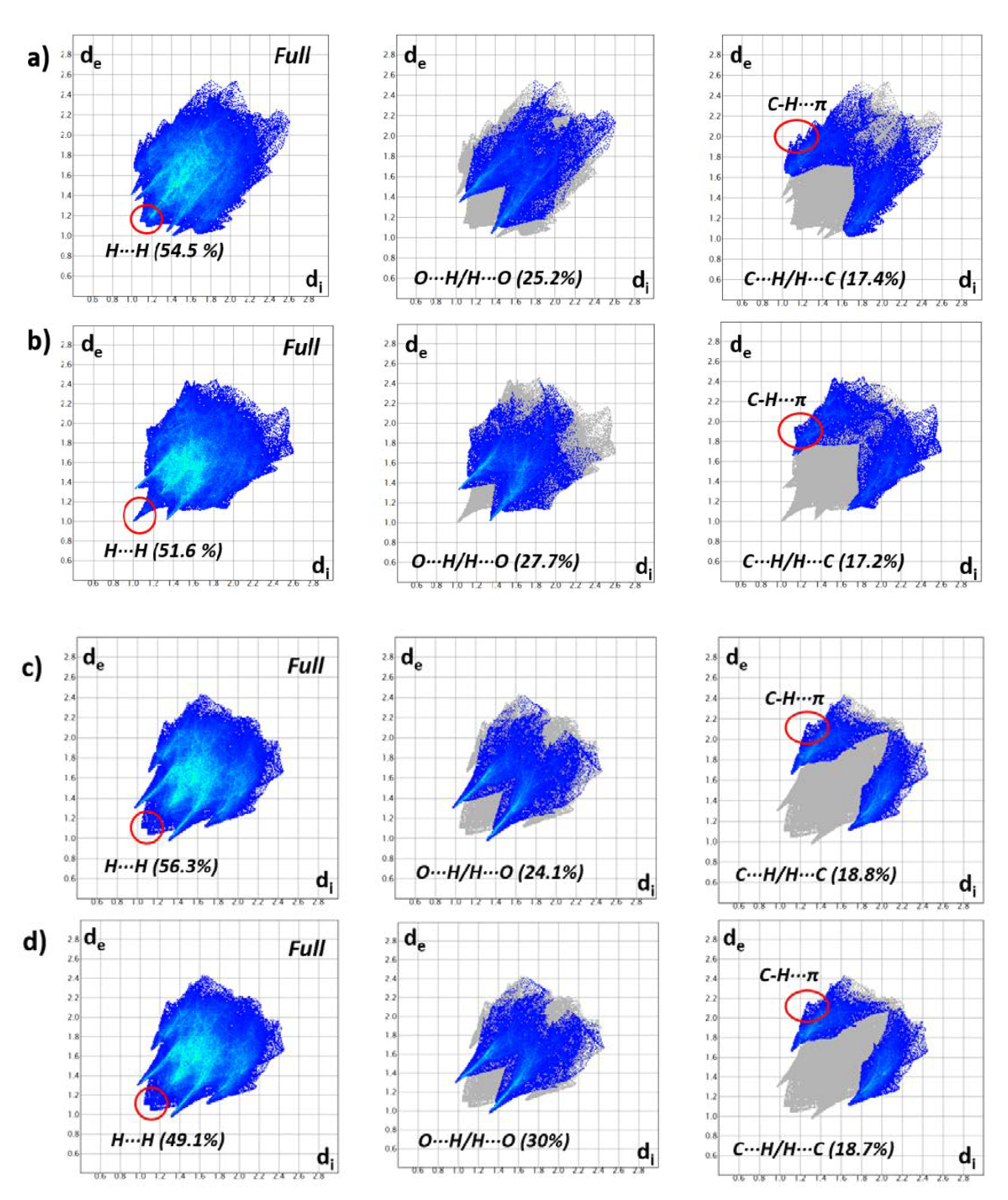
3.2. Hirshfeld Surfaces
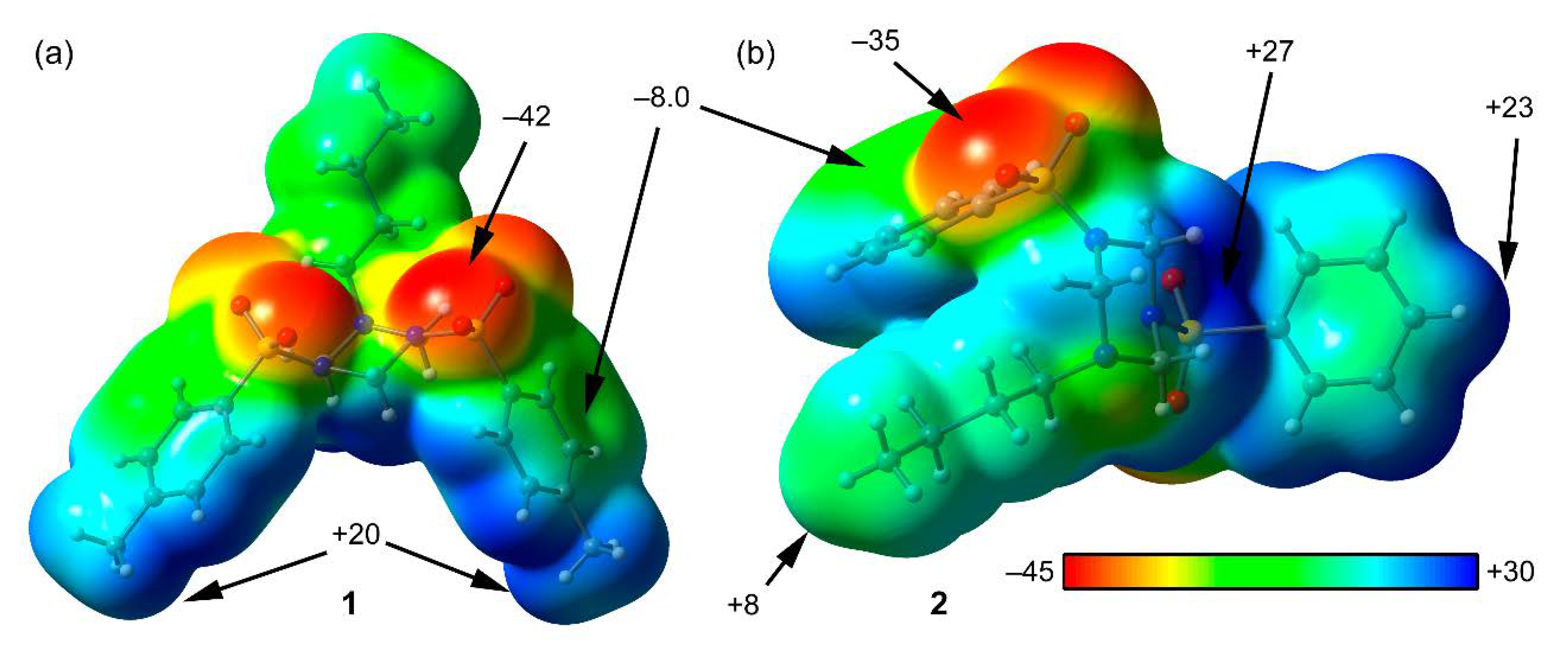
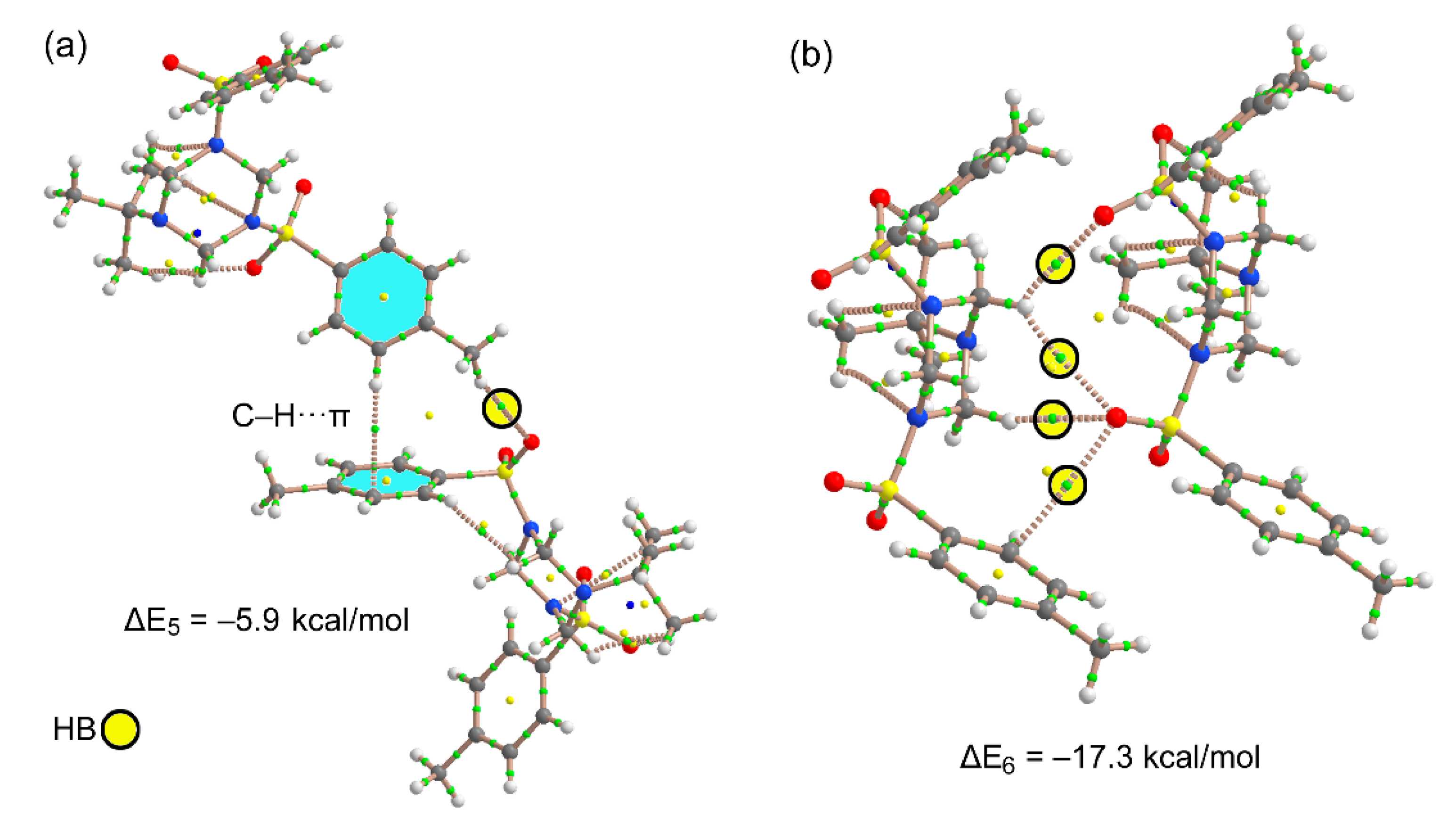
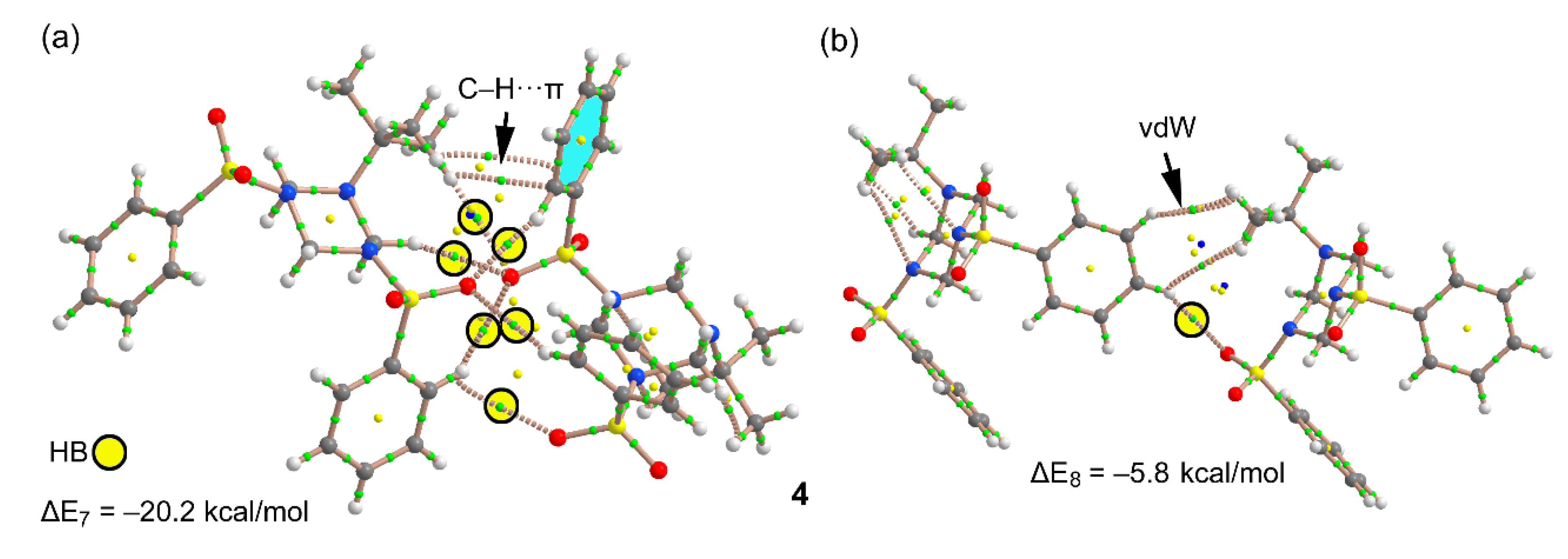
3.3. DFT Calculations
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ha, H.-J.; Koo Lee, W. Synthetic applications of Lewis acid-induced N-methyleneamine equivalents. Heterocycles 2002, 57, 1525–1538. [Google Scholar] [CrossRef]
- Ji, D.; Sun, J. [3+2]-Cycloaddition of azaoxyallyl cations with hexahydro-1,3,5-triazines: Access to 4-imidazolidinones. Org. Lett. 2018, 20, 2745–2748. [Google Scholar] [CrossRef]
- Ji, D.; Wang, C.; Sun, J. Asymmetric [4+2]-cycloaddition of copper–allenylidenes with hexahydro-1,3,5-triazines: access to chiral tetrahydroquinazolines. Org. Lett. 2018, 20, 3710–3713. [Google Scholar] [CrossRef]
- Chen, L.; Liu, K.; Sun, J. Catalyst-free synthesis of tetrahydropyrimidines via formal [3+3]-cycloaddition of imines with 1,3,5-hexahydro-1,3,5-triazines. RSC Adv. 2018, 8, 5532–5535. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Xu, G.; Sun, J. Gold-catalyzed formal [4 + 1]/[4 + 3] cycloadditions of diazo esters with triazines. Angew. Chem. Int. Ed. 2016, 55, 11867–11871. [Google Scholar] [CrossRef]
- Liu, S.; Yang, P.; Peng, S.; Zhu, C.; Cao, S.; Li, J.; Sun, J. Gold-catalyzed sequential annulations towards 3,4-fused bi/tri-cyclic furans involving a [3 + 2 + 2]-cycloaddition. Chem. Commun. 2017, 53, 1152–1155. [Google Scholar] [CrossRef]
