Green Synthesis, SC-XRD, Non-Covalent Interactive Potential and Electronic Communication via DFT Exploration of Pyridine-Based Hydrazone

 ,
,  , ,
, , 
Abstract
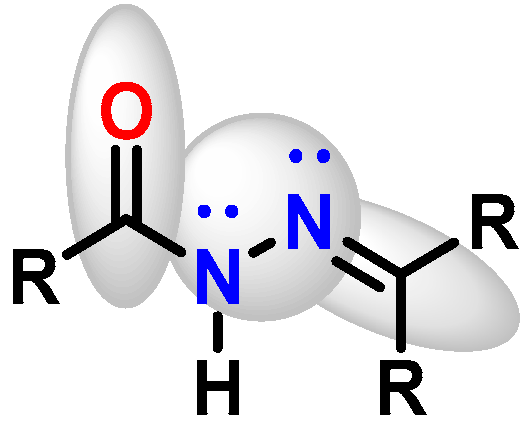
:1. Introduction
2. Materials and Methods
2.1. General
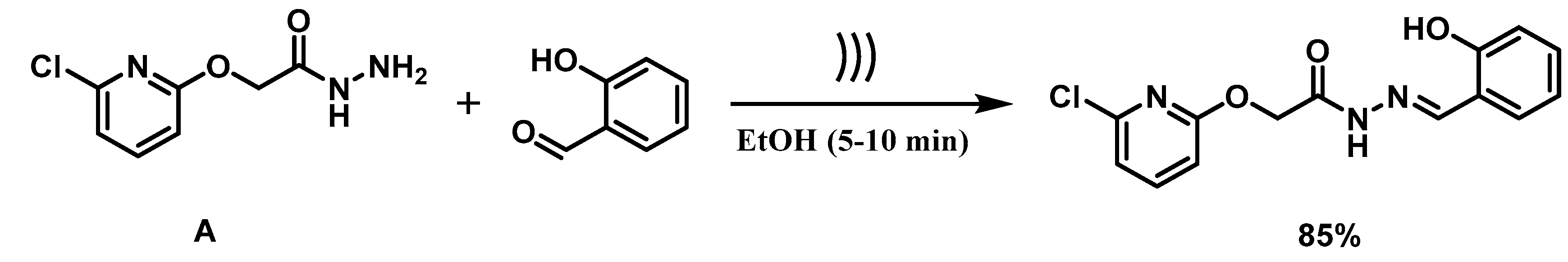
2.2. Synthesis of 2-(6′-Chloroazin-2′-yl) oxy-aceto-hydrazide (A)
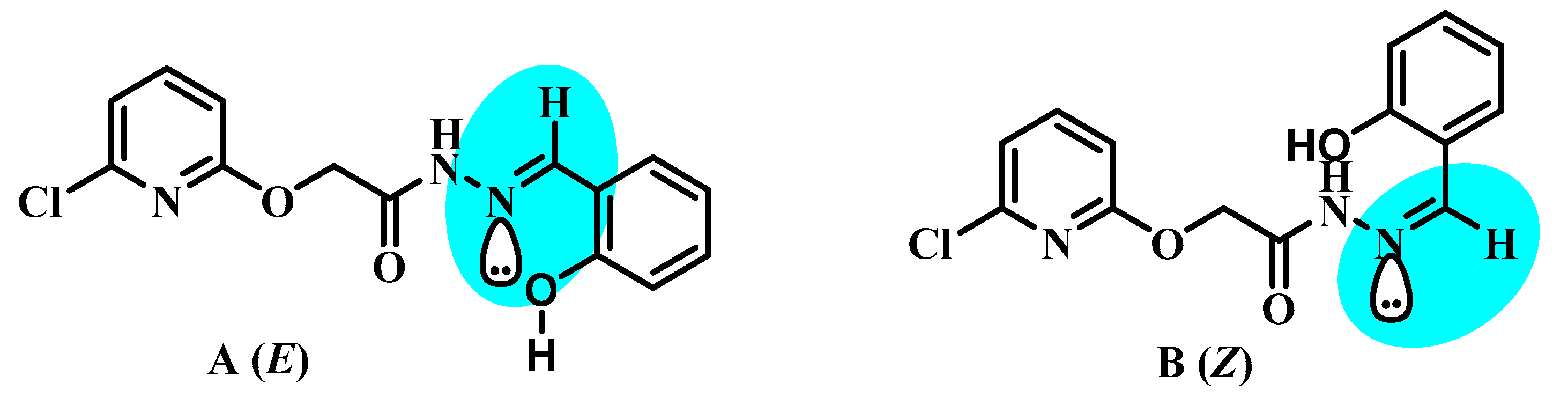
2.3. General Procedure for the Synthesis of (E)-2-((6-chloropyridin-2-yl)oxy)-N′-(2-hydroxybenzylidene)acetohydrazide
2.4. Computational Studies
3. Results and Discussion
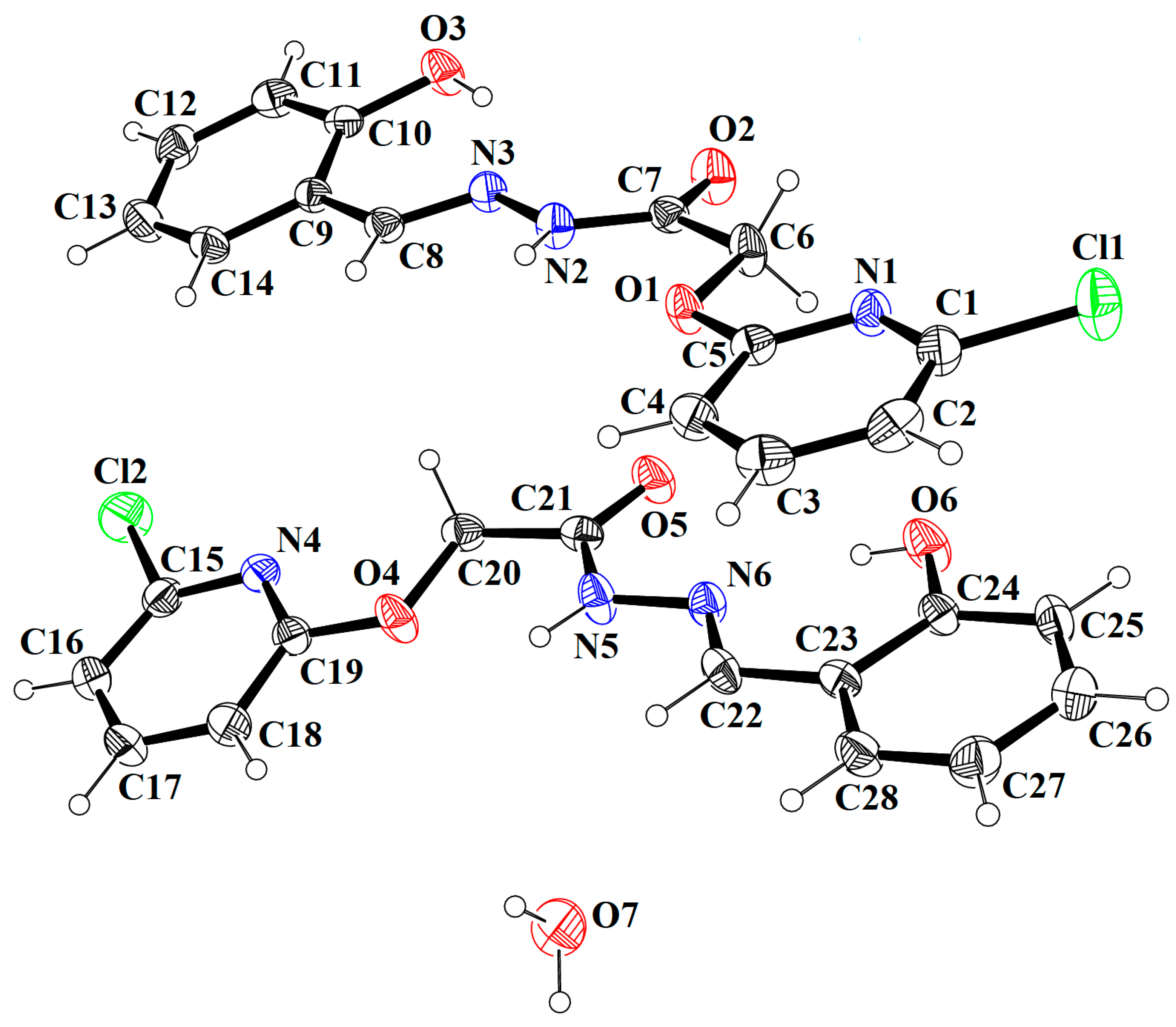
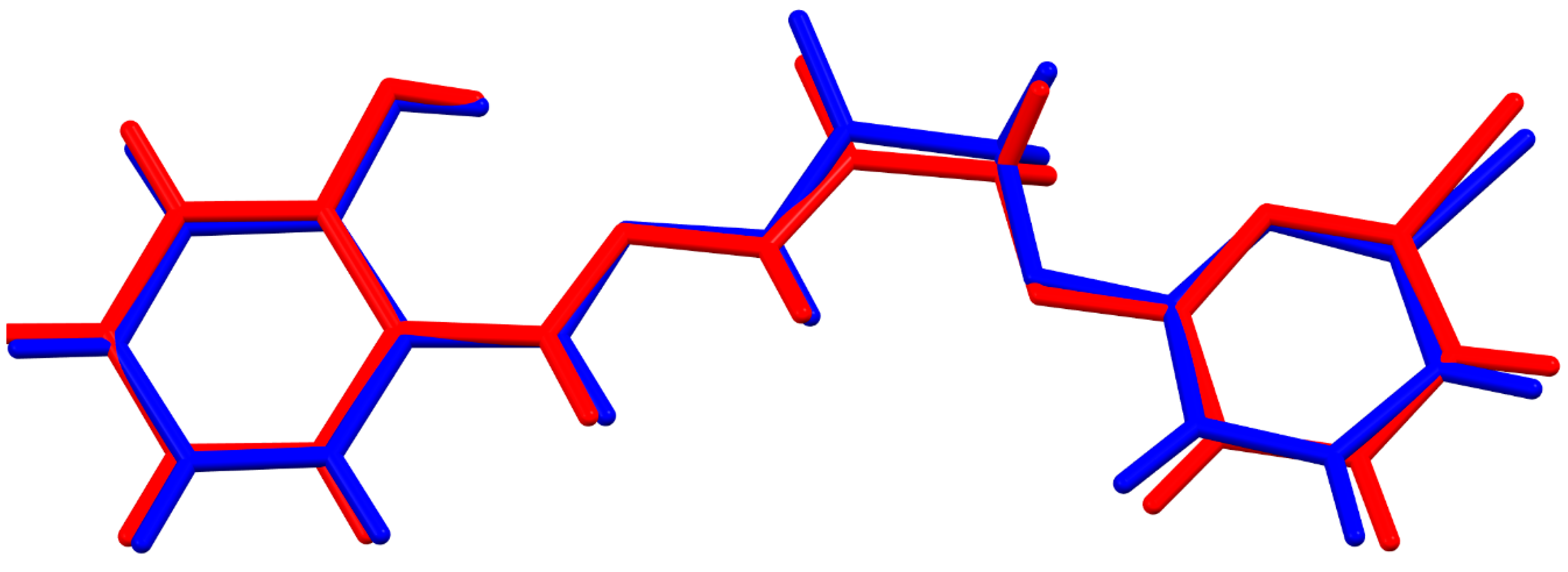
3.1. Comparative Structural Study
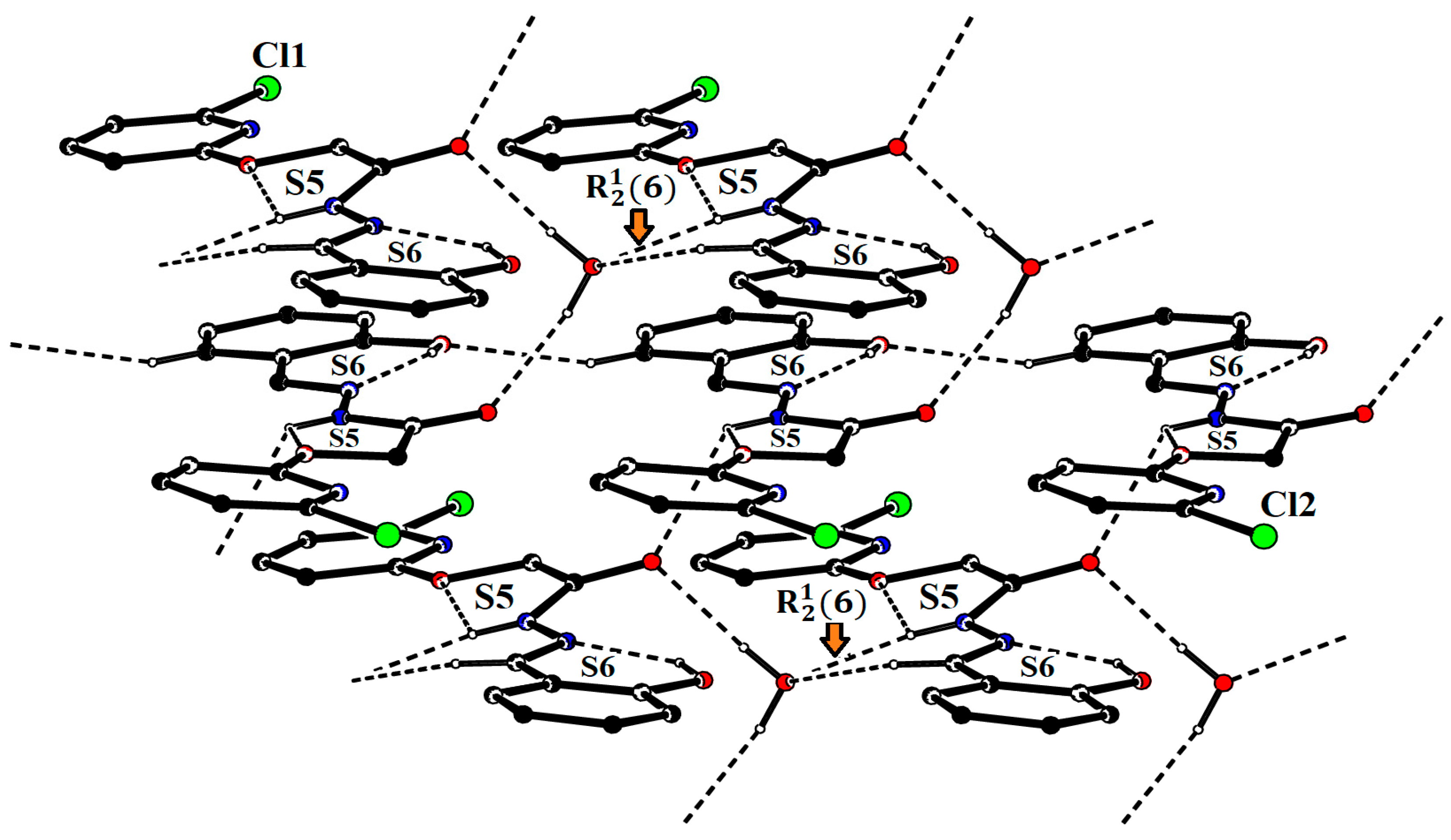
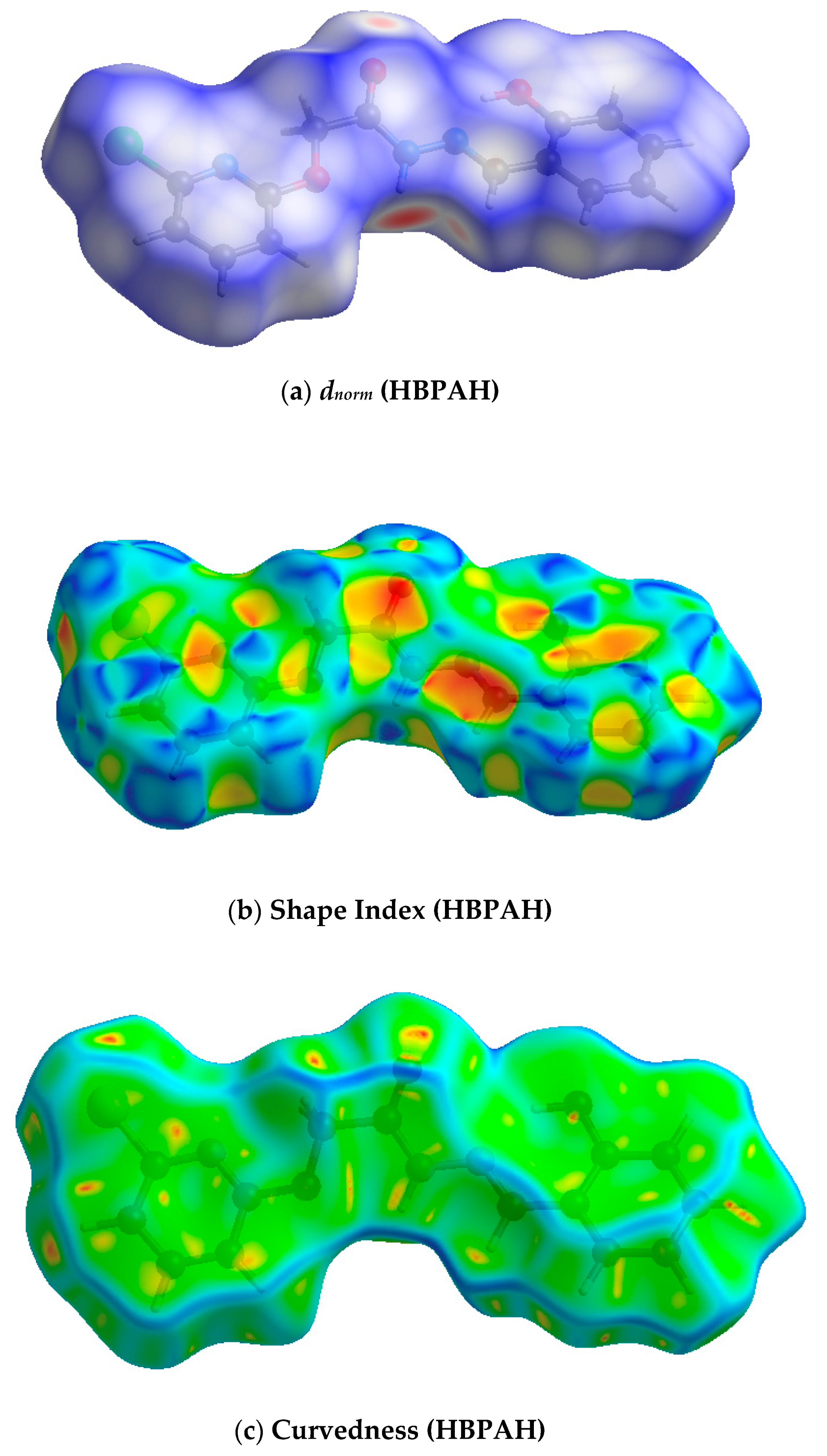
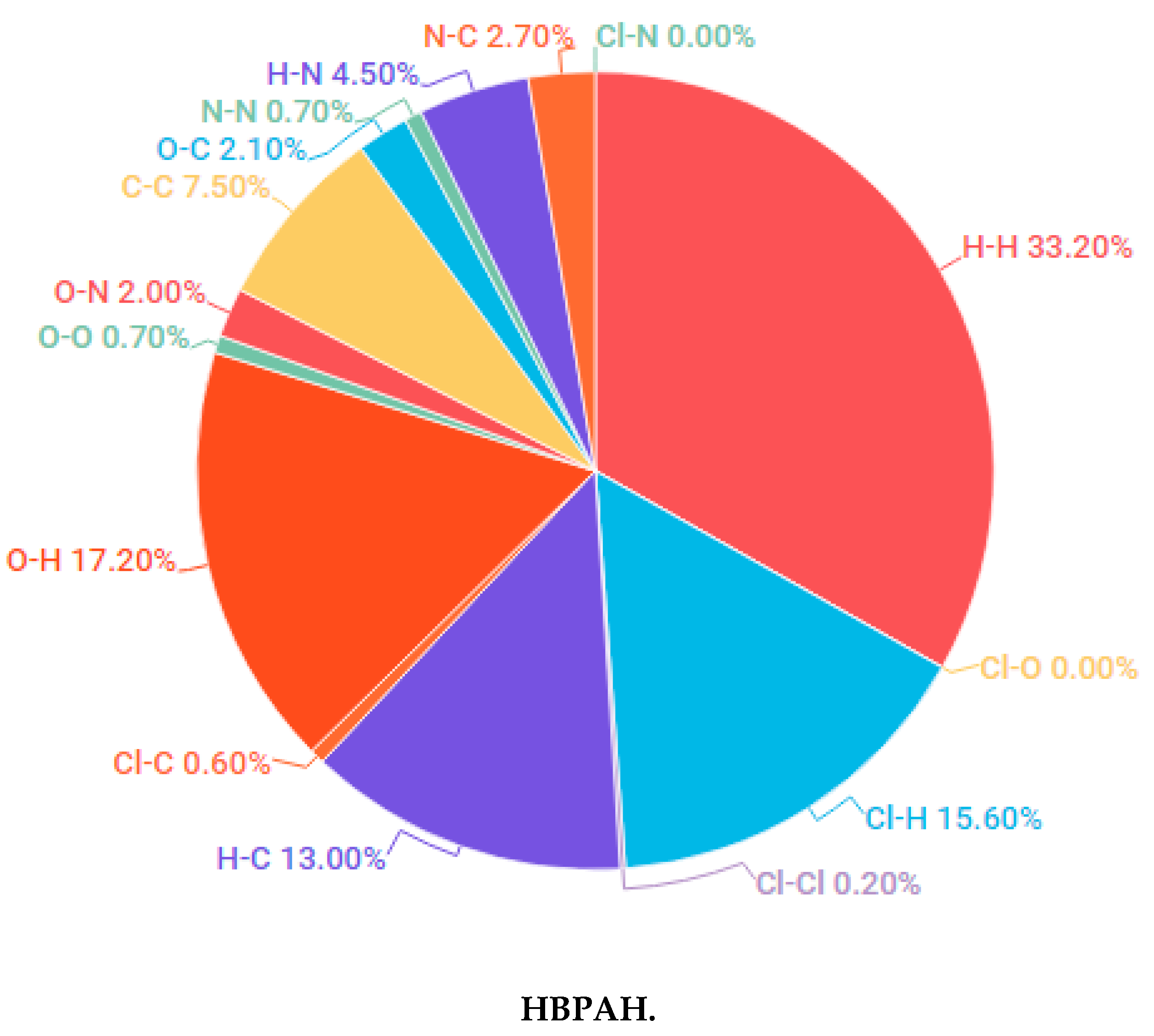
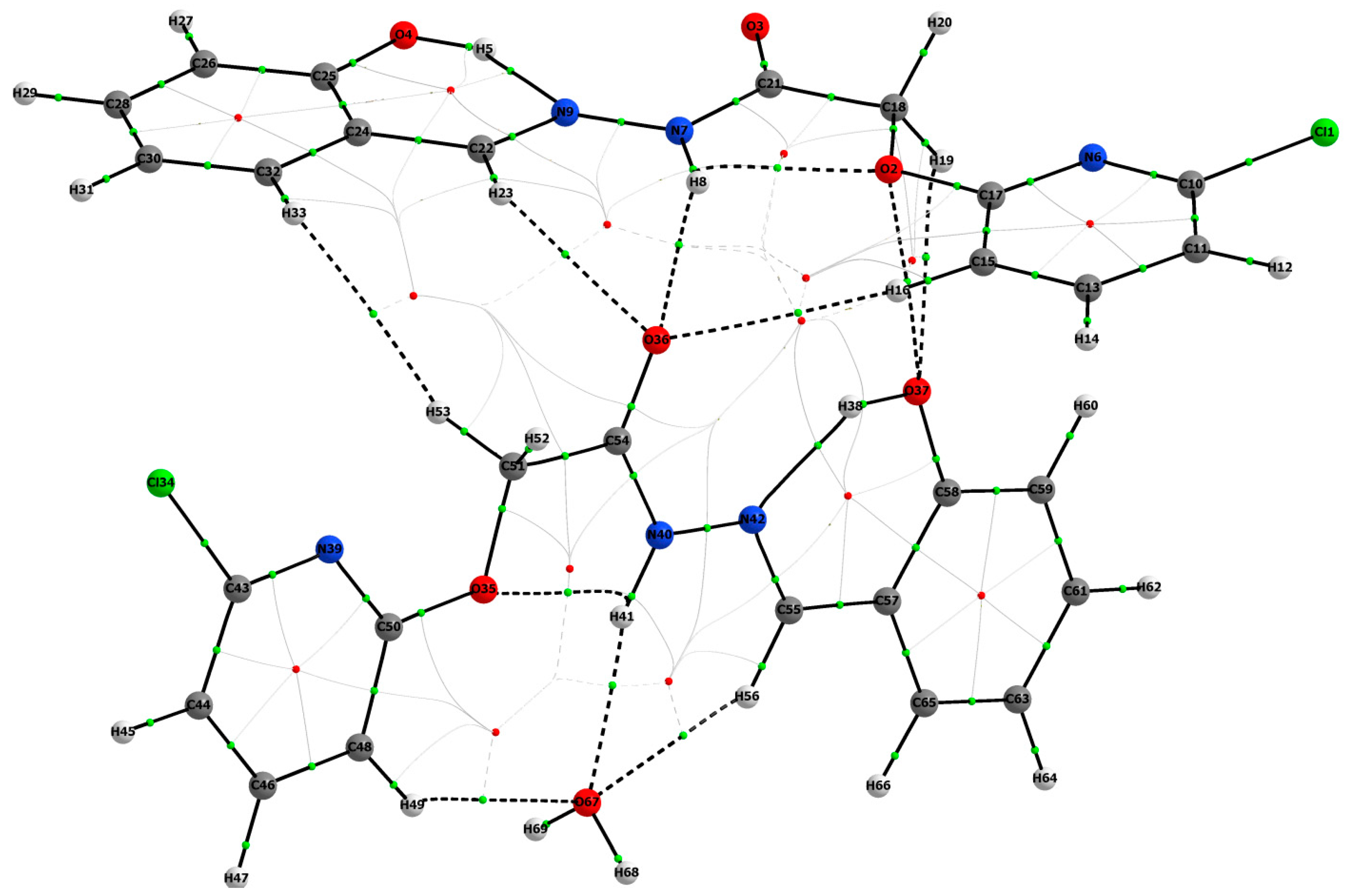
3.1.1. Hirshfeld Surface Analysis
3.1.2. QT-AIM Analysis
3.1.3. Natural Bonding Orbital (NBO) Analysis
3.1.4. Natural Population Analysis (NPA)
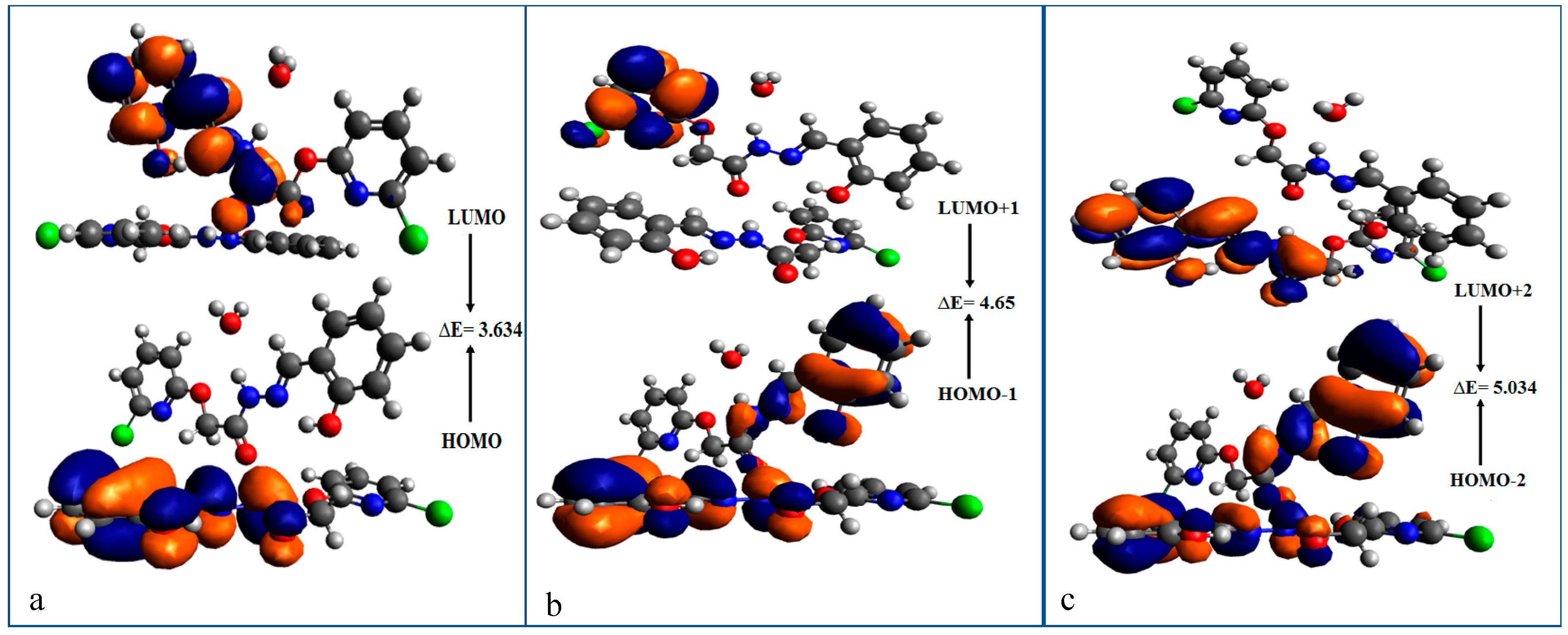
3.1.5. Frontier Molecular Orbital (FMO) Analysis
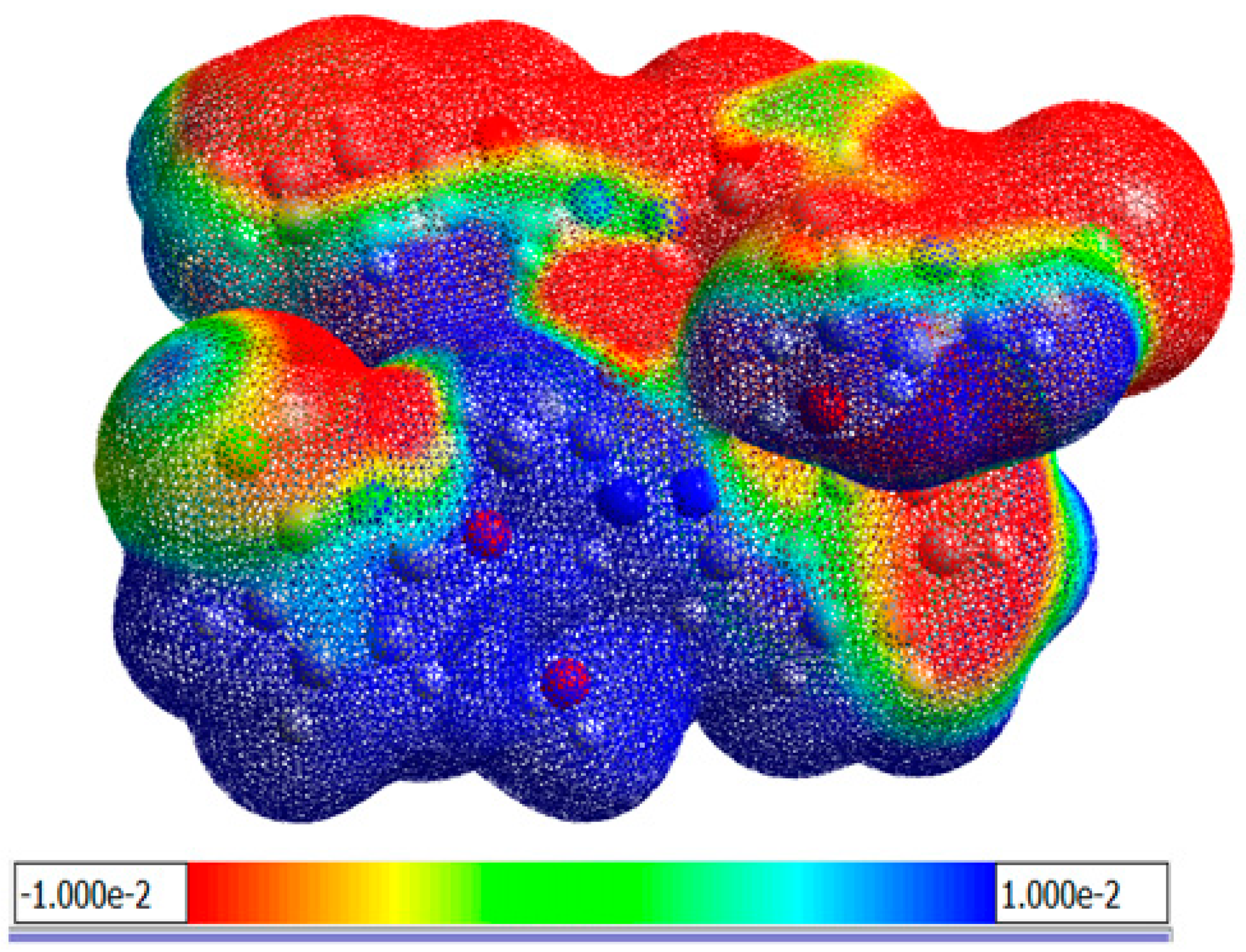
3.1.6. Molecular Electrostatic Potential (MEP)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rollas, S.; Küçükgüzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, G.; Marella, A.; Shaquiquzzaman, M.; Akhtar, M.; Ali, M.R.; Alam, M.M. A review exploring biological activities of hydrazones. J. Pharm. Bioallied Sci. 2014, 6, 69. [Google Scholar] [PubMed]
- Hussain, I.; Ali, A. Exploring the Pharmacological Activities of Hydrazone Derivatives: A Review. J. Phytochem. Biochem. 2017, 1, 1–5. [Google Scholar]
- Su, X.; Aprahamian, I. Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43, 1963–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, R.A.; Telvekar, V.N. Synthesis and evaluation of novel chloropyrrole molecules designed by molecular hybridization of common pharmacophores as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2010, 20, 5681–5685. [Google Scholar] [CrossRef] [PubMed]
