1. Introduction
Cemented carbides are one of the most widespread powder metallurgy products worldwide. The reason for this is that, compared with other cutting materials (such as diamond or high-speed steel), they have an excellent combination of hardness and toughness [
1,
2]. With rapid industrial development, the design and performance requirements imposed on cemented carbide tools are constantly becoming more onerous, and the performance of the ordinary grain size of cemented carbide cannot fully meet modern industrial demands. Ultrafine (nanocrystalline) cemented carbides have, in theory, superior mechanical properties to conventional coarse-grained cemented carbides [
3,
4], such as hardness, wear resistance, and flexural strength. Some studies have shown that when the content of Co-phase is constant and when the WC grain size reaches ultrafine or nanocrystalline, the hardness and toughness of cemented carbides are improved [
5,
6]. When the WC grain size is less than 500 nm, the hardness and toughness of the cemented carbides are greatly improved. Therefore, the development of ultrafine or nanocrystalline cemented carbides with high hardness and high strength has become a key research issue among those working with cemented carbide materials [
7].
In recent years, scholars have adopted many new technologies and methods to improve the mechanical properties of cemented carbides, prolong the service life of cemented carbide tools, and reduce the production costs thereof [
8,
9,
10]. The key to the preparation of ultrafine cemented carbides is the effective inhibition of WC grains during the preparation and sintering of the powdered raw materials [
11,
12]. Therefore, grain growth inhibitors (GGIs) and rapid sintering methods were used in ultrafine cemented carbides to mitigate and suppress the growth of grains at elevated sintering temperatures [
13].
In this experiment, WC-V
8C
7-Cr
3C
2-Co nanocomposites were used as raw materials, and ultrafine tungsten carbide-based cemented carbides were prepared by secondary pressing (moulding and cold isostatic pressing) and microwave sintering. Since the raw materials are nano-powders, with a high specific surface area, a large contact area between the particles, many interfacial atoms and a high atomic diffusion coefficient in the interfacial region, such nano-materials exhibit high chemical activity. In addition, nanocomposite GGIs can inhibit WC grain growth. Therefore, the sintering temperature can be lowered, the reaction time can be shortened, and the abnormal growth of the WC crystal grains can be effectively inhibited [
14,
15,
16]. Secondary pressing (moulding and cold isostatic pressing) is conducive to obtaining higher density green bodies, which is conducive to the rapid sintering and densification of composites. Furthermore, as a rapid sintering technology with a special mechanism of action, microwave sintering has received more attention in recent years. Microwaves refer to electromagnetic waves ranging from 300 MHz (1 m wavelength) to 300 GHz (1 mm wavelength) [
17]. Microwave sintering has many advantages that are difficult to achieve with conventional heating, such as high energy efficiency, no pollution, a low sintering temperature, uniform microstructure of materials thus produced, and materials that can acquire special structures or properties [
18,
19]. In this study, WC-V
8C
7-Cr
3C
2-Co nanocomposites were used as raw materials to prepare ultrafine cemented carbides by using microwave sintering technology and secondary pressing (moulding and cold isostatic pressing). The effects of sintering temperature and holding time on the microstructure and mechanical properties of the alloy were investigated.
2. Materials and Methods
The nano-WC (purity >99.9%, average particle size <200 nm, Shanghai Shuitian Material Technology Co., Ltd., Shanghai, China), nano-V8C7 (purity >99.9%, average particle size <200 nm, Shanghai Shuitian Material Technology Co., Ltd.), nano-Cr3C2 (purity >99.9%, average particle size <100 nm, Shanghai Shuitian Material Technology Co., Ltd.) and nano-Co (purity >99.9%, average particle size <50 nm, Shanghai Shuitian Material Technology Co., Ltd.) powders were used as raw materials. According to a certain ratio (WC:V8C7:Cr3C2:Co = 89.5%:0.25%:0.25%:10%), the raw materials were mixed in a planetary ball mill (QM-3SP2, Nanjing Laibe Industrial Co., Ltd., Nanjing, China) for ball milling (150 rpm), at a ball to powder weight ratio of 5:1, wherein the ball milling medium was anhydrous ethanol (liquid-solid ratio 350 mL/kg). After being milled for 8 h, the mixture was dried at 90 °C for 24 h. An appropriate amount of paraffin was added to these materials and mixed evenly. The mixed powders (after stuffing) were pressed into 6 mm × 6 mm × 28 mm cuboid specimens using a hydraulic press. They were then further pressed using a cold isostatic press, to increase the density of the green body. The pressed specimens were vacuum-dried at 100 °C for 24 h. Finally, the dried specimens were sintered in a microwave sintering furnace (RWS-3, Hunan Zhongsheng Thermal Energy Technology Co., Ltd.) at sintering temperatures of 1100, 1200, 1300, and 1400 °C, for holding times of 20, 40, 60, and 80 min. Before sintering, the sintering furnace was pumped out to a vacuum of 1 × 10−2 Pa, and then argon gas was introduced to form a protective atmosphere at a flow rate of 20 mL/min. The heating and cooling rates were 10–50 °C/min and 8–30 °C/min, respectively, and subsequent dewaxing was conducted at 610 °C for 30 min.
