
Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Combination of Polarized Light Microscope and Hot Stage
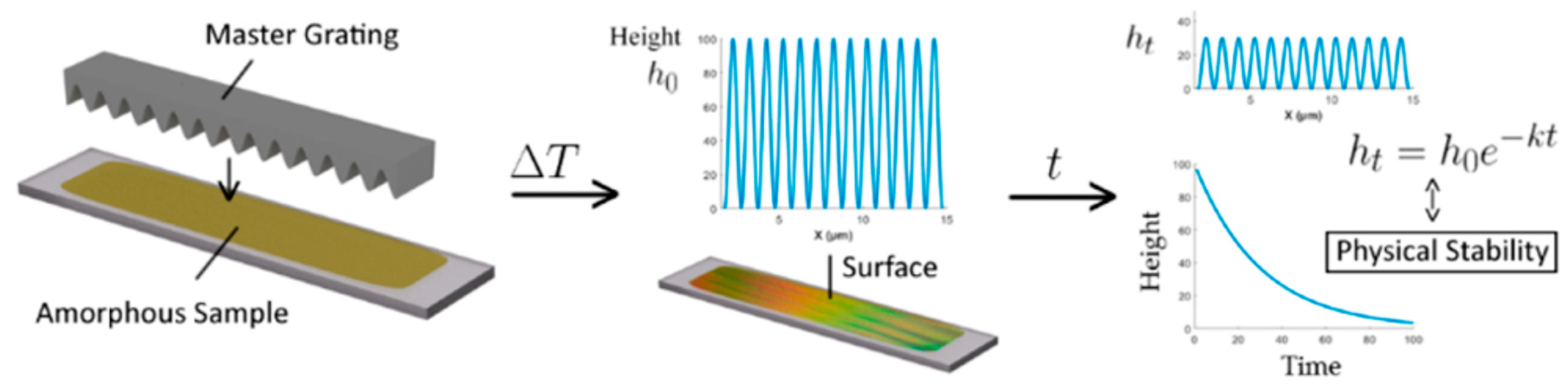
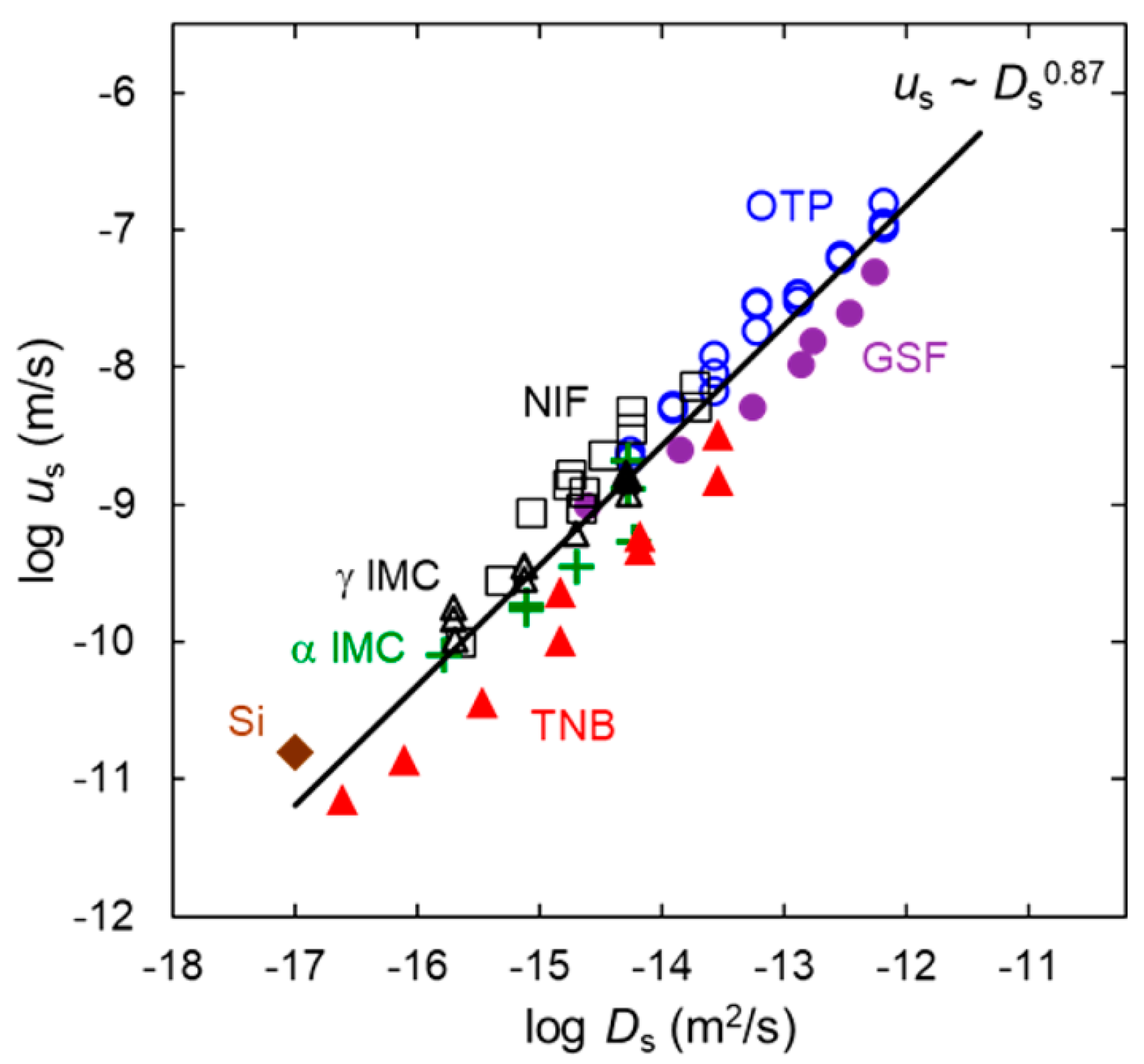
3. Surface Grating Decay and Surface Diffusion
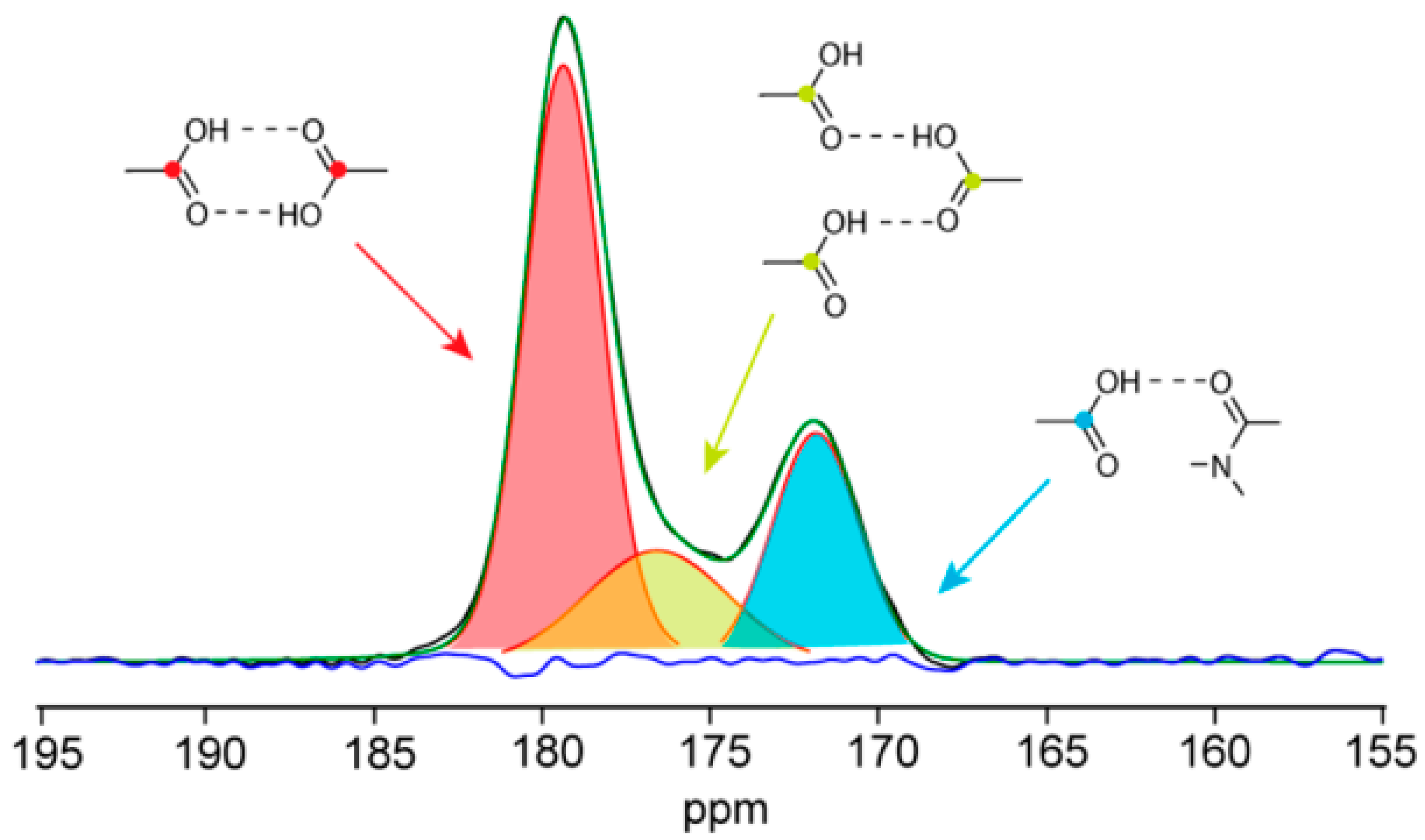
4. Solid-State Nuclear Magnetic Resonance (NMR)
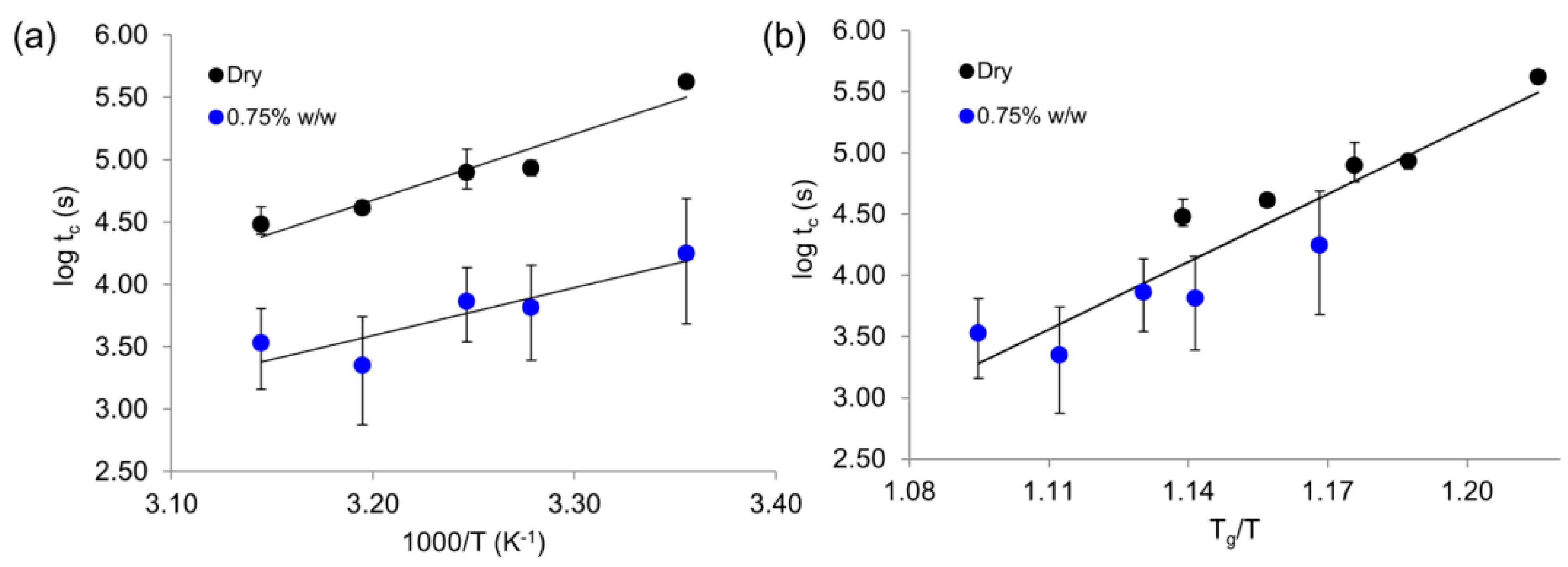
5. Broadband Dielectric Spectroscopy
6. Other Characterization Techniques
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Di, L.; Fish, P.V.; Mano, T. Bridging solubility between drug discovery and development. Drug Discov. Today 2012, 17, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J. Pharm. Sci. 2010, 99, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliver. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Brough, C.; Williams, R.O. Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. Int. J. Pharm. 2013, 453, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Mooter, G.V.D. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Tech. 2012, 9, 79–85. [Google Scholar] [CrossRef]
- Yan, H.; Chris, H. Amorphous solid dispersions: Utilization and challenges in drug discovery and development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar]
- Powell, C.T.; Cai, T.; Hasebe, M.; Gunn, E.M.; Gao, P.; Zhang, G.; Gong, Y.; Yu, L. Low-concentration polymers inhibit and accelerate crystal growth in organic glasses in correlation with segmental mobility. J. Phys. Chem. B 2013, 117, 10334–10341. [Google Scholar] [CrossRef]
- Kestur, U.S.; Taylor, L.S. Role of polymer chemistry in influencing crystal growth rates from amorphous felodipine. CrystEngComm 2010, 12, 2390–2397. [Google Scholar] [CrossRef]
- Cai, T.; Zhu, L.; Yu, L. Crystallization of organic glasses: Effects of polymer additives on bulk and surface crystal growth in amorphous nifedipine. Pharm. Res. 2011, 28, 2458–2466. [Google Scholar] [CrossRef]
- Huang, C.; Powell, C.T.; Sun, Y.; Cai, T.; Yu, L. Effect of low-concentration polymers on crystal growth in molecular glasses: A controlling role for polymer segmental mobility relative to host dynamics. J. Phys. Chem. B 2017, 121, 1963–1971. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Inhibition of solution crystal growth of ritonavir by cellulose polymers—Factors influencing polymer effectiveness. CrystEngComm 2012, 14, 6503–6514. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining supersaturation in aqueous drug solutions: Impact of different polymers on induction times. Cryst. Growth Des. 2013, 13, 740–751. [Google Scholar] [CrossRef]
- Schram, C.J.; Beaudoin, S.P.; Taylor, L.S. Polymer inhibition of crystal growth by surface poisoning. Cryst. Growth Des. 2016, 16, 2094–2103. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control Release 2018, 279, 114–125. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zhang, G.G. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef]