- El-Sabbagh, O.; Shabaan, M.A.; Kadry, H.H.; Al-Din, E.S. New octahydroquinazoline derivatives: Synthesis and hypotensive activity. Eur. J. Med. Chem. 2010, 45, 5390–5396. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.D.; Yang, S.Q.; Gu, S.X.; He, Q.Q.; Chen, F.E.; De Clercq, E.; Balzarini, J.; Pannecouque, C. Synthesis and anti-HIV activity of Aryl-2-[(4-cyanophenyl) amino]-4-pyrimidinone hydrazones as potent non-nucleoside reverse transcriptase inhibitors. ChemMedChem 2011, 6, 2225–2232. [Google Scholar] [CrossRef]
- Tributino, J.L.; Duarte, C.D.; Corrêa, R.S.; Doriguetto, A.C.; Ellena, J.; Romeiro, N.C.; Castro, N.G.; Miranda, A.L.P.; Barreiro, E.J.; Fraga, C.A. Novel 6-methanesulfonamide-3, 4-methylenedioxyphenyl-N-acylhydrazones: Orally effective anti-inflammatory drug candidates. Bioorg. Med. Chem. 2009, 17, 1125–1131. [Google Scholar] [CrossRef]
- Liu, W.-Y.; Li, H.-Y.; Zhao, B.-X.; Shin, D.-S.; Lian, S.; Miao, J.-Y. Synthesis of novel ribavirin hydrazone derivatives and anti-proliferative activity against A549 lung cancer cells. Carbohydr. Res. 2009, 344, 1270–1275. [Google Scholar] [CrossRef]
- Gil-Longo, J.; Laguna, M.D.L.R.; Verde, I.; Castro, M.E.; Orallo, F.; Fontenla, J.A.; Calleja, J.M.; Ravina, E.; Teran, C. Pyridazine derivatives. XI: Antihypertensive activity of 3-hydrazinocycloheptyl [1, 2-c] pyridazine and its hydrazone derivatives. J. Pharm. Sci. 1993, 82, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, A.; Kremer, L.; Louw, S.; Guéradel, Y.; Chibale, K.; Biot, C. Synthesis and in vitro antitubercular activity of ferrocene-based hydrazones. Bioorg. Med. Chem. Lett. 2011, 21, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- Fattorusso, C.; Campiani, G.; Kukreja, G.; Persico, M.; Butini, S.; Romano, M.P.; Altarelli, M.; Ros, S.; Brindisi, M.; Savini, L. Design, synthesis, and structure–activity relationship studies of 4-quinolinyl-and 9-acrydinylhydrazones as potent antimalarial agents. J. Med. Chem. 2008, 51, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, K.N.; Costa, P.; Santin, J.R.; Mazzambani, L.; Bürger, C.; Mora, C.; Nunes, R.J.; De Souza, M.M. Synthesis and antidepressant-like activity evaluation of sulphonamides and sulphonyl-hydrazones. Bioorg. Med. Chem. 2011, 19, 4295–4306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musad, E.A.; Mohamed, R.; Saeed, B.A.; Vishwanath, B.S.; Rai, K.L. Synthesis and evaluation of antioxidant and antibacterial activities of new substituted bis (1, 3, 4-oxadiazoles), 3, 5-bis (substituted) pyrazoles and isoxazoles. Bioorg. Med. Chem. Lett. 2011, 21, 3536–3540. [Google Scholar] [CrossRef]
