
Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis


Abstract
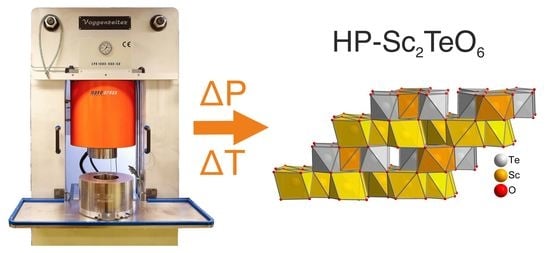
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
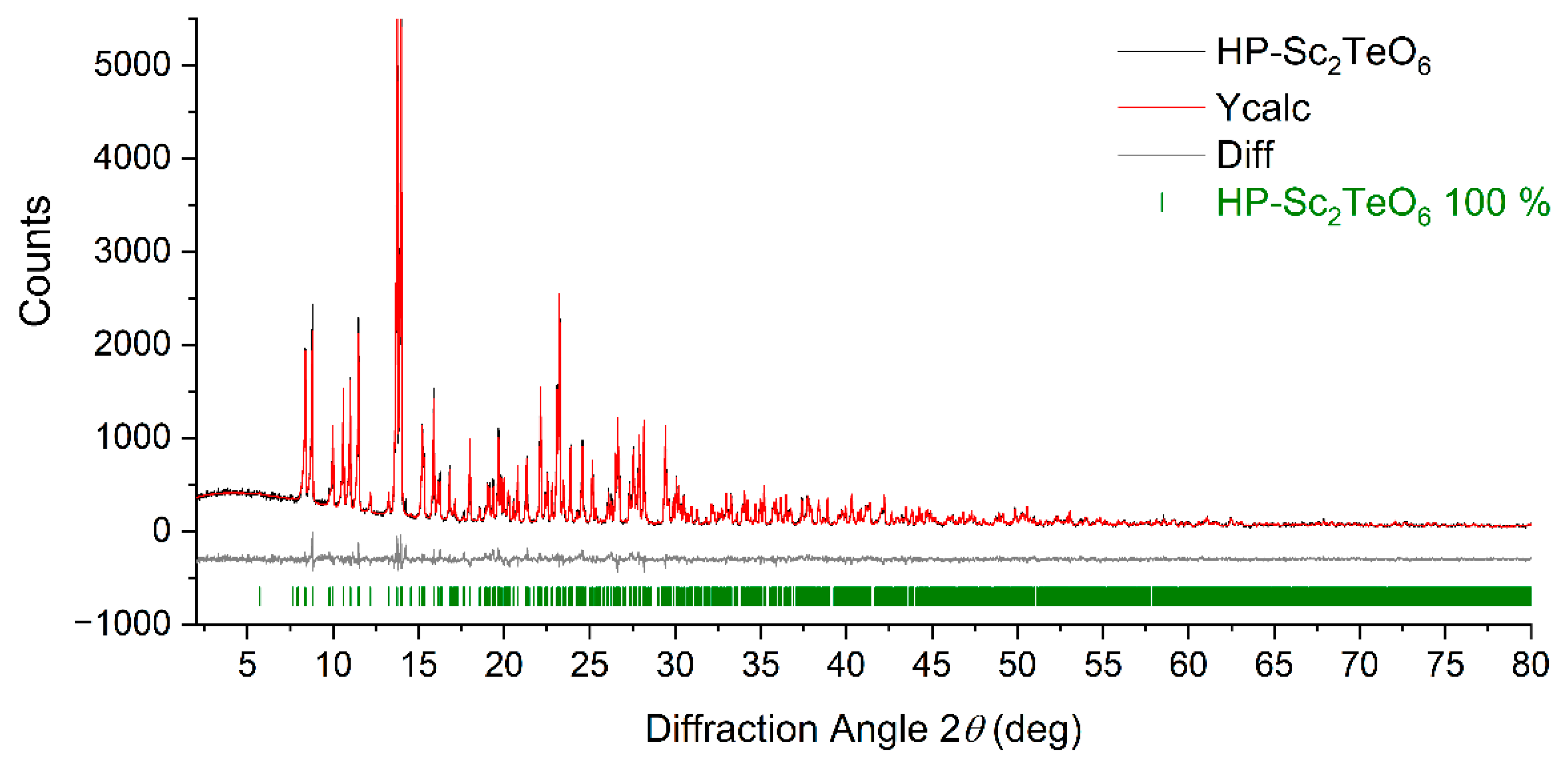
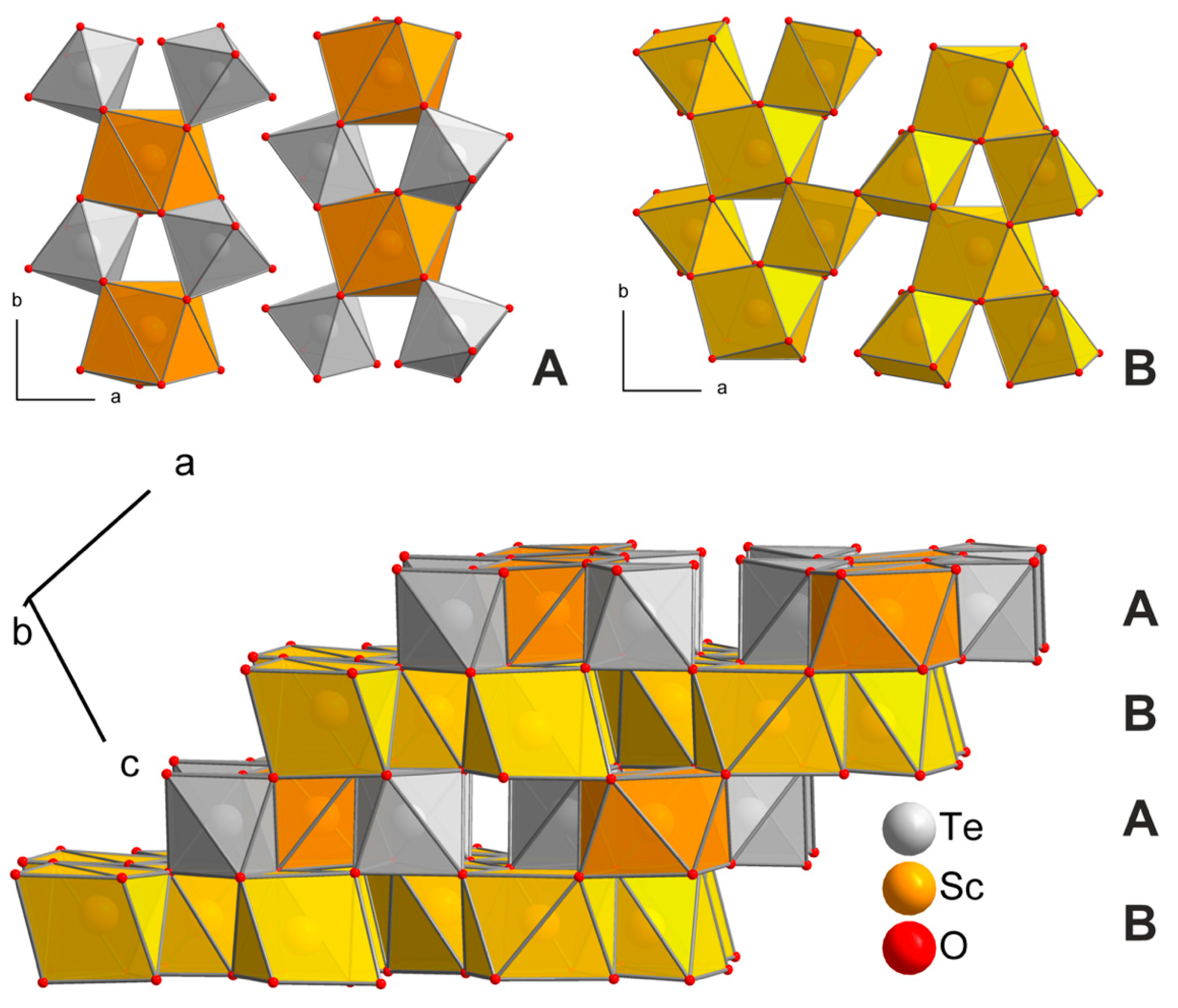
3.1. Structure Refinements
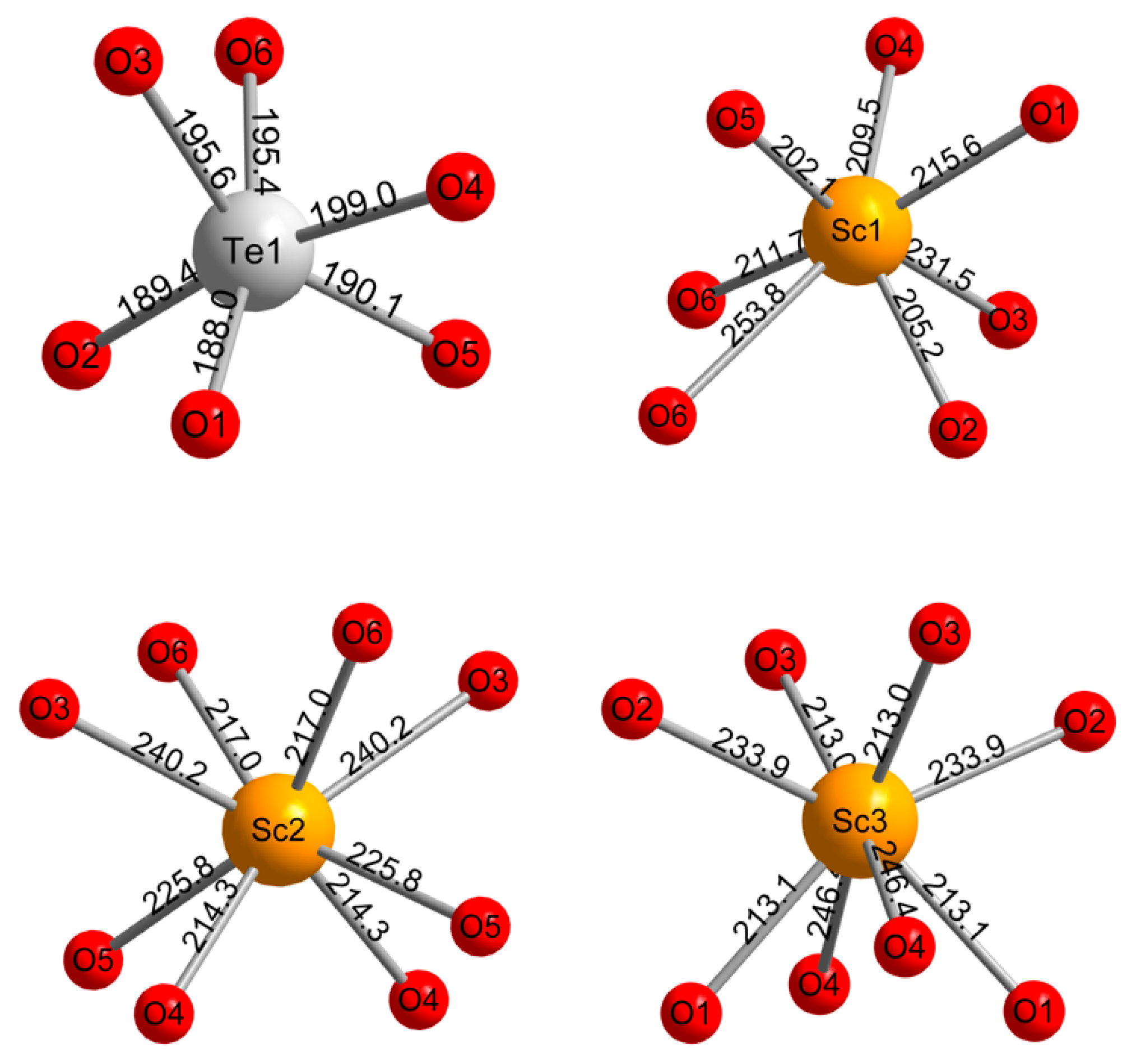
3.2. Crystal Chemistry
3.3. Theoretical Calculations
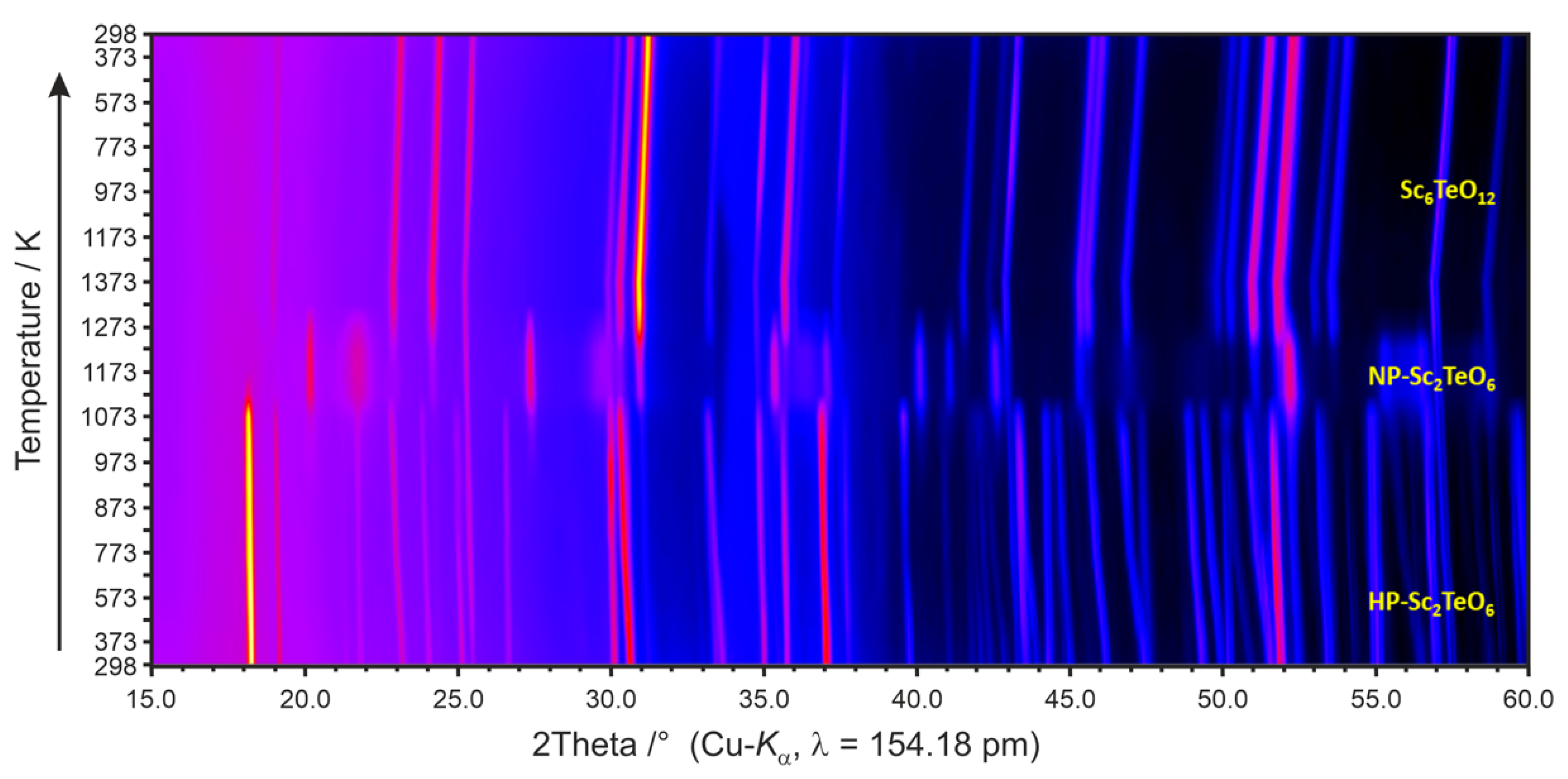
3.4. Temperature-Dependent X-ray Powder Diffractometry