- Zhou, D.; Grant, D.J.; Zhang, G.G.; Law, D.; Schmitt, E.A. A calorimetric investigation of thermodynamic and molecular mobility contributions to the physical stability of two pharmaceutical glasses. J. Pharm. Sci. 2010, 96, 71–83. [Google Scholar] [CrossRef]
- Laitinen, R.; Lobmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef]
- Grzybowska, K.; Capaccioli, S.; Paluch, M. Recent developments in the experimental investigations of relaxations in pharmaceuticals by dielectric techniques at ambient and elevated pressure. Adv. Drug Deliv. Rev. 2016, 100, 158–182. [Google Scholar] [CrossRef]
- Yu, L. Surface mobility of molecular glasses and its importance in physical stability. Adv. Drug Deliv. Rev. 2016, 100, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Chen, Z.; Gui, Y.; Shi, C.; Zhang, G.G.; Yu, L. Crystal nucleation rates in glass-forming molecular liquids: D-sorbitol, D-arabitol, D-xylitol, and glycerol. J. Chem. Phys. 2018, 149, 054503. [Google Scholar] [CrossRef]
- Yao, X.; Huang, C.; Benson, E.G.; Shi, C.; Zhang, G.G.; Yu, L. Effect of polymers on crystallization in glass-forming molecular liquids: Equal suppression of nucleation and growth and master curve for prediction. Cryst. Growth Des. 2019, 20, 237–244. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Wu, H.; Cai, T. Effect of polymeric excipients on nucleation and crystal growth kinetics of amorphous fluconazole. Biomater. Sci 2021, 9, 4308–4316. [Google Scholar] [CrossRef]
- Shi, Q.; Cai, T. Fast crystal growth of amorphous griseofulvin: Relations between bulk and surface growth modes. Cryst. Growth Des. 2016, 16, 3279–3286. [Google Scholar] [CrossRef]
- Shi, Q.; Tao, J.; Zhang, J.; Su, Y.; Cai, T. Crack- and bubble-induced fast crystal growth of amorphous griseofulvin. Cryst. Growth Des. 2020, 20, 24–28. [Google Scholar] [CrossRef]
- Srirambhatla, V.K.; Guo, R.; Dawson, D.M.; Price, S.L.; Florence, A.J. Reversible, two-step single-crystal to single-crystal phase transitions between desloratadine forms I., II, and III. Cryst. Growth Des. 2020, 20, 1800–1810. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Li, F.; Yeh, S.; Wang, Y.; Xin, J. Physical stability of amorphous pharmaceutical solids: Nucleation, crystal growth, phase separation and effects of the polymers. Int. J. Pharm. 2020, 590, 119925. [Google Scholar] [CrossRef]
- Wang, K.; Sun, C.C. Crystal growth of celecoxib from amorphous state: Polymorphism, growth mechanism, and kinetics. Cryst. Growth Des. 2019, 19, 3592–3600. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Q.; Tao, J.; Peng, Y.; Cai, T. Impact of polymer enrichment at the crystal-liquid interface on crystallization kinetics of amorphous solid dispersions. Mol. Pharm. 2019, 16, 1385–1396. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Q.; Guo, M.; Liu, Z.; Cai, T. Melt crystallization of indomethacin polymorphs in the presence of poly(ethylene oxide): Selective enrichment of the polymer at the crystal-liquid interface. Mol. Pharm. 2020, 17, 2064–2071. [Google Scholar] [CrossRef]