- Jain, J.; Kumar, Y.; Sinha, R.; Kumar, R.; Stables, J. Menthone aryl acid hydrazones: A new class of anticonvulsants. Med. Chem. 2011, 7, 56–61. [Google Scholar] [CrossRef]
- Mu, J.-X.; Shi, Y.-X.; Wu, H.-K.; Sun, Z.-H.; Yang, M.-Y.; Liu, X.-H.; Li, B.-J. Microwave assisted synthesis, antifungal activity, DFT and SAR study of 1,2,4-triazolo[4,3-a]pyridine derivatives containing hydrazone moieties. Chem. Cent. J. 2016, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, A.; Turan-Zitouni, G.; Asim kaplancikli, Z.; Demirci, F.; Iscan, G. Studies on hydrazone derivatives as antifungal agents. J. Enzym. Inhib. Med. Chem. 2008, 23, 470–475. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Watheyb, B.; Westmana, J. Microwave assisted organic synthesis: A review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Easmon, J.; Pürstinger, G.; Thies, K.-S.; Heinisch, G.; Hofmann, J. Synthesis, structure−activity relationships, and antitumor studies of 2-benzoxazolyl hydrazones derived from alpha-(N)-acyl heteroaromatics. J. Med. Chem. 2006, 49, 6343–6350. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chowdhury, S.; Paul, A.K.; Banerjee, R. Selective extraction of palladium (II) using hydrazone ligand: A novel fluorescent sensor. J. Lumin. 2011, 131, 2342–2346. [Google Scholar] [CrossRef]
- Chaouiki, A.; Chafiq, M.; Lgaz, H.; Al-Hadeethi, M.R.; Ali, I.H.; Masroor, S.; Chung, I.-M. Green Corrosion Inhibition of Mild Steel by Hydrazone Derivatives in 1.0 M HCl. Coatings 2020, 10, 640. [Google Scholar] [CrossRef]
- Chaston, T.B.; Richardson, D.R. Interactions of the pyridine-2-carboxaldehyde isonicotinoyl hydrazone class of chelators with iron and DNA: Implications for toxicity in the treatment of iron overload disease. JBIC J. Biol. Inorg. Chem. 2003, 8, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The whole palette of hydrogen bonds. Angew. Chem. Int. Ed. 2002, 41, 14–76. [Google Scholar]
