1. Introduction
Studies concerning the synthesis and applications of neutral organic molecules equipped with hydrogen-bond donors for binding or separating anions have become a topical area in supramolecular chemistry [
1,
2,
3,
4,
5,
6]. The use of pre-organized organic host molecules with cleft structures, such as calixarene [
7,
8,
9] functionalized with hydrogen bond donors and cyclic amides [
10,
11], is of interest due to their efficient binding and advantageous recognition properties.
Recently, incorporation of ligands possessing hydrogen-bond-donor moieties has also been a strategy of first choice to generate coordination frameworks capable of interactions with anions [
12,
13,
14]. The synthesis and properties of coordination polymers containing N–H-hydrogen-bond donor groups as the anion binding unit have been described in a number of independent studies [
15,
16,
17]. For example, Custelcean and co-workers have successfully synthesized coordination polymers from ligand-possessing urea moieties [
18]. These compounds form weak hydrogen-bonding interactions with the anions, through the urea moieties, allowing Custelcean et al. to examine selective anion separations via crystallization [
19]. The roles of non-covalent interactions in coordination compounds have been reported in many other relevant works [
20,
21,
22,
23,
24,
25,
26].
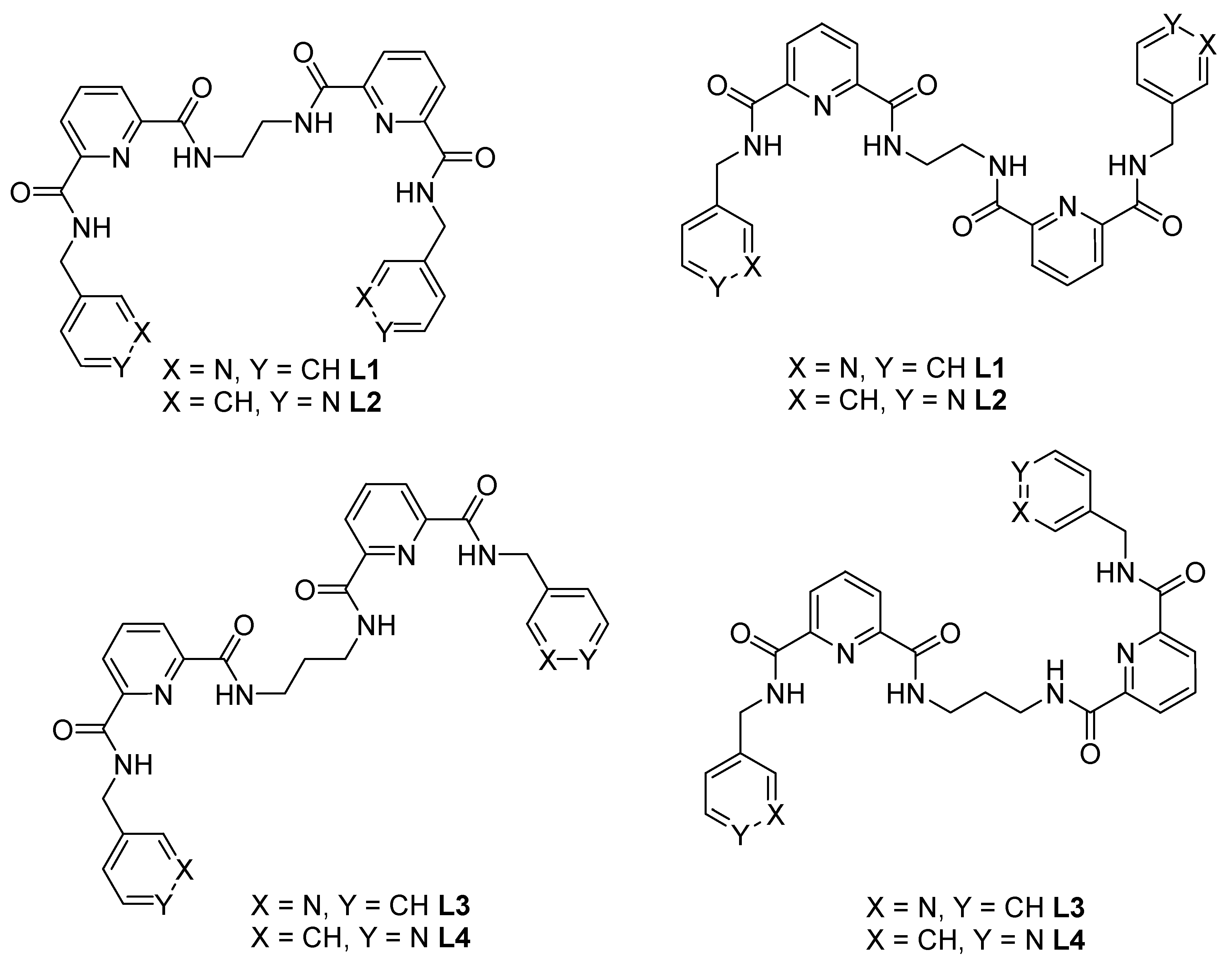
One of our goals is to synthesize coordination polymers with potentially larger anion pockets capable of accommodating larger anions within their extended structure. One design strategy uses pre-organized 2,6-pyridinedicarboxamide units separated by short yet flexible linkers. In the first attempt to generate such longer and more flexible tetraamide ligands, commercially and readily available linear aliphatic amines, such as 1,2-ethylenediamine and 1,3-propylenediamine, were used as spacers, incorporated between the two carboxamide units. By adding the alkyl spacer, an expanded pre-organized ligand and with potentially larger anion pockets could be obtained. In addition, by using combinations of these organic linkers and transition metal ions, new discrete or extended materials can also be prepared and their anion binding studied, especially in the solid state. Herein we report the synthesis of these new linkers, namely 1,2-bis[N,N′-6-(3-pyridylmethylamido)pyridyl-2-carboxyamido]ethane (L1), 1,2-bis[N,N′-6-(4-pyridylmethylamido)pyridyl-2-carboxyamido]ethane (L2), 1,2-bis[N,N′-6-(3-pyridylmethylamido)pyridyl-2-carboxyamido]propane (L3) and 1,2-bis[N,N′-6-(4-pyridylmethylamido)pyridyl-2-carboxyamido]propane (L4). We also examine the conformational flexibility of the new tetraamides and utilize compounds L2 and L4 for the synthesis of coordination polymers.