- Shi, Q.; Zhang, C.; Su, Y.; Zhang, J.; Zhou, D.; Cai, T. Acceleration of crystal growth of amorphous griseofulvin by low-concentration poly(ethylene oxide): Aspects of crystallization kinetics and molecular mobility. Mol. Pharm. 2017, 14, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, J.; Zhang, C.; Jiang, J.; Tao, J.; Zhou, D.; Cai, T. Selective acceleration of crystal growth of indomethacin polymorphs by low-concentration poly(ethylene oxide). Mol. Pharm. 2017, 14, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Nucleation of one polymorph by another. J. Am. Chem. Soc. 2003, 125, 6380–6381. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xi, H.; Yu, L. Cross-nucleation between ROY polymorphs. J. Am. Chem. Soc. 2005, 127, 17439–17444. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Xu, J.; Shi, Q.; Yu, L.; Cai, T. Polymorphism of griseofulvin: Concomitant crystallization from the melt and a single crystal structure of a metastable polymorph with anomalously large thermal expansion. Chem. Commun. 2018, 54, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Li, X.; Rong, H.; Yu, L.; Lu, M. A general method for cultivating single crystals from melt microdroplets. Chem. Commun. 2020, 56, 9950–9953. [Google Scholar] [CrossRef]
- Yang, F.; Su, Y.; Zhang, J.; DiNunzio, J.; Leone, A.; Huang, C.; Brown, C.D. Rheology guided rational selection of processing temperature to prepare copovidone/nifedipine amorphous solid dispersions via hot melt extrusion (HME). Mol. Pharm. 2016, 13, 3494–3505. [Google Scholar] [CrossRef]
- Chai, Y.; Salez, T.; Mcgraw, J.D.; Benzaquen, M.; Dalnokiveress, K.; Raphaël, E.; Forrest, J.A. A direct quantitative measure of surface mobility in a glassy polymer. Science 2014, 343, 994–999. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Fujii, Y.; Lee, F.K.; Lam, C.H.; Tsui, O.K. Glass transition dynamics and surface layer mobility in unentangled polystyrene films. Science 2010, 328, 1676–1679. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Yu, L.; Cai, T. Enhanced crystal nucleation in glass-forming liquids by tensile fracture in the glassy state. Cryst. Growth Des. 2018, 19, 40–44. [Google Scholar] [CrossRef]
- Swallen, S.F.; Kearns, K.L.; Mapes, M.K.; Kim, Y.S.; Mcmahon, R.J.; Ediger, M.D.; Wu, T.; Yu, L.; Satija, S. Organic glasses with exceptional thermodynamic and kinetic stability. Science 2007, 315, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Brian, C.W.; Zhu, L.; Yu, L. Effect of bulk aging on surface diffusion of glasses. J. Chem. Phys. 2014, 140, 054509. [Google Scholar] [CrossRef] [PubMed]