The phase composition of the specimen was measured using a D8 AA25 X-ray single crystal diffractometer (Bruker, Germany) with Cu-K
α radiation in the range 20° ≤ 2
θ ≤ 90°. The microstructure and grain size of the specimens were observed by INSPECT F50 scanning electron microscopy (FEI, Hillsboro, OR, USA). The sintered specimens were polished with diamond paste before scanning electron microscopy. The specimen density was measured by a digital solid densitometer using the Archimedes method. The hardness of the specimen was measured using an FM-700 Vickers microhardness tester (Toshiba Teli, Tokyo, Japan) under a load of 1 kgf and loading (displacement-controlled) rate of 50 μm/s. Indentation fracture toughness
KIC was estimated by applying the Palmqvist model to the cracks generated by indentation, using the Shetty equation [
20].
3. Results and Discussion
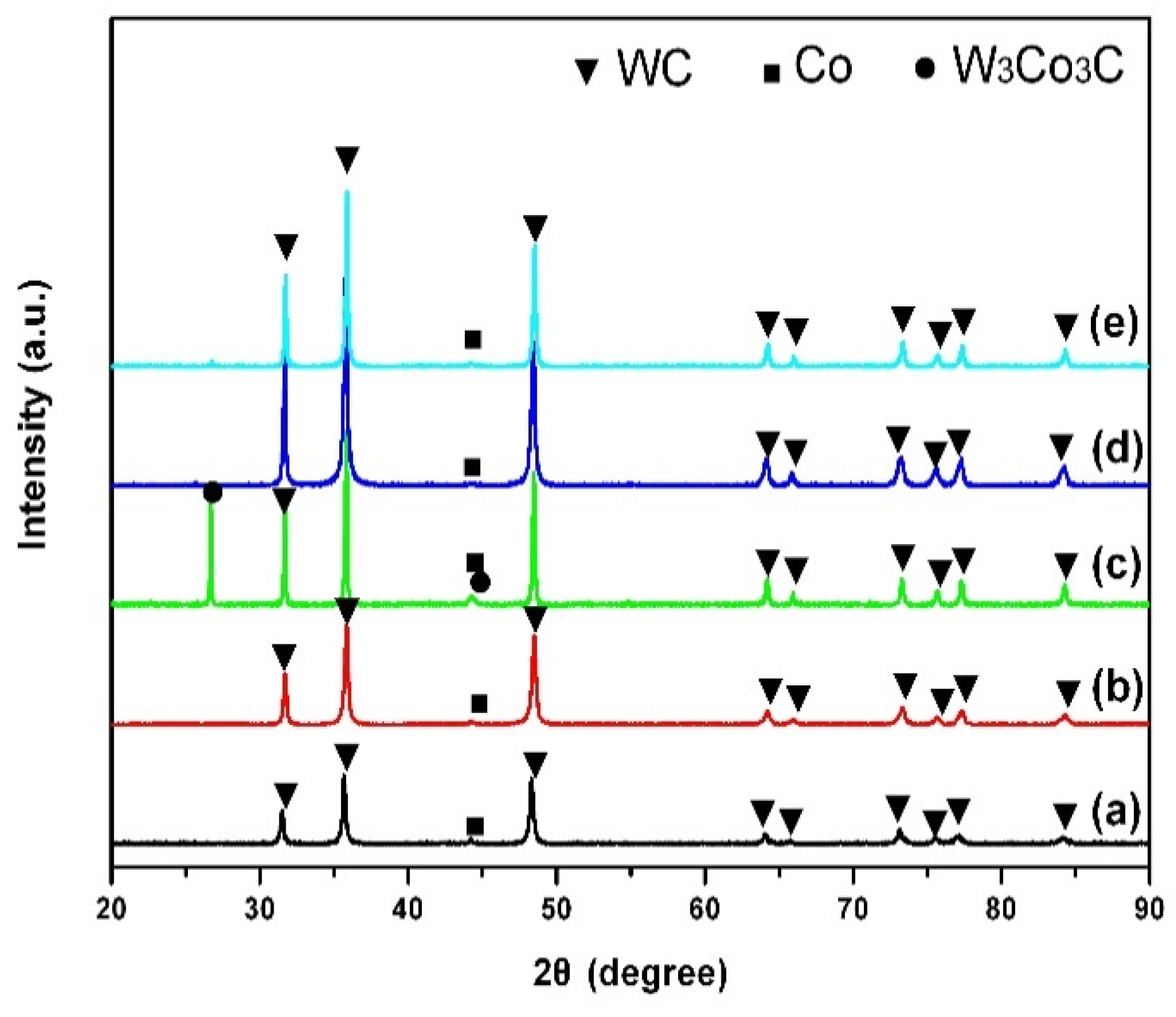
Figure 1 shows the X-ray diffraction (XRD) patterns of the raw materials and the specimens sintered at different temperatures for 40 min.