2. Experimental Section
2.1. Materials and Methods
All chemicals and reagents were used without further purification. Infrared spectra were collected on a Perkin Elmer Spectrum BX Infrared spectrometer as KBr disks. For X-ray diffraction datasets, the complexes were collected with Mo-Kα radiation (λ = 0.71073 Å), using an Oxford Diffraction X-Calibur single-crystal X-ray diffractometer at 150(2) K. All datasets were corrected for absorption, using a multi-scan method, and structures were solved directly by using SHELXS-97 [
27] and refined by full-matrix least-squares on F2 by SHELXL-97 [
28] interfaced through the program X-Seed [
29]. In general, all non-hydrogen atoms were refined anisotropically, and hydrogen atoms were included as invariants at geometrically estimated positions. The structure of {[Cd
3(L2)
4(H
2O)
10](NO
3)
6·12H
2O·CH
3OH}
n shows some disorder of the nitrate anions, requiring the use of DFIX, SIMU and RIGU restraints. Moreover, the hydrogen atoms on the coordinated and non-coordinated water molecules could not be located in the difference map; however, they are included in the formula. The remaining significant peaks in the difference map are close to the heavy atom Cd positions. Moreover, {[Cd(SO
4)(L4)(H
2O)
2]·4H
2O·CH
3OH}
n shows some disorder of the Cd position and the associated coordination sphere, which was partially modeled through the use of SIMU, RIGU and ISOR restraints. In general, the hydrogen atoms of the water molecules could not be located except where there were clear hydrogen-bonding interactions. Figures were produced by using the program POV-Ray [
30], interfaced through the program X-Seed, and the table was generated from CIFTAB software. Complete data on the crystal structure of L2 were deposited with the Cambridge Crystal Data Centre (CCDC), with reference number CCDC 1863040. Meanwhile, deposition reference numbers for {[Cd
3(L2)
4(H
2O)
10](NO
3)
6·12H
2O·CH
3OH}
n and {[Cd(SO
4)(L4)(H
2O)
2]·4H
2O·CH
3OH}
n are CCDC 1,867,235 and 2,051,544, respectively.
2.2. Density Functional Theory (DFT) Studies Using Gaussian 09 Software
The desktop is specified with a system that works in a 64-bit operating system with an x64-based processor. This computer has a 4.00 installed memory and 3.87 GB RAM. The molecule was optimized by density functional theory (DFT), B3LYP method. All calculations were made in vacuum by using 6–31 G (d,p) as a basis set for carbon, hydrogen, nitrogen and oxygen atoms, and GEN keyword with LanL2DZ for a metal atom such as anion chromate. The geometry of the molecule was optimized without any symmetry constraints [
31].
2.3. Synthesis of L1 and L2
A suspension of N-6-[(3-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (i) (0.50 g, 1.8 mmol) and a slight excess of ethylenediamine (0.065 mL, 0.96 mmol) in toluene (40 mL) was heated at reflux, for 36 h, under nitrogen. The solvent was removed in vacuo, to give a brown solid. The solid was purified by flash column chromatography on silica gel, eluting with 1:9 methanol–CH2Cl2, to give L1 as a cream solid (0.19 g, 35%). Mp 260–261 °C. The analysis found the following: C, 60.95; H, 4.80; N 20.07. C28H28N8O5 requires C, 60.41; H, 5.08; N 20.14%. 1H NMR (400 MHz) δ = 3.62 (4H, m, CH2NH), 4.77 (4H, d, J = 6.52 Hz, pyCH2NH), 7.35 (2H, m, pyH5′), 7.72 (2H, m, pyH4′), 8.22 (6H, m, pyH3, pyH5, pyH4), 8.46 (2H, m, pyH6′), 8.57 (2H, m, pyH2′), 9.07 (2H, m, CH2NH) and 9.14 (2H, m, pyCH2NH). 13C NMR (75.1 MHz) δ = 165.8, 164.90, 164.19, 160.39, 148.93, 148.69, 148.11, 139.36, 136.48, 132.65, 126.22, 125.56, 124.51, 41.19., m/z (ESI-MS) 539.2 (MH+, 100%), 561.2. Selected IR bands (KBr disk, cm−1): 3292 (m), 1663 (s), 1524 (s), 1418 (m), 1419 (m) and 1001 (m). Crystals were obtained by slow evaporation of a methanol solution of L1 and copper acetate.
To synthesize L2, N-6-[(3-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (i) was replaced with N-6-[(4-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (ii). L2 was isolated as a cream solid (0.21 g, 41%). Mp 262–264 °C. The analysis found the following: C, 60.78; H, 4.79; N 20.15. C28H28N8O5 requires C, 60.41; H, 5.08; N 20.14%. 1H NMR (400 MHz) δ = 2.85 (4H, m, CH2NH), 4.61 (4H, d, J = 6.45 Hz, pyCH2NH), 7.29 (4H, m, pyH3′, pyH5′), 8.20 (2H, m, pyH5), 8.48 (2H, m, pyH4), 9.60 (4H, m, pyH2′, pyH6′) and 9.95 (2H, m, NH). 13C NMR (75.1 MHz) δ = 163.72, 163.69, 150.27, 149.66, 148.10, 148.00, 139.44, 125.77, 125.75, 121.97, 42.64, 41.45, 21.08. m/z (ESI-MS) 539.30 (MH+, 65%), 561.3, (MNa+, 100%), 1076.7 (M2H+, 5%), 1098.8 (M2Na+, 15%), Selected IR bands (KBr disk, cm−1 ): 3354 (s), 3292 (m), 1663 (s), 1529 (s), 1418 (m), 1307 (w), 1001 (m) and 846 (m). Crystals were obtained by slow evaporation of a methanol solution of L2 and cobalt acetate.