- Bannow, J.; Karl, M.; Larsen, P.E.; Hwu, E.T.; Rades, T. Direct measurement of lateral molecular diffusivity on the surface of supersaturated amorphous solid dispersions by atomic force microscopy. Mol. Pharm. 2020, 17, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Brian, C.W.; Swallen, S.F.; Straus, P.T.; Ediger, M.D.; Yu, L. Surface self-diffusion of an organic glass. Phys. Rev. Lett. 2011, 106, 256103. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Ruan, S.; Cai, T.; Yu, L. Fast surface diffusion and crystallization of amorphous griseofulvin. J. Phys. Chem. B 2017, 121, 9463–9468. [Google Scholar] [CrossRef]
- Brian, C.W.; Yu, L. Surface self-diffusion of organic glasses. J. Phys. Chem. A 2013, 117, 13303–13309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Brian, C.W.; Yu, L. Fast surface diffusion of amorphous o-terphenyl and its competition with viscous flow in surface evolution. J. Phys. Chem. B 2015, 119, 5071–5078. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, W.; Yu, L. Hydrogen bonding slows down surface diffusion of molecular glasses. J. Phys. Chem. B 2016, 120, 8007–8015. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, L. Surface diffusion of polymer glasses. Macromolecules 2016, 49, 731–735. [Google Scholar] [CrossRef]
- Barták, J.; Málek, J.; Bagchi, K.; Ediger, M.D.; Li, Y.; Yu, L. Surface mobility in amorphous selenium and comparison with organic molecular glasses. J. Chem. Phys. 2021, 154, 074703. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Z.; Tylinski, M.; Ediger, M.D.; Yu, L. Effect of molecular size and hydrogen bonding on three surface-facilitated processes in molecular glasses: Surface diffusion, surface crystal growth, and formation of stable glasses by vapor deposition. J. Chem. Phys. 2019, 150, 024502. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, W.; Bishop, C.; Huang, C.; Ediger, M.D.; Yu, L. Surface diffusion in glasses of rod-like molecules posaconazole and itraconazole: Effect of interfacial molecular alignment and bulk penetration. Soft Matter 2020, 16, 5062–5070. [Google Scholar] [CrossRef]
- Mokshin, A.V.; Galimzyanov, B.N.; Yarullin, D.T. Scaling relations for temperature dependences of the surface self-diffusion coefficient in crystallized molecular glasses. JETP Lett. 2019, 110, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Paudel, A.; Geppi, M.; Mooter, G.V.D. Structural and dynamic properties of amorphous solid dispersions: The role of solid-state nuclear magnetic resonance spectroscopy and relaxometry. J. Pharm. Sci. 2014, 103, 2635–2662. [Google Scholar] [CrossRef] [PubMed]