- Liu, P.; Xu, G.; Sun, J. Metal-free [2 + 1 + 2]-cycloaddition of tosylhydrazones with hexahydro-1,3,5-triazines to form imidazolidines. Org. Lett. 2017, 19, 1858–1861. [Google Scholar] [CrossRef]
- Peng, S.; Ji, D.; Sun, J. Gold-catalyzed [2 + 2 + 2 + 2]-annulation of 1,3,5-hexahydro-1,3,5-triazines with alkoxyallenes. Chem. Commun. 2017, 53, 12770–12773. [Google Scholar] [CrossRef]
- Zeng, Z.; Jin, H.; Song, X.; Wang, Q.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Gold-catalyzed intermolecular cyclocarboamination of ynamides with 1,3,5-triazinanes: En route to tetrahydropyrimidines. Chem. Commun. 2017, 53, 4304–4307. [Google Scholar] [CrossRef] [Green Version]
- Garve, L.K.B.; Jones, P.G.; Werz, D.B. Ring-opening 1-amino-3-aminomethylation of donor-acceptor cyclopropanes via 1,3-diazepanes. Angew. Chem. Int. Ed. 2017, 56, 9226–9230. [Google Scholar] [CrossRef]
- Yin, B. Synergistic Antimicrobial Composition. WO Patent 2,011,049,761, 28 April 2011. [Google Scholar]
- Kletskov, A.V.; Frontera, A.; Sinelshchikova, A.A.; Grigoriev, M.S.; Zaytsev, V.P.; Grudova, M.V.; Bunev, A.S.; Presnukhina, S.; Shetnev, A.; Zubkov, F.I. Straightforward three-component synthesis of N-alkyl-N′,N″-substituted hexahydro-1,3,5-triazines. Synlett 2020. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Cryst. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolf, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 13.05.06); TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Breneman, C.M.; Weber, L.W. Charge and Energy Redistribution in Sulfonamides Undergoing Conformational Changes. Hybridization as a Controlling Influence over Conformer Stability. Can. J. Chem. 1996, 74, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.C.; Geist, V. International Tables for Crystallography. Volume C: Mathematical, Physical and Chemical Tables (See, Table 9.5.1.1.). Cryst. Res. Technol. 1993, 28, 110. [Google Scholar] [CrossRef]
- Caine, B.A.; Bronzato, M.; Popelier, P.L.A. Experiment Stands Corrected: Accurate Prediction of the Aqueous pKa Values of Sulfonamide Drugs Using Equilibrium Bond Lengths. Chem. Sci. 2019, 10, 6368–6381. [Google Scholar] [CrossRef] [Green Version]
- Habgood, M.; Grau-Crespo, R.; Price, S.L. Substitutional and Orientational Disorder in Organic Crystals: A Symmetry-Adapted Ensemble Model. Phys. Chem. Chem. Phys. 2011, 13, 9590. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
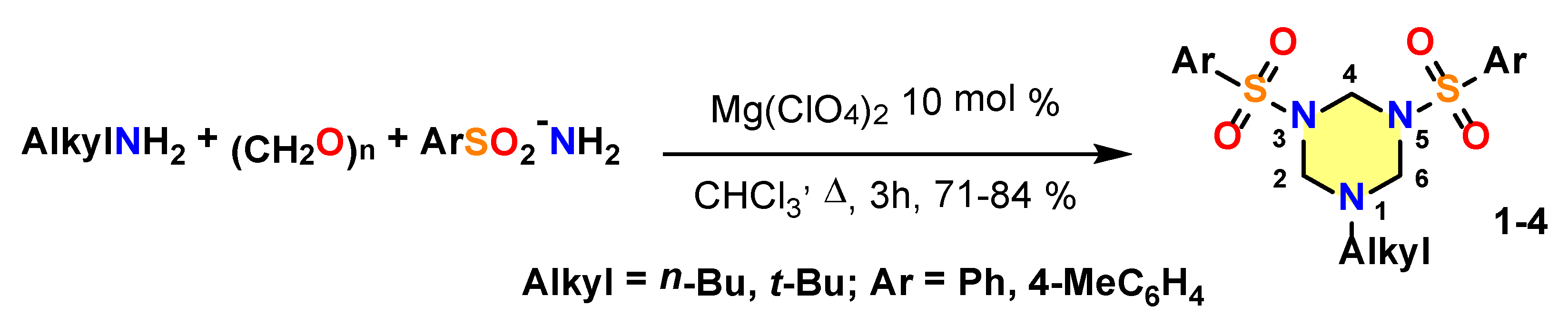{kind=link}
| Identification Code | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| CCDC number | 1992667 | 1992668 | 1992669 | 1992670 |
| Empirical formula | C21H29N3O4S2 | C19H25N3O4S2 | C21H29N3O4S2 | C19H25N3O4S2 |
| Formula weight | 451.59 | 423.54 | 451.59 | 423.54 |