- Gerlt, J.A.; Kreevoy, M.M.; Cleland, W.; Frey, P.A. Understanding enzymic catalysis: The importance of short, strong hydrogen bonds. Chem. Biol. 1997, 4, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Perrin, C.L.; Nielson, J.B. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Zundel, G. Hydrogen bonds with large proton polarizability and proton transfer processes in electrochemistry and biology. Adv. Chem. Phys. 1999, 111, 1–217. [Google Scholar] [CrossRef]
- Bernstein, J.; Etter, M.C.; Leiserowitz, L. The role of hydrogen bonding in molecular assemblies. Struct. Correl. 1994, 431–507. [Google Scholar] [CrossRef]
- Pokorna, P.; Krepl, M.; Kruse, H.; Sponer, J. MD and QM/MM Study of the Quaternary HutP Homohexamer Complex with mRNA, L-Histidine Ligand, and Mg2+. J. Chem. Theory Comput. 2017, 13, 5658–5670. [Google Scholar] [CrossRef]
- Zhang, Z.; Schreiner, P.R. (Thio) urea organocatalysis—What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef]
- Pihko, P.M. Activation of carbonyl compounds by double hydrogen bonding: An emerging tool in asymmetric catalysis. Angew. Chem. Int. Ed. 2004, 43, 2062–2064. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Khalid, M.; Abid, S.; Iqbal, J.; Tahir, M.N.; Rauf Raza, A.; Zukerman-Schpector, J.; Paixão, M.W. Facile synthesis, crystal growth, characterization and computational study of new pyridine-based halogenated hydrazones: Unveiling the stabilization behavior in terms of noncovalent interactions. Appl. Organomet. Chem. 2020, 34, e5399. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, M.; Hu, B.; Sun, Z.; Liu, X.; Weng, J.; Tan, C. Microwave-assisted synthesis of novel 8-chloro-[1, 2, 4] triazolo [4, 3- alpha ] pyridine derivatives. Turk. J. Chem. 2015, 39, 867–873. [Google Scholar] [CrossRef]
- De la Hoz, A.; Diaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Amariucai-Mantu, D.; Mangalagiu, V.; Danac, R.; Mangalagiu, I.I. Microwave Assisted Reactions of Azaheterocycles Formedicinal Chemistry Applications. Molecules 2020, 25, 716. [Google Scholar] [CrossRef] [Green Version]
- APEX2 (Version 1.22) and SAINT-Plus (Version 7.06a); Bruker: Billica, MA, USA, 2009.