- Thrane, L.W.; Berglund, E.A.; Wilking, J.N.; Vodak, D.; Seymour, J.D. NMR relaxometry to characterize the drug structural phase in a porous construct. Mol. Pharm. 2018, 15, 2614–2620. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Su, Y.; Zhu, L.; Brown, C.D.; Rosen, L.A.; Rosenbery, K.J. Rheological and solid-state NMR assessments of copovidone/clotrimazole model solid dispersions. Int. J. Pharm. 2016, 500, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, L.; Decressain, R.; Gusseme, A.; Neves, C.; Descamps, M. Molecular mobility in glass forming fananserine: A dielectric, NMR, and TMDSC investigation. Pharm. Res. 2006, 23, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.K.; Kamierski, S.; Potrzebowski, M.J. Fast and very fast MAS solid state NMR studies of pharmaceuticals. Annu. Rep. NMR Spectro. 2021, 103, 97–189. [Google Scholar]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating miscibility and molecular mobility of nifedipine-PVP amorphous solid dispersions using solid-state NMR spectroscopy. Mol. Pharm. 2014, 11, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Sarpal, K.; Tower, C.W.; Munson, E.J. Investigation into intermolecular interactions and phase behavior of binary and ternary amorphous solid dispersions of ketoconazole. Mol. Pharm. 2020, 17, 787–801. [Google Scholar] [CrossRef]
- Lu, X.; Huang, C.; Lowinger, M.B.; Yang, F.; Xu, W.; Brown, C.D.; Hesk, D.; Koynov, A.; Schenck, L.; Su, Y. Molecular Interactions in Posaconazole Amorphous Solid Dispersions from Two-Dimensional Solid-State NMR Spectroscopy. Mol. Pharm. 2019, 16, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, M.; Huang, C.; Lowinger, M.B.; Xu, W.; Yu, L.; Byrn, S.R.; Templeton, A.C.; Su, Y. Atomic-Level Drug Substance and Polymer Interaction in Posaconazole Amorphous Solid Dispersion from Solid-State NMR. Mol. Pharm. 2020, 17, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Huang, J.; Zhu, Q.; Haddadin, R.; Gawel, J.; Garmise, R.; Hussain, M. Is a distinctive single Tg a reliable indicator for the homogeneity of amorphous solid dispersion? Int. J. Pharm. 2010, 395, 232–235. [Google Scholar] [CrossRef]
- Paudel, A.; Van Humbeeck, J.; Van den Mooter, G. Theoretical and experimental investigation on the solid solubility and miscibility of naproxen in poly(vinylpyrrolidone). Mol. Pharm. 2010, 7, 1133–1148. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Guns, S.; Adriaensens, P.; Scholtens, B.J.; Quaedflieg, M.P.; Carleer, R.; Van den Mooter, G. Solid state solubility of miconazole in poly[(ethylene glycol)-g-vinyl alcohol] using hot-melt extrusion. Mol. Pharm. 2012, 9, 2924–2932. [Google Scholar] [CrossRef]
- Sarpal, K.; Delaney, S.; Zhang, G.G.; Munson, E.J. Phase Behavior of Amorphous Solid Dispersions of Felodipine: Homogeneity and Drug−Polymer Interactions. Mol. Pharm. 2019, 16, 4836–4851. [Google Scholar] [CrossRef]
- Sarpal, K.; Munson, E.J. Amorphous Solid Dispersions of Felodipine and Nifedipine with Soluplus®: Drug-Polymer Miscibility and Intermolecular Interactions. J. Pharm. Sci. 2021, 110, 1457–1469. [Google Scholar] [CrossRef]