2.4. Synthesis of L3 and L4
A suspension of N-6-[(3-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (L2) (0.50 g, 1.8 mmol) and a slight excess of ethylenediamine (0.065 mL, 0.96 mmol) in toluene (40 mL) was heated at reflux for 36 h under nitrogen. The solvent was removed in vacuo to give a brown solid. The solid was purified by flash column chromatography on silica gel, eluting with 1:9 methanol–CH2Cl2, to give L3 as a cream solid (0.32 g, 62%). Mp 171–172 °C. The analysis found the following: C, 61.80; H, 5.00; and N, 19.64. C28H30N8O5 requires C, 61.03; H, 5.31; N 19.64%. 1H NMR (400 MHz) δ = 3.49 (2H, m, CH2CH2NH), 3.97 (2H, m, CH2CH2NH), 4.68 (4H, d, J = 6.45 Hz, pyCH2NH), 7.26 (4H, m, pyH3′, pyH5′), 7.75 (2H, 2d, J = 7.87, 4.80 Hz, pyH3), 8.06 (2H, m, pyH5), 8.23 (2H, m, pyH4), 8.40 (2H, m, pyH6′), 8.45 (2H, m, pyH2′), 9.10 (4H, m, pyH2′, pyH6′), and 9.33 (2H, m, NH). 13C NMR (75.1 MHz) δ = 165.04, 163.91, 149.99, 149.43, 149.03, 146.69, 138.87, 135.86, 134.02, 127.68, 125.78, 123.76, 53.14, 41.12. m/z (ESI-MS) 553.0, (MH +, 65%), 1106.1 (M2H +, 10%). Selected IR bands (KBr disk, cm−1): 3296 (m), 1624 (s), 1523 (s), 1417 (m), 1309 (m), 1069 (m), 1009 (m) and 743 (s).
To synthesis L4, N-6-[(3-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (i) was replaced with N-6-[(4-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (ii). L4 was isolated as a cream solid (0.29 g, 55%). Mp 145–46 °C. The analysis found the following: C, 59.28; H, 5.12; and N, 19.21. Moreover, (C29H32N8O6) requires C, 59.16; H, 5.49; N 19.04%. 1H (400 MHz) δ = 3.50 (2H, m, CH2CH2NH), 3.98 (2H, m, CH2CH2NH), 4.69 (4H, m, pyCH2NH), 7.34 (4H, m, pyH3′, pyH5′), 7.99 (2H, m, pyH3), 8.01 (2H, m, pyH5), 8.09 (2H, m, pyH4), 8.89 (4H, m, pyH2′, pyH6′) and 9.24 (2H, m, NH). 13C (75.1 MHz; CDCl3; Me4Si) δ = 164.47, 164.31, 149.93, 149.76, 148.59, 148.17, 139.38, 125.45, 124.87, 122.51, 42.42, 34.94. m/z (ESI-MS) 553 (MH +, 100%). Selected IR bands (KBr disk, cm−1) were as follows: 3387 (m), 3295 (m), 1623 (s), 1520 (s), 1414 (m), 1237 (m) and 1009 (m).
2.5. Synthesis of Coordination Polymers
Cd(NO3)2·4H2O (0.0064 g, 0.021 mmol) was dissolved in methanol (5 mL), and then it was heated for a few minutes, before being added dropwise to a solution of L2 (0.0223 g, 0.0414 mmol), which was dissolved in 1:1 methanol–acetonitrile (15 mL). The resulting colorless solution was heated for 45 min and left to evaporate at room temperature. After two months, the solution afforded {[Cd3(L2)4(H2O)10](NO3)6·12H2O·CH3OH}n as colorless crystals (0.008 g, 36%). Mp 240–245 °C. Selected IR bands (KBr disk, cm−1) were as follows: 3294 (s), 1654 (s), 1541 (s), 1315 (s, N–O stretch), 1002 (s) and 744 (m).
Cd(SO4)·H2O (0.0023 g, 0.010 mmol) was dissolved in methanol (5 mL), and then it was heated for a few minutes, before being added dropwise to a solution of L4 (0.0114 g, 0.021 mmol) which was also dissolved in methanol (15 mL). The addition gave a colorless solution that was heated for 45 min and left to evaporate at room temperature. After two months, the solution afforded {[Cd(SO4)(L4)(H2O)2]·4H2O·CH3OH}n as colorless block-shaped crystals (0.003 g, 32%). Mp 250–252 °C. Selected IR bands (UATR, cm−1) were as follows: 3236 (s), 1647 (s), 1535 (m), 1432 (w), 1468 (m), 1305 (m), and 1022 and 670 (s, S–O stretch).
3. Results and Discussion
A survey of the literature indicated that cyclic tetraamides have been commonly studied for anion recognition [
32], but the investigation of acyclic tetraamide compounds is still limited and in need of exploration. A simple strategy to generate longer and more flexible tetraamide ligands involves using commercial and readily available linear aliphatic amines, such as 1,2-ethylenediamine and 1,3-propylenediamine, as spacers between pre-organized 2,6-pyridine dicarboxamide units. These new symmetrical bridging ligands should exhibit far greater flexibility than previously reported macrocyclic tetraamides, owing to the greater degree of freedom in the aliphatic chains and the acyclic structures. Thus, the potential for formation of a larger anion pocket created by two 2,6-pyridine dicarboxamide units was proposed to be a source of interesting anion-encapsulation behavior.