- Higashino, T.; Akiyama, Y.; Kojima, H.; Kawamoto, T.; Mori, T. Organic semiconductors and conductors with tert-butyl substituents. Crystals 2012, 2, 1222–1238. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Delgado, G.; Henao, J.; Quintana, J.; Al-Maqtari, H.; Jamalis, J.; Sirat, H. Structural Characterization of a New Chalcone Compound Containing a Thiophene Moiety:(E)-3-(5-Bromothiophen-2-YL)-1-(2, 5-Dichlorothiophen-3-YL)-2-Propen-1-One. J. Struct. Chem. 2018, 59, 1440–1445. [Google Scholar] [CrossRef]
- Thomassen, I.K.; McCormick, L.J.; Ghosh, A. Molecular Structure of a Free-Base β-Octaiodo-meso-tetraarylporphyrin. A Rational Route to cis Porphyrin Tautomers? Cryst. Growth Des. 2018, 18, 4257–4259. [Google Scholar] [CrossRef]
- Ali, A.; Badawy, M.; Shah, R.; Rehman, W.; El kilany, Y.; El Ashry, E.S.H.; Tahir, N. Synthesis, characterization and in-silico admet screening of mono-and dicarbomethoxylated 6, 6′-methylenebis (2-cyclohexyl-4-methylphenol) and their hydrazides and hydrazones. Chim. Sin. 2017, 8, 446–460. [Google Scholar]
- Braga, A.A.C.; Ujaque, G.; Maseras, F. A DFT Study of the Full Catalytic Cycle of the Suzuki−Miyaura Cross-Coupling on a Model System. Organometallics 2006, 25, 3647–3658. [Google Scholar] [CrossRef]
- García-Melchor, M.; Braga, A.A.C.; Lledós, A.; Ujaque, G.; Maseras, F. Computational Perspective on Pd-Catalyzed C–C Cross-Coupling Reaction Mechanisms. Acc. Chem. Res. 2013, 46, 2626–2634. [Google Scholar] [CrossRef]
- Braga, A.A.; Morgon, N.H.; Ujaque, G.; Maseras, F. Computational characterization of the role of the base in the Suzuki−Miyaura cross-coupling reaction. J. Am. Chem. Soc. 2005, 127, 9298–9307. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. D. 0109, Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Chemcraft. Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 31 March 2020).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, TK Gristmill Software; AIMAll: Overland Park, KS, USA, 2012. [Google Scholar]
- Wolff, S.; Grimwood, D.; McKinnon, J.; Turner, M.; Jayatilaka, D.; Spackman, M. CrystalExplorer; Version 3.1; University of Western Australia: Crawley, Australia, 2012. [Google Scholar]
- Glendenning, E.; Reed, A.; Carpenter, J.; Weinhold, F. NBO; Version 3.1; 2001; Available online: http://www.ccl.net/cca/software/MS-WIN95-NT/mopac6/nbo.htm (accessed on 31 August 2020).