- Duan, P.; Lamm, M.S.; Yang, F.; Xu, W.; Skomski, D.; Su, Y.; Schmidt-Rohr, K. Quantifying molecular mixing and heterogeneity in pharmaceutical dispersions at sub-100 nm resolution by spin diffusion NMR. Mol. Pharm. 2020, 17, 3567–3580. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, R.G.; Van Zee, N.J.; Li, Z.; Johnson, L.M.; Lodge, T.P.; Hillmyer, M.A. Recent advances in understanding the micro- and nanoscale phenomena of amorphous solid dispersions. Mol. Pharm. 2019, 16, 4089–4103. [Google Scholar] [CrossRef]
- Knapik-Kowalczuk, J.; Rams-Baron, M.; Paluch, M. Current research trends in dielectric relaxation studies of amorphous pharmaceuticals: Physical stability, tautomerism, and the role of hydrogen bonding. TrAC-Trend Anal. Chem. 2021, 134, 116097. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. Influence of molecular mobility on the physical stability of amorphous pharmaceuticals in the supercooled and glassy States. Mol. Pharm. 2014, 11, 3048–3055. [Google Scholar] [CrossRef]
- Bhardwaj, S.P.; Arora, K.K.; Kwong, E.; Templeton, A.; Clas, S.D.; Suryanarayanan, R. Mechanism of amorphous itraconazole stabilization in polymer solid dispersions: Role of molecular mobility. Mol. Pharm. 2014, 11, 4228–4237. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, S.P.; Suryanarayanan, R. Molecular mobility as an effective predictor of the physical stability of amorphous trehalose. Mol. Pharm. 2012, 9, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Dantuluri, A.K.; Amin, A.; Puri, V.; Bansal, A.K. Role of alpha-relaxation on crystallization of amorphous celecoxib above T(g) probed by dielectric spectroscopy. Mol. Pharm. 2011, 8, 814–822. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. The role of polymer concentration on the molecular mobility and physical stability of nifedipine solid dispersions. Mol. Pharm. 2015, 12, 1477–1484. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. The role of drug-polymer hydrogen bonding interactions on the molecular mobility and physical stability of nifedipine solid dispersions. Mol. Pharm. 2015, 12, 162–170. [Google Scholar] [CrossRef]
- Mohapatra, S.; Samanta, S.; Kothari, K.; Mistry, P.; Suryanarayanan, R. Effect of polymer molecular weight on the crystallization behavior of indomethacin amorphous solid dispersions. Cryst. Growth Des. 2017, 17, 3142–3150. [Google Scholar] [CrossRef]
- Mistry, P.; Suryanarayanan, R. Strength of drug-polymer interactions: Implications for crystallization in dispersions. Cryst. Growth Des. 2016, 16, 5141–5149. [Google Scholar] [CrossRef]
- Mehta, M.; Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. Effect of water on molecular mobility and physical stability of amorphous pharmaceuticals. Mol. Pharm. 2016, 13, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Suryanarayanan, R. Accelerated physical stability testing of amorphous dispersions. Mol. Pharm. 2016, 13, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Fung, M.H.; Suryanarayanan, R. Use of a plasticizer for physical atability prediction of amorphous solid dispersions. Cryst. Growth Des. 2017, 17, 4315–4325. [Google Scholar] [CrossRef]