Through the implementation of a previously reported synthetic approach, Reference [
33] allowed compounds L1–L4 to be successfully synthesized, with some minor modifications to the original procedure. Compound 1,2-bis[
N,N′-6-(3-pyridylmethylamido)pyridyl-2-carboxyamido]ethane (L1) was synthesized by reaction between 1,2-ethylenediamine and
N-6-[(3-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (i). The reaction of 1,2-ethylenediamine and
N-6-[(4-pyridylmethylamino)carbonyl]-pyridine-2-carboxylic acid methyl ester (ii) generated 1,2-bis[
N,N′-6-(4-pyridylmethylamido)pyridyl-2-carboxyamido]ethane (L2). Finally, reaction of (i) and (ii) with 1,3-propylenediamine gave 1,2-bis[
N,N′-6-(3-pyridylmethylamido)pyridyl-2-carboxyamido] propane (L3) and 1,2-bis[
N,N′-6-(4-pyridylmethylamido)pyridyl-2-carboxyamido] propane (L4), respectively (
Scheme 1).
Taking L1 as a typical example, precursor (i) and 1,2-ethylenediamine were combined in the necessary 2:1 molar ratio, by suspension in toluene, and heated at reflux, for 36 h; meanwhile, for L2, it took an additional 24 h for the reaction to be completed. After purification, these small-scale procedures yielded both L1 and L2 in ca. 30–40% yield (mainly due to losses during purification and isolation), while a larger-scale synthesis gave a considerably improved yield of ca. 60% for each product. Both compounds were characterized by NMR and FTIR spectroscopy and mass spectrometry. The NH amides of compound L1 in deuterated dimethylsulfoxide are defined by the resonances in the
1H NMR spectrum at 9.14 and 9.07 ppm, respectively. The methylene spacer of ethylenediamine is observed at 3.62 ppm, while the other methylene spacer that links the amide and pendant pyridine is observed at 4.77 ppm. The FTIR spectrum of the compound indicates strong N–H, C=O, C=N and C=C stretches at 3292 (m), 1663 (s), 1418 (m) and 1001 (m) cm
−1, respectively. As for L2, IR spectroscopy indicates strong N–H, and C=N stretches at approximately, 3292 and 1663 cm
−1, respectively (
Figure S1).
Compound L2 was anticipated to display simpler
1H and
13C NMR spectra due to the use of the higher symmetry
para-substituted pyridyl pendant arms (
Figure S2). This has given signals for 4-substituted pyridine and central 2,6-pyridinecarboxamide resonances at 7.29 (2H, d, pyH8), 8.20 (3H, m, pyH3, pyH4, pyH5), 8.48 (2H, d, pyH9). The methylene protons that are adjacent to the pendant pyridine ring were observed at 4.61 (4H, d, J = 6.45 Hz). The signals which are corresponded to the amide NH moieties appear as separate broad triplets at 9.60 and 9.95 ppm. Like L1, it is expected that compound L2 can adopt a conformation possessing either one or two anion pockets; however, these conformers are free to interconvert in solution. The
13C NMR spectrum of L2 shows that the resonances of the methylene signals are observed at 21.08 and 41.45 ppm (
Figure S3). The electronegativity of NH of the amide group leads to the methylene group being de-shielded, similar to previous reported results for related amide compounds [
33]. Meanwhile, the resonances of aromatic carbon are observed in the range of 121.97–149.66 ppm, and the signals of the two C=O resonances were observed on 163.69 and 163.72 ppm. We also provide the figure containing enlargement of the NH amide and pyridine dicarboxamide moieties in the
1H NMR spectra for L1, L3 and L4 as a reference (
Figure S4).
Further confirmation of the successful syntheses of L1 and L2 was achieved by using Electron Spray Ionization Mass Spectrometry (ESI-MS). ESI-MS of L2 provided two major species, which appeared as [L2 + Na]
+ and [L]
+ observed at
m/z 561.3 (100%) and
m/z 539.3 (65%), respectively. A protonated dimer [(L2)
2 + H]
+ and a dimer with a sodium cation [(L2)
2 + Na]
+ were also observed, at
m/z 1076.7 and 1098.8, respectively. In the ESI-MS spectrum of L1 (from methanol), a protonated species [L1 + H]
+, was identified at
m/z 539.2, and [L1 + Na]
+ was observed at
m/z 561.2. A protonated species bound to water, [L1 + H
2O + H]
+ (
m/z 557.2), was also observed in relatively low abundance, and dimers [(L1)
2 + H]
+ and [(L1)
2 + Na]
+ were observed at
m/z 1081 and 1145.7, respectively. The mass spectrometry data for L1 and L2 were similar to that reported for related diamide compounds [
19], which also showed that hydrogen-bonded aggregates were encountered.