| Temperature/K | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | monoclinic | monoclinic | monoclinic | orthorhombic |
| Space group | P21/c | P21/n | P21/n | P212121 |
| a/Å | 13.2871(4) | 8.4284(2) | 5.955(4) | 10.7298(3) |
| b/Å | 10.3261(3) | 25.9248(8) | 15.378(12) | 11.1010(3) |
| c/Å | 15.9595(4) | 9.5601(3) | 23.915(19) | 16.9303(5) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 90.511(2) | 106.639(1) | 90.968(16) | 90 |
| γ/° | 90 | 90 | 90 | 90 |
| Volume/Å3 | 2189.62(11) | 2001.46(10) | 2190(3) | 2016.59(10) |
| Z | 4 | 4 | 4 | 4 |
| ρcalcg/cm3 | 1.370 | 1.406 | 1.370 | 1.395 |
| μ/mm−1 | 0.276 | 0.297 | 0.276 | 0.295 |
| F(000) | 960.0 | 896.0 | 960.0 | 896.0 |
| Crystal size/mm3 | 0.440 × 0.360 × 0.320 | 0.400 × 0.320 × 0.260 | 0.500 ×0.180 × 0.030 | 0.420 × 0.400 × 0.360 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 7.126 to 59.998 | 7.392 to 59.994 | 8.476 to 55 | 8.16 to 69.998 |
| Index ranges | −18 ≤ h ≤ 18, −14 ≤ k ≤ 14, −20 ≤ l ≤ 22 | −6 ≤ h ≤ 11, −35 ≤ k ≤ 36, −13 ≤ l ≤ 13 | −4 ≤ h ≤ 7, −19 ≤ k ≤ 19, −31 ≤ l ≤ 31 | −17 ≤ h ≤ 16, −17 ≤ k ≤ 16, −19 ≤ l ≤ 27 |
| Reflections collected | 33914 | 28404 | 14104 | 35553 |
| Independent reflections | 6383 [Rint = 0.0390, Rsigma = 0.0303] | 5835 [Rint = 0.0351, Rsigma = 0.0280] | 4940 [Rint = 0.1432, Rsigma = 0.1883] | 8855 [Rint = 0.0303, Rsigma = 0.0303] |