- Madejczyk, O.; Kaminska, E.; Tarnacka, M.; Dulski, M.; Jurkiewicz, K.; Kaminski, K.; Paluch, M. Studying the crystallization of various polymorphic forms of nifedipine from binary mixtures with the use of different experimental techniques. Mol. Pharm. 2017, 14, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Suryanarayanan, R. Local mobility in amorphous pharmaceuticals-characterization and implications on stability. J. Pharm. Sci. 2009, 98, 2935–2953. [Google Scholar] [CrossRef]
- Mehta, M.; Ragoonanan, V.; McKenna, G.B.; Suryanarayanan, R. Correlation between molecular mobility and physical stability in pharmaceutical glasses. Mol. Pharm. 2016, 13, 1267–1277. [Google Scholar] [CrossRef]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Jurkiewicz, K.; Tajber, L.; Paluch, M. Molecular dynamics and physical stability of coamorphous ezetimib and indapamide mixtures. Mol. Pharm. 2015, 12, 3610–3619. [Google Scholar] [CrossRef]
- Knapik-Kowalczuk, J.; Wojnarowska, Z.; Rams-Baron, M.; Jurkiewicz, K.; Cielecka-Piontek, J.; Ngai, K.L.; Paluch, M. Atorvastatin as a promising crystallization inhibitor of amorphous probucol: Dielectric studies at ambient and elevated pressure. Mol. Pharm. 2017, 14, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Knapik-Kowalczuk, J.; Tu, W.; Chmiel, K.; Rams-Baron, M.; Paluch, M. Co-stabilization of amorphous pharmaceuticals-The case of nifedipine and nimodipine. Mol. Pharm. 2018, 15, 2455–2465. [Google Scholar] [CrossRef]
- Fung, M.H.; Berzins, K.; Suryanarayanan, R. Physical stability and dissolution behavior of ketoconazole-organic acid coamorphous systems. Mol. Pharm. 2018, 15, 1862–1869. [Google Scholar] [CrossRef]
- Fung, M.H.; DeVault, M.; Kuwata, K.T.; Suryanarayanan, R. Drug-excipient interactions: Effect on molecular mobility and physical stability of ketoconazole-organic acid coamorphous systems. Mol. Pharm. 2018, 15, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Jurkiewicz, K.; Stankiewicz, A.; Paluch, M. Stabilization of the amorphous ezetimibe drug by confining its dimension. Mol. Pharm. 2016, 13, 1308–1316. [Google Scholar] [CrossRef]
- Zhang, C.; Sha, Y.; Zhang, Y.; Cai, T.; Li, L.; Zhou, D.; Wang, X.; Xue, G. Nanostructures and dynamics of isochorically confined amorphous drug mediated by cooling rate, interfacial, and intermolecular interactions. J. Phys. Chem. B 2017, 121, 10704–10716. [Google Scholar] [CrossRef]
- Thakral, S.; Terban, M.W.; Thakral, N.K.; Suryanarayanan, R. Recent advances in the characterization of amorphous pharmaceuticals by X-ray diffractometry. Adv. Drug Deliv. Rev. 2016, 100, 183–193. [Google Scholar] [CrossRef]