The crystal structure of L1 has been previously reported [
34], but given our interest in the anion binding potential of these ligands as components of coordination polymers, we also attempted crystallization of L2. Crystals of L2 were obtained from a solution containing cobalt acetate, yielding colorless rectangular-block-shaped crystals after several days of standing at room temperature. These crystals were subjected to single crystal X-ray diffraction, to provide the solid-state structure of L2. Compound L2 crystallizes in the triclinic space group
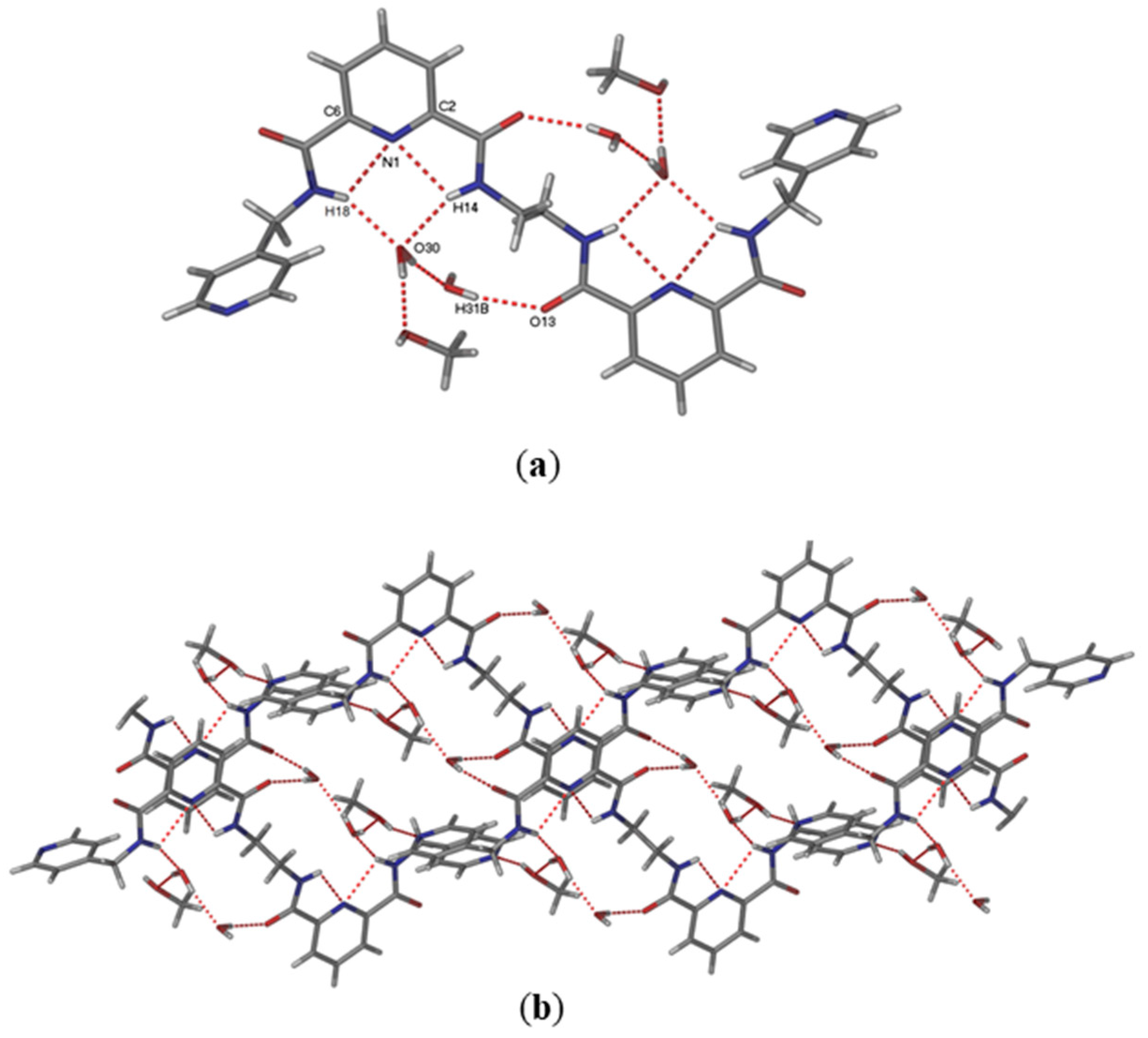
P-1, but with half a molecule of L2, two water molecules and one methanol in the asymmetric unit. One water molecule acts as the hydrogen bond acceptor for the amide N–H donors in L2 (NH
…O,
d = 2.083 and 2.115 Å). The presence of other hydrogen bond is shown in
Table 1. This water molecule is, in turn, hydrogen bonded to a methanol molecule and to a second water molecule, which is further hydrogen bonded to the carbonyl group of L2 (
Figure 1).
The crystal structure of L2 reveals that this compound adopts quite a different conformation to L1 [
34]. Due to free rotation provided by the ethylene spacer and the intermolecular hydrogen bonding interactions with solvate molecules (C=O
…H-OH), the two pyridine dicarboxamide cores are directed almost opposite one another. Thus, as expected, it appears that either one or two anion pockets could be observed for compounds L1 and L2.
As outlined earlier, to synthesize 1,2-bis[
N,N′-6-(3-pyridylmethylamido)pyridyl-2-carboxyamido] propane (L3) and 1,2-bis[
N,N′-6-(4-pyridylmethylamido)pyridyl-2-carboxyamido] propane (L4), we required 1,3-propylenediamine to be employed as the spacer. These two compounds were synthesized based on the method described for L1 and L2, as shown in
Scheme 1. The synthesis of L3 is straightforward, with the product being obtained directly from the reaction; a cream solid began to precipitate from the reaction mixture after 36 h of heating at reflux, in toluene, to give L3 in 62% yield. In contrast to the synthesis of L3, the reaction of L2 with 1,3-propylenediamine to form L4 required considerably longer periods for the reaction to reach completion, typically taking approximately 96 h of heating at reflux. The crude product was purified via column chromatography, to give L4 in 55% yield.
The syntheses of both compounds were confirmed by the observation of characteristic resonances in the 1H NMR spectra. The N–H amide signals of L3 and L4 were observed at 9.10 and 8.89 ppm, and 9.33 and 9.24 ppm, respectively. While the 1H NMR spectrum displayed readily noticeable differences between the resonances for amide protons in both compounds, only small differences were observed in the resonances for the propylene and methylene linkers. Compound L3 has signals for the propylene spacer at 3.49 and 3.97 ppm, compared to 3.50 and 3.98 ppm in L4. Similarly, a peak for the methylene linker was observed at 4.68 ppm for L3 and 4.69 ppm for L4.