| Data/restraints/parameters | 6383/0/273 | 5835/0/253 | 4940/6/274 | 8855/0/253 |
| Goodness-of-fit on F2 | 1.029 | 1.035 | 1.049 | 1.042 |
| Final R indexes [I >= 2σ (I)] | R1 = 0.0350, wR2 = 0.0877 | R1 = 0.0334, wR2 = 0.0824 | R1 = 0.1398, wR2 = 0.3472 | R1 = 0.0283, wR2 = 0.0698 |
| Final R indexes [all data] | R1 = 0.0457, wR2 = 0.0939 | R1 = 0.0423, wR2 = 0.0873 | R1 = 0.2253, wR2 = 0.4075 | R1 = 0.0316, wR2 = 0.0715 |
| Largest diff. peak/hole/e Å−3 | 0.38/−0.37 | 0.39/−0.37 | 0.94/−0.55 | 0.36/−0.28 |
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Alkyl | Bu | Bu | t-Bu | t-Bu |
| SO2Ar | SO2C6H4Me | SO2Ph | SO2C6H4Me | SO2Ph |
| Sum of angles around N3 (°) | 359.0 | 359.7 | 359.8 | 345.0 |
| Sum of angles around N5 (°) | 344.2 | 347.4 | 350.7 | 345.6 |
| S1–N3 distance (Å) | 1.642(1) | 1.630(1) | 1.632(9) | 1.640(1) |
| S2–N5 distance (Å) | 1.632(1) | 1.614(1) | 1.629(9) | 1.638(1) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kletskov, A.V.; Gil, D.M.; Frontera, A.; Zaytsev, V.P.; Merkulova, N.L.; Beltsova, K.R.; Sinelshchikova, A.A.; Grigoriev, M.S.; Grudova, M.V.; Zubkov, F.I. Intramolecular sp2-sp3 Disequalization of Chemically Identical Sulfonamide Nitrogen Atoms: Single Crystal X-Ray Diffraction Characterization, Hirshfeld Surface Analysis and DFT Calculations of N-Substituted Hexahydro-1,3,5-Triazines. Crystals 2020, 10, 369. https://doi.org/10.3390/cryst10050369