- Weinhold, F.; Glendening, E.D. NBO 5.0 Program Manual; Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin: Madison, WI, USA, 2001; Volume 53706, p. 101. [Google Scholar]
- Gross, E.K.U.; Kohn, W. Time-dependent density-functional theory. In Advances in Quantum Chemistry; Löwdin, P.-O., Ed.; Academic Press: Cambridge, MA, USA, 1990; Volume 21, pp. 255–291. [Google Scholar]
- Burke, K.; Werschnik, J.; Gross, E. Time-dependent density functional theory: Past, present, and future. J. Chem. Phys. 2005, 123, 062206. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s theory of atoms in molecules (AIM) and its applications to chemical bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity Index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity Index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Saravanan, S.; Balachandran, V. Quantum chemical studies, natural bond orbital analysis and thermodynamic function of 2, 5-dichlorophenylisocyanate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 120, 351–364. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Sheela, N.; Muthu, S.; Sampathkrishnan, S. Molecular orbital studies (hardness, chemical potential and electrophilicity), vibrational investigation and theoretical NBO analysis of 4-4′-(1H-1, 2, 4-triazol-1-yl methylene) dibenzonitrile based on abinitio and DFT methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 120, 237–251. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Padmanabhan, J.; Elango, M.; Subramanian, V.; Chattaraj, P. Intermolecular reactivity through the generalized philicity concept. Chem. Phys. Lett. 2004, 394, 225–230. [Google Scholar] [CrossRef]
- Politzer, P.; Truhlar, D.G. Introduction: The role of the electrostatic potential in chemistry. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Springer: Boston, MA, USA, 1981; pp. 1–6. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.v.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Roy, D.; Sarkar, U.; Chattaraj, P.; Mitra, A.; Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Van Damme, S.; Bultinck, P. Analyzing toxicity through electrophilicity. Mol. Divers. 2006, 10, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lesar, A.; Milošev, I. Density functional study of the corrosion inhibition properties of 1, 2, 4-triazole and its amino derivatives. Chem. Phys. Lett. 2009, 483, 198–203. [Google Scholar] [CrossRef]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Desiraju, G.R. The CH. cntdot. cntdot. cntdot. O hydrogen bond in crystals: What is it? Acc. Chem. Res. 1991, 24, 290–296. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Kishan, K.R. Crystal chemistry of some (alkoxyphenyl) propiolic acids. The role of oxygen and hydrogen atoms in determining stack structures of planar aromatic compounds. J. Am. Chem. Soc. 1989, 111, 4838–4843. [Google Scholar] [CrossRef]
- Çakmak, O.; Ökten, S.; Alımlı, D.; Ersanlı, C.C.; Taslimi, P.; Koçyiğit, Ü.M. Novel piperazine and morpholine substituted quinolines: Selective synthesis through activation of 3, 6, 8-tribromoquinoline, characterization and their some metabolic enzymes inhibition potentials. J. Mol. Struct. 2020, 128666. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Zukerman-Schpector, J.; Dallasta Pedroso, S.; Sousa Madureira, L.; Weber Paixão, M.; Ali, A.; Tiekink, E.R. 4-Benzyl-1-(4-nitrophenyl)-1H-1, 2, 3-triazole: Crystal structure and Hirshfeld analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2017, 73, 1716–1720. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Wang, H.; Yin, Z. Crystal structure and Hirshfeld surface analysis of dibutyl 5, 5′-(pentane-3, 3-diyl) bis (1H-pyrrole-5-carboxylate). Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 711. [Google Scholar] [CrossRef] [Green Version]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Pook, N.-P.; Adam, A.; Gjikaj, M. Crystal structure and Hirshfeld surface analysis of (μ-2-{4-[(carboxylatomethyl) carbamoyl] benzamido} acetato-κ2O: O′) bis [bis (1, 10-phenanthroline-κ2N, N′) copper (II)] dinitrate N, N′-(1, 4-phenylenedicarbonyl) diglycine monosolvate octahydrate. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 667–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangeetha, K.; Rajina, S.; Marchewka, M.; Binoy, J. The study of inter and intramolecular hydrogen bonds of NLO crystal melaminium hydrogen malonate using DFT simulation, AIM analysis and Hirshfeld surface analysis. Mater. Today Proc. 2020, 25, 307–315. [Google Scholar] [CrossRef]
- Toronto, S.T. Artificial Intelligence for Engineering Design, Analysis and Manufacturing; AIE, Cambridge University Press: Cambridge, UK, 1989; Volume 3, pp. B1–B8. [Google Scholar] [CrossRef] [Green Version]