ESI-MS of L3 conducted in methanol led to observation of peaks for [L3 + H]
+ (
m/z 553.0, 65%) and a dimer [(L3)
2 + H]
+ (
m/z 1106.1, 10%). The ESI-MS of compound L4, also conducted in methanol, showed peaks for [L4 + H]
+ (
m/z 553.3, 100%), a dimer [(L4)
2 + H]
+ (
m/z 1104.7, 10%) and a trimer [(L4)
3 + H]
+ (
m/z 1656.8, 5%). The presence of dimeric or trimeric species indicates that these compounds appear to readily form hydrogen-bonded aggregates in the gas phase and that the solution chemistry may be affected by such interactions. The IR spectra showed a distinctive C=O stretching vibration at approximately 1624–1654 cm
−1 for both compounds. Attempts to crystallize compounds L3 and L4 with a view to obtaining crystals suitable for structure studies were unsuccessful.
Table 2 and
Table 3 summarize the spectroscopic data obtained for L1–L4.
The structure studies indicate that compounds L1–L4 can adopt two distinct conformations.
Figure 2 shows the two types of conformations, adopted by these four tetraamide compounds, either having a U-shaped structure (syn arrangement), as proposed earlier (having two pre-organized amides facing inwards, towards the molecule cavities), or, as seen for L2, having an S-shaped conformation (anti-arrangement). To understand whether there is a preference for one conformer over another, we undertook computational studies to determine the stability of the two conformers. Thus, the DFT studies were carried out to determine the relative energy of the conformers, in which L2 was chosen as an example.
The calculations reveal that, for L2, the S-shaped conformer was more stable (−4,782,816.68 kJ/mol) compared to the U-shaped arrangement (−4,782,810.91 kJ/mol) (
Table S1). This is consistent with the solid-state structure of L2, which shows the S-shaped conformation rather than the U–shaped conformation. However, given that the difference in energy between S- and U-shaped conformers is very small (ca. 6 kJ/mol), this suggests that rotation of one 2,6-pyridine dicarboxamide entity with respect to the other can be easily influenced by intermolecular interactions with solvent molecules or anions.
Coordination Polymers of L2 and L4
To investigate the coordination chemistry and the possibility to bind anions within the resulting coordination frameworks, the tetraamide ligands were reacted with an array of first- and second-row transition metals. Specifically, the tetraamide ligands, L1–L4, were reacted with MX
n (where M=Cu, Cd, Co, Zn, Ag; X=ClO
4, NO
3, CH
3COO, Cl, Br; n=1 or 2) metal salts in 1:2 metal-to-ligand ratio in methanol. The extended amide ligands incorporating 4-pyridyl donors (L2 and L4) showed a propensity to form 1D coordination polymers when reacted with cadmium(II) salts (CdX
2 where X=NO
3, X
2=SO
4). This is similar to the coordination chemistry observed for the structurally related but simpler monoamide [
35] and diamide ligands [
36,
37]. The reaction of L1 with the metal salts in methanol solution gave clear solutions, which then evaporated to give oils that could not be characterized. The oils obtained from these reactions were triturated with warm ethanol or diethyl ether, but all attempts failed to provide reasonable quantities of any solid. The reaction of L3, which also incorporated 3-pyridyl donors as the metal coordinating sites, also failed to give isolable (crystalline, microcrystalline or amorphous solid) compounds, and it resulted in products that were oils. Repeated attempts to optimize the reaction conditions (different solvents, crystallization conditions) and also to repeat the reaction with different metal–ligand ratios were not successful in providing analytically pure solids.
In contrast, reaction of the tetraamide ligands incorporating pendant 4-pyridyl donors as the metal coordinating sites (L2 and L4), with cadmium salts, gave colorless solutions from which were obtained two new coordination polymers, {[Cd
3(L2)
4(H
2O)
10](NO
3)
6·12H
2O·CH
3OH}
n and {[Cd(SO
4)(L4)(H
2O)
2]·4H
2O·CH
3OH}
n. For these compounds, {[Cd
3(L2)
4(H
2O)
12](NO
3)
6·10H
2O·CH
3OH}
n and {[Cd(SO
4)(L4)(H
2O)
2]·4H
2O·CH
3OH}
n, distinctive stretches were observed in their IR spectra, an N–O stretch at 1315 cm
−1 and two S–O stretches at 1022 and 670 cm
−1, respectively. X-ray crystallography revealed that these two compounds crystallized in the triclinic space group
P-1 but with different structures and crystal packing.
Table 4 lists the crystal data for all crystals obtained from this study.