Kletskov AV, Gil DM, Frontera A, Zaytsev VP, Merkulova NL, Beltsova KR, Sinelshchikova AA, Grigoriev MS, Grudova MV, Zubkov FI. Intramolecular sp2-sp3 Disequalization of Chemically Identical Sulfonamide Nitrogen Atoms: Single Crystal X-Ray Diffraction Characterization, Hirshfeld Surface Analysis and DFT Calculations of N-Substituted Hexahydro-1,3,5-Triazines. Crystals. 2020; 10(5):369. https://doi.org/10.3390/cryst10050369
Chicago/Turabian StyleKletskov, Alexey V., Diego M. Gil, Antonio Frontera, Vladimir P. Zaytsev, Natalia L. Merkulova, Ksenia R. Beltsova, Anna A. Sinelshchikova, Mikhail S. Grigoriev, Mariya V. Grudova, and Fedor I. Zubkov. 2020. "Intramolecular sp2-sp3 Disequalization of Chemically Identical Sulfonamide Nitrogen Atoms: Single Crystal X-Ray Diffraction Characterization, Hirshfeld Surface Analysis and DFT Calculations of N-Substituted Hexahydro-1,3,5-Triazines" Crystals 10, no. 5: 369. https://doi.org/10.3390/cryst10050369
APA StyleKletskov, A. V., Gil, D. M., Frontera, A., Zaytsev, V. P., Merkulova, N. L., Beltsova, K. R., Sinelshchikova, A. A., Grigoriev, M. S., Grudova, M. V., & Zubkov, F. I. (2020). Intramolecular sp2-sp3 Disequalization of Chemically Identical Sulfonamide Nitrogen Atoms: Single Crystal X-Ray Diffraction Characterization, Hirshfeld Surface Analysis and DFT Calculations of N-Substituted Hexahydro-1,3,5-Triazines. Crystals, 10(5), 369. https://doi.org/10.3390/cryst10050369