- Koenderink, J.J.; Van Doorn, A.J. The singularities of the visual mapping. Biol. Cybern. 1976, 24, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Zukerman-Schpector, J.; Weber Paixão, M.; Jotani, M.M.; Tiekink, E.R. 7-Methyl-5-[(4-methylbenzene) sulfonyl]-2H, 5H-[1, 3] dioxolo [4, 5-f] indole: Crystal structure and Hirshfeld analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2018, 74, 184–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebbar, N.K.; Hni, B.; Hökelek, T.; Jaouhar, A.; Labd Taha, M.; Mague, J.T.; Essassi, E.M. Crystal structure, Hirshfeld surface analysis and interaction energy and DFT studies of 3-{(2Z)-2-[(2, 4-dichlorophenyl) methylidene]-3-oxo-3, 4-dihydro-2H-1, 4-benzothiazin-4-yl} propanenitrile. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Tahir, M.N.; Ashfaq, M.; Alexander, F.; Caballero, J.; Hernández-Rodríguez, E.W.; Ali, A. Rationalizing the stability and interactions of 2, 4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium 2-hydroxy-3, 5-dinitrobenzoate salt. J. Mol. Struct. 2019, 1193, 185–194. [Google Scholar] [CrossRef]
- Khalid, M.; Ali, A.; Rehman, M.F.U.; Mustaqeem, M.; Ali, S.; Khan, M.U.; Asim, S.; Ahmad, N.; Saleem, M. Exploration of Noncovalent Interactions, Chemical Reactivity, and Nonlinear Optical Properties of Piperidone Derivatives: A Concise Theoretical Approach. ACS Omega 2020, 5, 13236–13249. [Google Scholar] [CrossRef]
- Thakur, T.S.; Dubey, R.; Desiraju, G.R. Intermolecular atom–atom bonds in crystals–a chemical perspective. IUCrJ 2015, 2, 159–160. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.; Nguyen-Dang, T. Quantum theory of atoms in molecules–Dalton revisited. In Advances in Quantum Chemistry; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 1981; Volume 14, pp. 63–124. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: A brief overview. J. Chem. Sci. 2010, 122, 667–675. [Google Scholar] [CrossRef]
- Hernández-Paredes, J.; Carrillo-Torres, R.C.; López-Zavala, A.A.; Sotelo-Mundo, R.R.; Hernández-Negrete, O.; Ramírez, J.Z.; Alvarez-Ramos, M.E. Molecular structure, hydrogen-bonding patterns and topological analysis (QTAIM and NCI) of 5-methoxy-2-nitroaniline and 5-methoxy-2-nitroaniline with 2-amino-5-nitropyridine (1: 1) co-crystal. J. Mol. Struct. 2016, 1119, 505–516. [Google Scholar] [CrossRef]
- Tamer, Ö.; Avcı, D.; Atalay, Y. Quantum chemical characterization of N-(2-hydroxybenzylidene) acetohydrazide (HBAH): A detailed vibrational and NLO analysis. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 117, 78–86. [Google Scholar] [CrossRef]
- Szafran, M.; Komasa, A.; Bartoszak-Adamska, E. Crystal and molecular structure of 4-carboxypiperidinium chloride (4-piperidinecarboxylic acid hydrochloride). J. Mol. Struct. 2007, 827, 101–107. [Google Scholar] [CrossRef]
- James, C.; Raj, A.A.; Reghunathan, R.; Jayakumar, V.; Joe, I.H. Structural conformation and vibrational spectroscopic studies of 2, 6-bis (p-N, N-dimethyl benzylidene) cyclohexanone using density functional theory. J. Raman Spectrosc. Int. J. Orig. Work All Asp. Raman Spectrosc. Incl. High. Order Process. Also Brillouin Rayleigh Scatt. 2006, 37, 1381–1392. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1960; Volume 260. [Google Scholar]
- Mulliken, R. Overlap populations, bond orders and covalent bond energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Javed, I.; Khurshid, A.; Arshad, M.N.; Wang, Y. Photophysical and electrochemical properties and temperature dependent geometrical isomerism in alkyl quinacridonediimines. New J. Chem. 2014, 38, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Khalid, M.; Ali, A.; Jawaria, R.; Asghar, M.A.; Asim, S.; Khan, M.U.; Hussain, R.; ur Rehman, M.F.; Ennis, C.J.; Akram, M.S. First principles study of electronic and nonlinear optical properties of A–D–π–A and D–A–D–π–A configured compounds containing novel quinoline–carbazole derivatives. RSC Adv. 2020, 10, 22273–22283. [Google Scholar] [CrossRef]
- Ali, A.; Khalid, M.; Rehman, M.F.u.; Haq, S.; Ali, A.; Tahir, M.N.; Ashfaq, M.; Rasool, F.; Braga, A.A.C. Efficient Synthesis, SC-XRD, and Theoretical Studies of O-Benzenesulfonylated Pyrimidines: Role of Noncovalent Interaction Influence in Their Supramolecular Network. ACS Omega 2020, 5, 15115–15128. [Google Scholar] [CrossRef]
- Luque, F.J.; López, J.M.; Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | HBPAH |
|---|---|
| CCDC* number | 2012169 |
| Chemical formula | 2(C14H12ClN3O3)·H2O |
| Mr | 629.45 |
| Crystal system, space group | Triclinic, P |
| Temperature (K) | 296 |
| a, b, c (Å) | 6.6987 (8), 7.3628 (9), 31.513 (4) |
| α, β, γ (°) | 90.978 (7), 93.508 (6), 113.954 (7) |
| V (Å3) | 1416.2 (3) |
| Z | 2 |
| Density (calculated) | 1.476 Mg/m3 |
| F(000) | 652 |
| Radiation type | Mo Kα |
| Wavelength (λ) | 0.71073 Å |
| µ (mm−1) | 0.288 |
| Crystal shape | Needle |
| Crystal Color | Colorless |