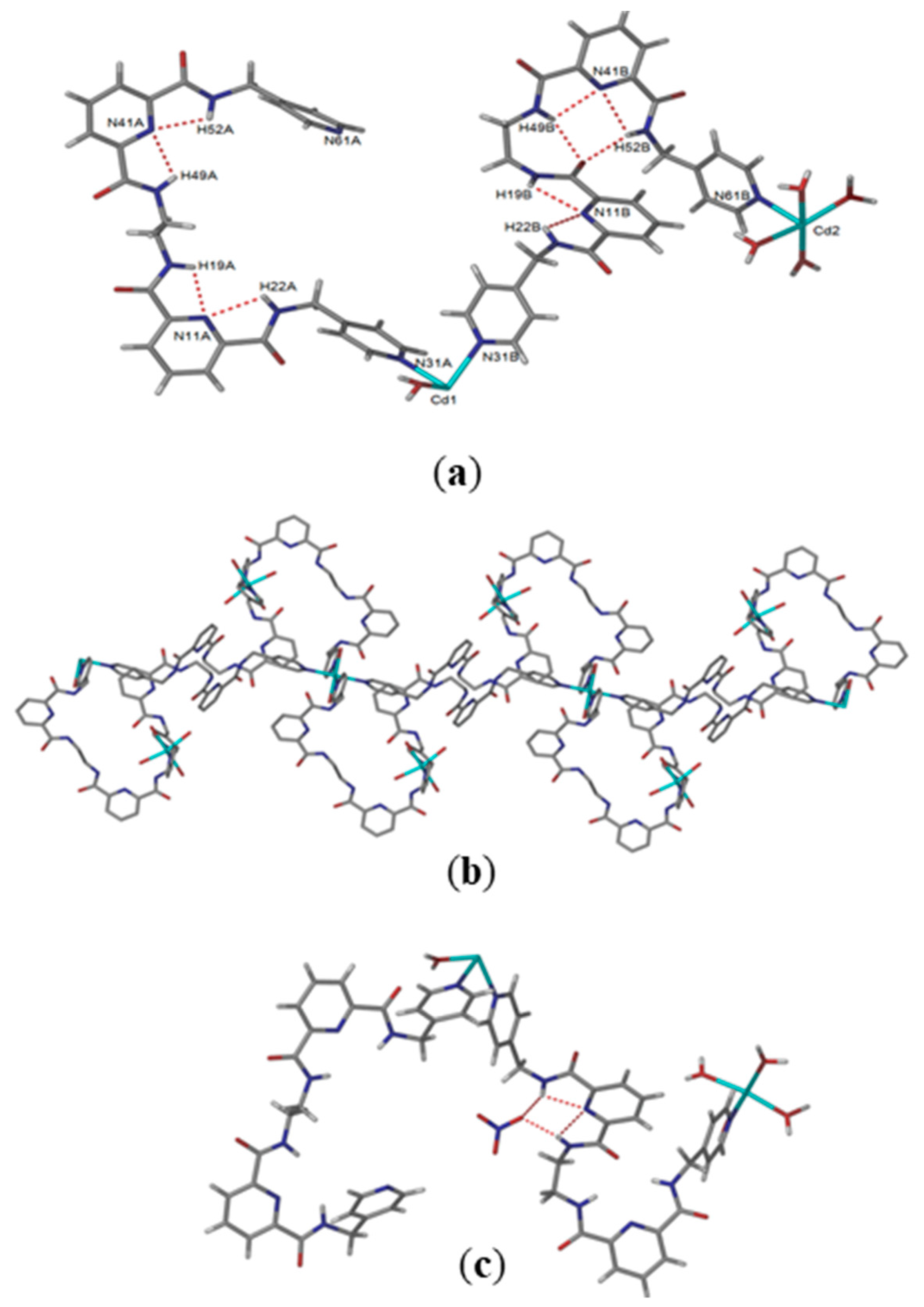
Reaction of L2 with cadmium(II) nitrate in methanol, followed by slow evaporation of the solvent, yielded colorless rectangular-block-shaped crystals in 36% yield. The complex crystallizes have an asymmetric unit containing two molecules of ligand L2, two cadmium positions (one on a centre of inversion), five coordinated and six non-coordinated water molecules, one non-coordinated methanol and three non-coordinated nitrate anions (two of which are disordered over three positions). Despite the lack of coordination by nitrate anions, both cadmium centers have octahedral geometries but different coordination environments. One cadmium (Cd1) atom is coordinated by two coordinated water molecules and four pyridyl groups from four different molecules of L2, while the second cadmium atom (Cd2) is coordinated by four water molecules and two nitrogen donors from pyridyl units of ligand L2. The cadmium centers have Cd–O bond lengths of 2.273(8) –2.338(5) Å and Cd–N bond lengths in the range 2.292(8)–2.309(8) Å, which is typical [
35]. The distances between the Cd–Cd atoms are in the range of 10.605 and 19.914 Å.
The presence of weak intramolecular hydrogen-bonding interactions which pre-organize the NH donors of the ligand into a central pocket are indicated (
d = 2.233–2.384 Å;
D = 2.642–2.733 Å; N-H···N angles = 104.33–108.88°). In one ligand, the ethylene spacer (-N-CH
2-CH
2-N- torsion angle = 69.4°) of L2 acts as a hinge to allow the second part of the ligand to interact with the second cadmium atom and form a metallo-macrocyclic structure. As a consequence of the flexibility provided by the ethylene spacer, the two pre-organized 2,6-pyridine dicarboxamide moieties form independent pockets, but one is occupied by the carbonyl oxygen of the other amide moiety (N-H··O=C,
d = 2.137, 2.204 Å,
D = 3.188, 2.731 Å (
Figure 3a). The second molecule of L2 adopts the single pocket conformation with both spacers in a syn arrangement.
The extended structure of {[Cd
3(L2)
4(H
2O)
10](NO
3)
6·12H
2O·CH
3OH}
n is shown in
Figure 3b, with the [M
4L
4] metallo-macrocyclic repeating units arranged into 1D coordination polymers. Taking the CdN
4O
2 centers as the axis of the 1D coordination polymer, adjacent CdN
4O
2 nodes in the structure are connected by two loops, each formed from two molecules of L2 and a CdN
2O
4 center. These are related by a center of inversion at the centroid between two CdN
4O
2 centers. Again, the NH amide moieties of the 2,6-pyridine dicarboxamide units are directed inwards, facing the core of the 1D coordination polymers, and the C=O groups are directed outward. There are moderately strong hydrogen bonds interactions between the amide hydrogen bond donors with the nitrate anions (
d = 2.029, 2.146 Å,
D = 2.861, 2.959). The arrangement of the 1D coordination polymers in the crystal packing is shown in
Figure 3c, with hydrogen atoms omitted for the sake of clarity.