As shown in
Figure 1a, the diffraction peaks of the composite powders are mainly composed of WC and Co-phases, and there are no diffraction peaks representing either V
8C
7 or Cr
3C
2. This is mainly because the amount of grain inhibitors added in this experiment was small (a mass percentage of 0.5%) and not within the detection range of the X-ray diffractometer (mass percentage >1%). When the sintering temperature is 1100 °C, the product is mainly composed of WC, and the diffraction peak intensity of the Co-phase is very weak (
Figure 1b). This indicates that a liquid phase has appeared in the specimen sintered at 1100 °C, and some WC begins to dissolve in the liquid Co, resulting in the reduction in Co content. Compared with traditional sintering methods [
21,
22], the liquid phase can appear at a lower sintering temperature. This is mainly because microwave sintering is rapid and relies on a unique heating mechanism. This method is a new sintering method, which uses the dielectric loss of materials to absorb microwave energy directly through the interaction between the microwave and material particles (molecules or ions). In addition, the microwave field can enhance the ionic conductivity. The high-frequency electric field can promote the migration of charged vacancies in the grain surface, thus resulting in plastic deformation, similar to diffusion creep, and promoting sintering [
23]. With the increase in sintering temperature, an η-phase W
3Co
3C occurs when the sintering temperature is 1200 °C (
Figure 1c). This is mainly because, with the increase in sintering temperature, a large amount of WC dissolves in the Co solution and a small amount of the W
3Co
3C phase dissolves and precipitates. When the sintering temperature is 1300 °C, the reaction products mainly consist of WC, and the W
3Co
3C phase disappears (
Figure 1d). Moreover, the diffraction peak representing WC has a higher intensity than that at 1200 °C, mainly due to the decomposition of the W
3Co
3C phase at this temperature, and the high solubility of nano-inhibitors V
8C
7 and Cr
3C
2 in Co solution. This hinders the dissolution of WC in Co, so that the diffraction peak of WC has a high intensity [
24]. When the sintering temperature is 1400 °C, the composition of the reaction product remains unchanged, but the diffraction peak of WC becomes sharper (
Figure 1e). This is mainly because, when the temperature is too high, the solubility of the nano-inhibitor in the Co liquid reaches saturation, and it is difficult to inhibit the dissolution and growth of the WC particles in the Co liquid. This results in the increase in WC grain size and the narrowing of its diffraction peak.
Figure 2 shows the XRD patterns of the specimens sintered at 1300 °C for different holding times. As shown in
Figure 2, the specimens are mainly composed of WC and contain a small amount of Co-phase. As shown in
Figure 2, the diffraction peaks of WC gradually shift to smaller angles with increased holding time. This is mainly due to the increase in the interplanar spacing and the decrease of the diffraction angles of WC with increased holding time [
25].
To observe the surface morphology and pore distribution of the sintered specimens, scanning electron microscopy (SEM) measurements were conducted on specimens prepared at different sintering temperatures and holding times (
Figure 3;
Figure 4).
As shown in
Figure 3a, when the sintering temperature is 1100 °C, there are many pores on the surface of the specimen (the residual porosity is 8.36%), most of which are 5–50 μm in size. With the increase in the sintering temperature, the number and size of the voids on the surface of the specimens decrease gradually. When the sintering temperature is 1300 °C, the specimen contains fewer pores and these are smaller (the residual porosity is 0.93%), as shown in
Figure 3c. This indicates that the grain size of WC can be effectively inhibited at the sintering temperature of 1300 °C, which is beneficial to the rearrangement and densification of the particles, thus reducing the number and size of the pores, and improving the mechanical properties of the specimen. It can be seen in
Figure 4 that the trend in the surface structure of the specimens under different holding time conditions is similar to that seen in
Figure 3. When the holding time is 60 min, the number of pores on the surface of the specimen is lower (the residual porosity is 0.18%), the pores are smaller, and the microstructure of the specimen is relatively uniform, as shown in
Figure 4c.