| Crystal size (mm) | 0.38 × 0.22 × 0.18 |
| Data Collection | HBPAH |
| Diffractometer | Bruker APEXII CCD diffractometer |
| Absorption correction | multi-scan (SADABS; Bruker, 2007) |
| No. of measured, independent and observed [I> 2s(I)] reflections | 15,051, 5452, 2920 |
| Rint | 0.070 |
| Theta range for data collection | 0.648 to 26.000° |
| Index ranges | −8 ≤ h ≤ 7, -9 ≤ k ≤ 9, −37 ≤ l ≤ 38 |
| (sin θ/λ)max (Å−1) | 0.617 |
| Refinement | HBPAH |
| R[F2 > 2σ(F2)], wR(F2), S | 0.083, 0.195, 1.05 |
| No. of reflections | 5452 |
| No. of parameters | 396 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.24, −0.30 |
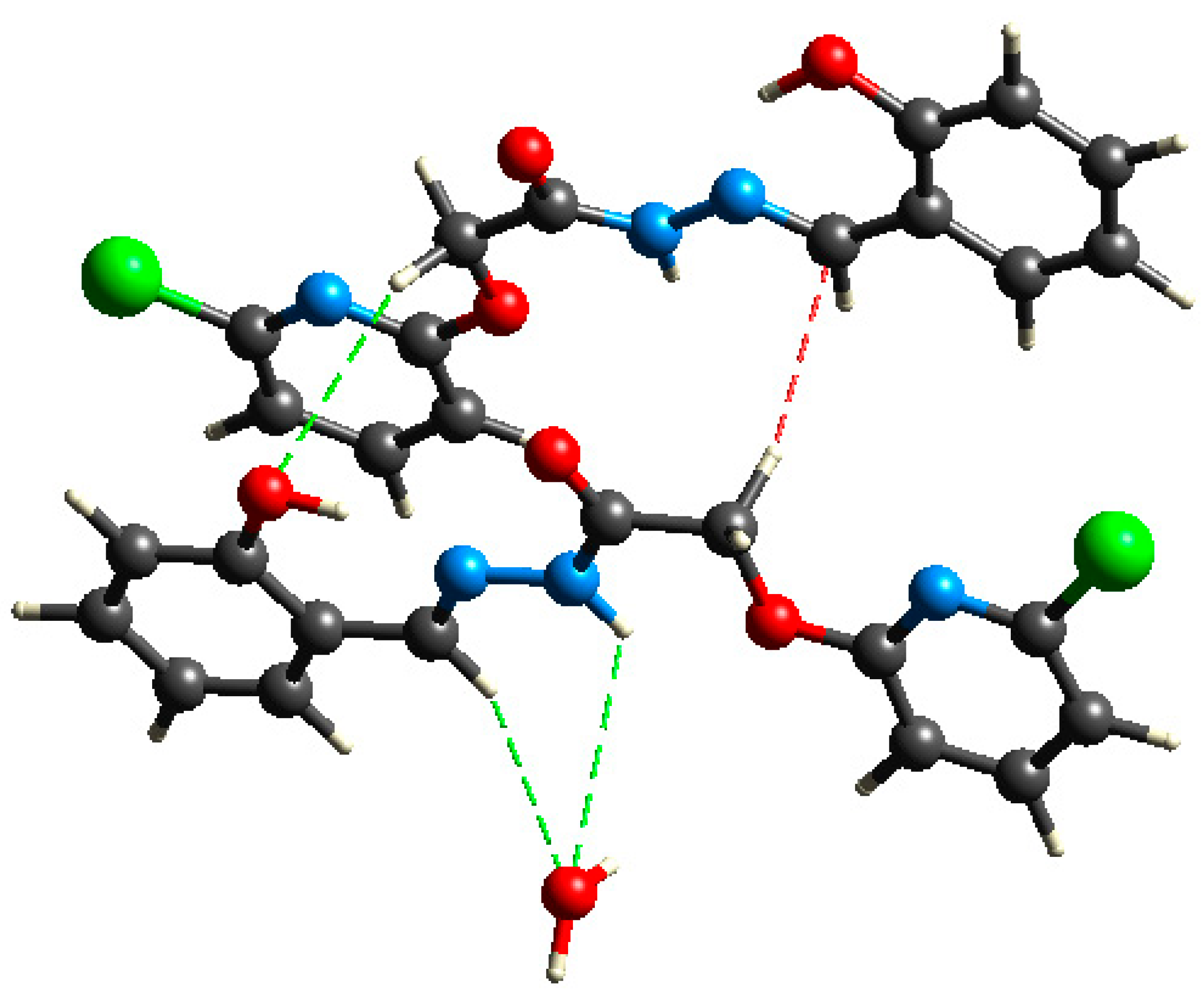
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|
| O3–H3A⋯N3 | 0.82 | 1.95 | 2.659 (5) | 145 |
| N2–H2A⋯O1 | 0.86 | 2.235 | 2.655 | 105.34 |
| N2–H2A⋯O7 | 0.86 | 2.08 | 2.902 (5) | 160 |
| O6–H6⋯N6 | 0.82 | 1.88 | 2.587 (5) | 145 |
| N5–H5⋯O4 | 0.86 | 2.170 | 2.571 | 108.18 |
| N5–H5⋯O2i | 0.86 | 2.48 | 3.029 (5) | 122 |
| O7–H7A⋯O5ii | 0.91 (6) | 2.03 (6) | 2.877 (5) | 155 (6) |
| O7–H7B⋯O2ii | 0.89 (6) | 1.95 (7) | 2.826 (6) | 168 (6) |
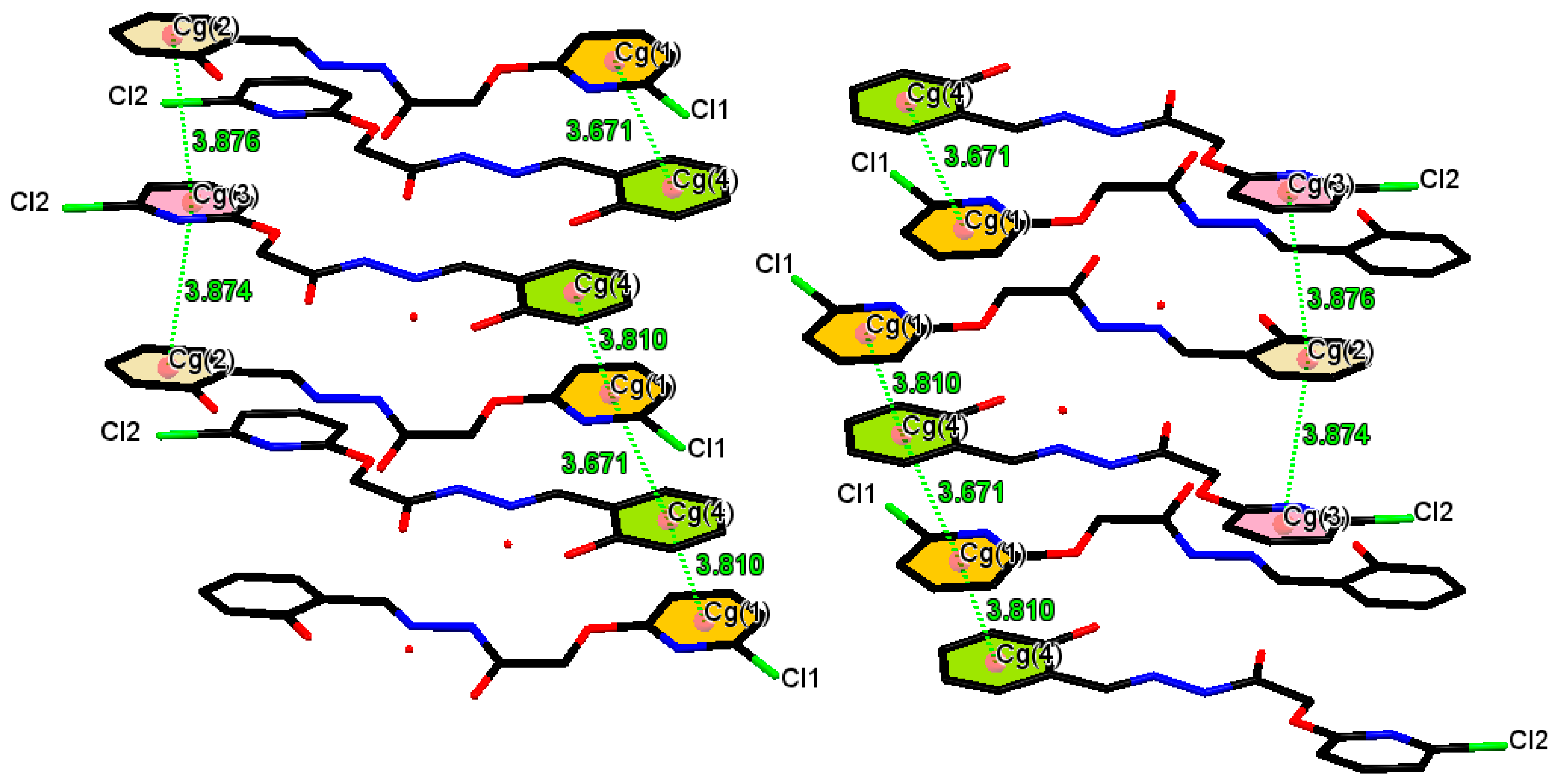
| Cg(e)–Cg(f) | Def | DAef | De (f) | Df (e) | Ring Off-Set |
|---|---|---|---|---|---|
| Cg(1)–Cg(4)iii | 3.671 | 1.2(2) | 3.4556(19) | 3.460(2) | - |
| Cg(1)–Cg(4)iv | 3.810 | 1.2(2) | 3.4495(19) | 3.479(2) | - |
| Cg(2)–Cg(3)iv | 3.874 | 2.7(2) | 3.4350(19) | 3.3581(19) | - |
| Cg(2)–Cg(3)v | 3.876 | 2.7(2) | 3.4495(19) | 3.4066(19) | - |
| BCP | Bonds | ρ (e/a3) | ε | Va | |
|---|---|---|---|---|---|
| 4 | O2–H8 | +0.0181 | +0.0878 | +0.5847 | −0.0144 |
| 5 | H8–O36 | +0.0127 | +0.0487 | +0.0339 | −0.0081 |
| 8 | H5–N9 | +0.0436 | +0.1157 | +0.0397 | −0.0392 |
| 18 | H16–O36 | +0.0029 | +0.0107 | +0.0863 | −0.0017 |
| 26 | H23–O36 | +0.0091 | +0.0279 | +0.0808 | −0.0054 |
| 42 | O2–O37 | +0.0066 | +0.0267 | +0.2101 | −0.0052 |
| 43 | H19–O37 | +0.0073 | +0.0263 | +0.1561 | −0.0047 |
| 49 | O35–H41 | +0.0179 | +0.0868 | +0.6171 | −0.0143 |
| 51 | H38–N42 | +0.0421 | +0.1147 | +0.0433 | −0.0375 |
| 55 | H41–O67 | +0.0148 | +0.0553 | +0.1317 | −0.0097 |
| 65 | H49–O67 | +0.0062 | +0.0181 | +0.0638 | −0.0035 |
| 66 | H33–H53 | +0.0016 | +0.0052 | +0.1869 | −0.0007 |
| 70 | H56–O67 | +0.0114 | +0.0324 | +0.0980 | −0.0068 |
| Compound | Donor(i) | Type | Acceptor(j) | Type | Ea(2) | E(j)E(i)b(a.u) | F(i,j)c(a.u) |
|---|---|---|---|---|---|---|---|
| C13-H14 | ∂ | C11-C13 | ∂ * | 0.51 | 1.09 | 0.021 | |
| C46-C48 | π | N39-C50 | π * | 30.35 | 0.26 | 0.082 | |
| C13-C15 | π | N6-C17 | π * | 29.62 | 0.26 | 0.082 | |
| N39-C50 | π | C43-C44 | π * | 28.32 | 0.33 | 0.088 | |
| C43-C44 | π | C45-C48 | π * | 22.45 | 0.30 | 0.074 | |
| C30-C32 | π | C26-C28 | π * | 21.46 | 0.29 | 0.071 | |
| HBPAH | C13-C15 | π | C10-C11 | π * | 16.09 | 0.27 | 0.060 |
| C43-C44 | π | N39-C50 | π * | 14.78 | 0.27 | 0.058 | |
| N40 | LP(1) | O36-C54 | π * | 62.83 | 0.29 | 0.121 | |
| N7 | LP(1) | O3-C21 | π * | 56.48 | 0.29 | 0.117 | |
| O35 | LP(2) | N39-C50 | π * | 35.80 | 0.32 | 0.103 | |
| Cl1 | LP(2) | N6-C10 | ∂ * | 5.79 | 0.85 | 0.063 | |
| N9 | LP(1) | N7-H8 | ∂ * | 7.04 | 0.81 | 0.069 | |
| O2 | LP(1) | N6-C17 | ∂ * | 6.61 | 1.08 | 0.076 | |
| O36 | LP(1) | N7-H8 | ∂ * | 2.22 | 1.13 | 0.045 |
| HBPAH | ||
|---|---|---|
| MO(s) | Energy | ∆E(eV) |
| HOMO | −5.600 | 3.634 |
| LUMO | −1.966 | |
| HOMO-1 | −6.266 | 4.65 |
| LUMO+1 | −1.616 | |
| HOMO-2 | −6.274 | 5.034 |
| LUMO+2 | −1.240 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Khalid, M.; Abid, S.; Tahir, M.N.; Iqbal, J.; Ashfaq, M.; Kanwal, F.; Lu, C.; Rehman, M.F.u. Green Synthesis, SC-XRD, Non-Covalent Interactive Potential and Electronic Communication via DFT Exploration of Pyridine-Based Hydrazone. Crystals 2020, 10, 778. https://doi.org/10.3390/cryst10090778
Ali A, Khalid M, Abid S, Tahir MN, Iqbal J, Ashfaq M, Kanwal F, Lu C, Rehman MFu. Green Synthesis, SC-XRD, Non-Covalent Interactive Potential and Electronic Communication via DFT Exploration of Pyridine-Based Hydrazone. Crystals. 2020; 10(9):778. https://doi.org/10.3390/cryst10090778
Chicago/Turabian StyleAli, Akbar, Muhammad Khalid, Saba Abid, Muhammad Nawaz Tahir, Javed Iqbal, Muhammad Ashfaq, Fariha Kanwal, Changrui Lu, and Muhammad Fayyaz ur Rehman. 2020. "Green Synthesis, SC-XRD, Non-Covalent Interactive Potential and Electronic Communication via DFT Exploration of Pyridine-Based Hydrazone" Crystals 10, no. 9: 778. https://doi.org/10.3390/cryst10090778
APA StyleAli, A., Khalid, M., Abid, S., Tahir, M. N., Iqbal, J., Ashfaq, M., Kanwal, F., Lu, C., & Rehman, M. F. u. (2020). Green Synthesis, SC-XRD, Non-Covalent Interactive Potential and Electronic Communication via DFT Exploration of Pyridine-Based Hydrazone. Crystals, 10(9), 778. https://doi.org/10.3390/cryst10090778