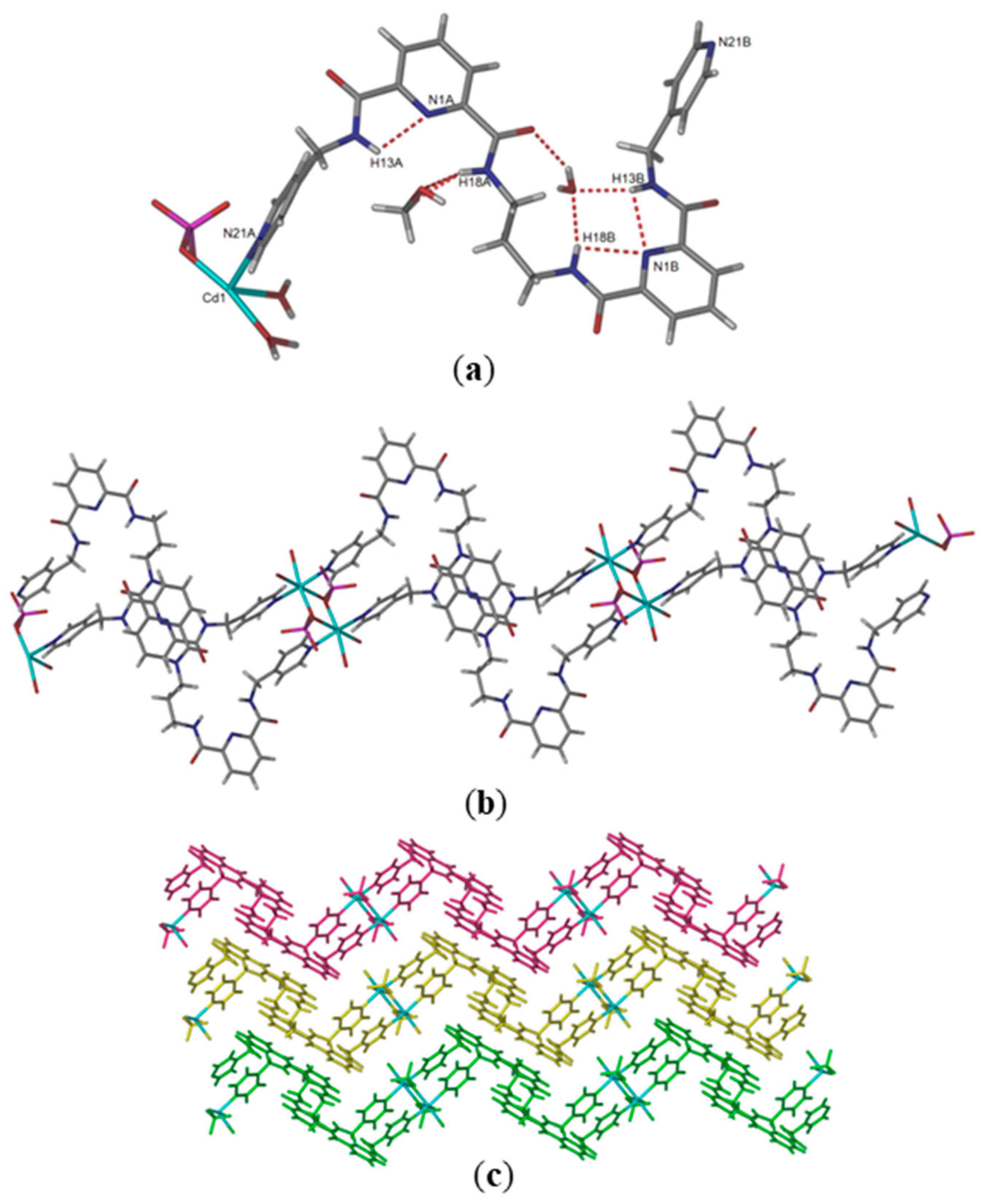
Reaction of L4 with cadmium sulfate yielded rectangular-block-shaped crystals in 32% yield. Like the coordination polymer described above, this compound also crystallized in the triclinic space group
P-1 but with quite a different structure. The asymmetric unit consists of one molecule of ligand L4, one cadmium atom, one methanol solvate molecule, one coordinated sulfate, two coordinated water molecules and four non-coordinated water solvates, disordered over six sites (
Figure 4a). The cadmium atom in this complex adopts an octahedral geometry, coordinated by two μ
2-oxygen atoms from two bridging sulfate anions, two water molecules and two pyridyl donors from two separate molecules of L4. The Cd–N and Cd–O distances are in the range of 2.204(5)–2.465(6) Å. This structure is stabilized by intramolecular hydrogen-bonding interactions between the pre-organized amide NH donors with water and methanol solvate molecules. There are two intermolecular hydrogen-bonding interactions between water molecules to carbonyl groups (O–H
…C=O,
d = 2.003,
D = 2.913 Å and
d = 2.006,
D = 2.874 Å). As noted, the anions are involved in bridging the di-cadmium units and not hydrogen bonded to the 2,6-pyridine dicarboxamide moiety. An extended view of compound {[Cd(SO
4)(L4)(H
2O)
2]·4H
2O·CH
3OH}
n is shown in
Figure 4b. The repeating unit in this 1D coordination polymer is a [M
4L
2] metallo-macrocyclic building block, where two cadmium atoms are bridged by two sulfate oxygen donors. These di-cadmium units are then coordinated by the nitrogen donors of four ligands, to form the 1D necklace-type coordination polymer. The distance between the bridged Cd–Cd subunit is 3.697 Å and the distance between two separated cadmium atoms is 19.685 Å. The adjacent polymer chains of this compound are arranged with all NH amide units directed inwards, facing the cavity of the coordination polymer and the C=O groups directed outward. The packing of the 1D coordination polymers are stabilized by face-to-face π-stacking interactions between the pyridyl cores (centroid–centroid distance 3.60 Å; angle 78.02°, centroid offset 1.38 Å) and the two pendant pyridines (centroid–centroid distance 3.80 Å; angle 81.95°, centroid offset 1.39 Å) (
Figure 4c).

 and
and 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}