Figure 5 shows the SEM images of the composite powders and specimens sintered at different temperatures for 40 min. As shown in
Figure 5a, the particles are spherical or quasi-spherical, and the average particle size is about 200 nm; however, a small amount of agglomeration occurs after ball-milling. This is mainly due to the small grain size of the raw materials, their high specific surface area, and high activity. After high-energy ball-milling, a small amount of soft agglomeration readily occurs [
26]. As shown in
Figure 5b,c, the distribution of binder phase Co is not uniform at 1100 and 1200 °C. This shows that WC is dissolving and becoming rearranged in the Co-phase, which leads to the uneven distribution of the Co-phase. When the sintering temperature is 1300 °C, WC grains are polygonal or spherical, with an average size of about 500 nm, and these are uniformly distributed in the Co-phase, as shown in
Figure 5d. When the sintering temperature is 1400 °C, the WC grains increase in size, showing quadrilateral or polygonal forms, and the average grain size is about 1 μm (
Figure 5e). This is mainly due to the dissolution, growth, and re-precipitation of the WC particles with increasing temperature, resulting in the increase in grain size.
Figure 6 shows the backscatter electron (BSE) images of specimens sintered at 1300 °C for different holding times, and the EDS spectrum of the sintered specimens (60 min). The trend in
Figure 6 is similar to that in
Figure 5. When the holding time is 60 min, the structure of the specimen is more uniform, as shown in
Figure 6c. The WC grains are polygonal or quasi-spherical, and the grain size is 300–500 nm. The sintering temperature (1300 °C) is about 100 °C lower than that used in traditional sintering processes [
21,
22], mainly because microwave sintering can reduce the activation energy, accelerate diffusion, and increase the rate of densification [
27]. In addition, nanocomposite GGIs have a high solubility and mobility in liquid cobalt, which can prevent the dissolution and growth of the WC particles and further optimise the microstructure of the alloy [
28]. Another mechanism causing the aforementioned inhibition is that nanocomposite GGIs segregate to the WC-Co grain boundaries and their triple junctions, which hinders WC grain growth by exerting a pinning force (Zener-drag) on the moving grain boundaries [
13]. As shown in
Figure 6e, the main components of the selected area are C, W, and Co, and contain a small amount of V, Cr, and O. The reason for the presence of a small amount of O may be that nano-Co powders have high activity and are easily oxidised in air; this may lead to a small amount of Co being oxidised during mixing, moulding, and the like.
Figure 7 and
Table 1 show the relative density, Vickers hardness, and fracture toughness of ultrafine cemented carbides at different sintering temperatures (1100, 1200, 1300, and 1400 °C) for 40 min.
Figure 7 and
Table 1 show that the relative density, Vickers hardness, and fracture toughness of cemented carbide specimens first increase, then decrease with the increasing sintering temperature or prolonged holding time [
29]. When the sintering temperature is 1300 °C, the relative density, Vickers hardness, and fracture toughness of the specimen reach the maximum values of 98.38%, 1759 kg/mm
2, and 12.2 MPa·m
1/2, respectively. The hardness and fracture toughness of the WC-V
8C
7-Cr
3C
2-Co ultrafine cemented carbides sintered at 1400 °C decrease (albeit slightly). The main reason for this is that, when the sintering temperature reaches 1400 °C, an excess amount of liquid phase is produced, too much WC dissolution and precipitation occurs, and WC grains grow abnormally, resulting in the decrease in hardness and fracture toughness of the specimen. In addition, the relative density of the WC-V
8C
7-Cr
3C
2-Co ultrafine cemented carbide sintered at 1400 °C is decreased. This is mainly attributed to the over-burning of the specimen caused by too high a sintering temperature, resulting in swelling and the reduction in the density of the specimen.
Figure 8 and
Table 2 show the relative density, Vickers hardness, and fracture toughness of ultrafine cemented carbides sintered at 1300 °C for different holding times (20, 40, 60, and 80 min)—the trend is similar to the effect of sintering temperature on the relative density, Vickers hardness, and fracture toughness of the specimens (
Figure 7 and
Table 1). When the holding time is 60 min, the relative density, Vickers hardness, and fracture toughness of the specimens reach the maximum values of 99.79%, 1842 kg/mm
2, and 12.6 MPa·m
1/2, respectively. This is mainly because the power of the microwave field between the particles is almost 30 times than that of the external field, which leads to the enhancement of surface ionisation. Consequently, the rate of ionic diffusion and ion kinetic energy increases across the entire area, particularly at grain boundaries. This results in the formation of more uniformly distributed grain sizes and a denser body [
17,
30]. When the holding time is prolonged to 80 min, the relative density, Vickers hardness, and fracture toughness of the specimen begin to decrease, which is similar to the decrease in the properties of the specimen caused by the increase in sintering temperature (i.e., to 1400 °C).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}