- Hedoux, A. Recent developments in the Raman and infrared investigations of amorphous pharmaceuticals and protein formulations: A review. Adv. Drug Deliv. Rev. 2016, 100, 133–146. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Li, J.; Kumar, N.S.K.; Gopinath, T.; Suryanarayanan, R. A supramolecular synthon approach to design amorphous solid dispersions with exceptional physical stability. Chem. Commun. 2019, 55, 5551–5554. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Kumar, N.S.K.; Suryanarayanan, R. Crosslinking: An avenue to develop stable amorphous solid dispersion with high drug loading and tailored physical stability. J. Control Release 2019, 311–312, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Taylor, L.S. Nanoscale infrared, thermal, and mechanical characterization of telaprevir-polymer miscibility in amorphous solid dispersions prepared by solvent evaporation. Mol. Pharm. 2016, 13, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Purohit, H.S.; Taylor, L.S. Miscibility of itraconazole-hydroxypropyl methylcellulose blends: Insights with high resolution analytical methodologies. Mol. Pharm. 2015, 12, 4542–4553. [Google Scholar] [CrossRef]
- Sibik, J.; Zeitler, J.A. Direct measurement of molecular mobility and crystallisation of amorphous pharmaceuticals using terahertz spectroscopy. Adv. Drug Deliv. Rev. 2016, 100, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Yang, K.; Huang, C.; Zhu, A.; Yu, L.; Qian, F. Surface enrichment and depletion of the active ingredient in spray dried amorphous solid dispersions. Pharm. Res. 2018, 35, 38. [Google Scholar] [CrossRef]
- Rautaniemi, K.; Vuorimaa-Laukkanen, E.; Strachan, C.J.; Laaksonen, T. Crystallization kinetics of an amorphous pharmaceutical compound using fluorescence lifetime imaging microscopy. Mol. Pharm. 2018, 15, 1964–1971. [Google Scholar] [CrossRef] [Green Version]
- Bhujbal, S.V.; Zemlyanov, D.; Cavallaro, A.A.; Mangal, S.; Taylor, L.S.; Zhou, Q.T. Qualitative and quantitative characterization of composition heterogeneity on the surface of spray dried amorphous solid dispersion particles by an advanced surface analysis platform with high surface-sensitivity and superior spatial resolution. Mol. Pharm. 2018, 15, 2045–2053. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, Y.; Cheng, J.; Chen, H.; Xu, J.; Liu, Z.; Shi, Q.; Zhang, C. Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids. Crystals 2021, 11, 1440. https://doi.org/10.3390/cryst11121440
Wang Y, Wang Y, Cheng J, Chen H, Xu J, Liu Z, Shi Q, Zhang C. Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids. Crystals. 2021; 11(12):1440. https://doi.org/10.3390/cryst11121440
Chicago/Turabian StyleWang, Yanan, Yong Wang, Jin Cheng, Haibiao Chen, Jia Xu, Ziying Liu, Qin Shi, and Chen Zhang. 2021. "Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids" Crystals 11, no. 12: 1440. https://doi.org/10.3390/cryst11121440
APA StyleWang, Y., Wang, Y., Cheng, J., Chen, H., Xu, J., Liu, Z., Shi, Q., & Zhang, C. (2021). Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids. Crystals, 11(12), 1440. https://doi.org/10.3390/cryst11121440