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lass, J.; Andersen, C.R.; Leerberg, H.K.; Birkemose, S.; Toth, S.; Stuhr, U.; Bartkowiak, M.; Niedermayer, C.; Lu, Z.; Toft-Petersen, R.; et al. Field-induced magnetic incommensurability in multiferroic Ni3TeO6. Phys. Rev. B Condens. Matter 2020, 101, 054415. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Wang, C.-W.; Zhao, Y.; Li, W.-H.; Lynn, J.W.; Harris, A.B.; Rule, K.; Yang, H.-D.; Berger, H. Complex magnetic incommensurability and electronic charge transfer through the ferroelectric transition in multiferroic Co3TeO6. Sci. Rep. 2017, 7, 6437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selb, E.; Buttlar, T.; Janka, O.; Tribus, M.; Ebbinghaus, S.G.; Heymann, G. Multianvil high-pressure/high-temperature synthesis and characterization of magnetoelectric HP-Co3TeO6. J. Mater. Chem. C 2021, 9, 5486–5496. [Google Scholar] [CrossRef]
- Liu, L.; Skogby, H.; Ivanov, S.; Weil, M.; Mathieu, R.; Lazor, P. Bandgap engineering in Mn3TeO6: Giant irreversible bandgap reduction triggered by pressure. Chem. Commun. 2019, 55, 12000–12003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, R.; Ivanov, S.A.; Nordblad, P.; Weil, M. Enhancement of antiferromagnetic interaction and transition temperature in M3TeO6 systems (M = Mn, Co, Ni, Cu). Eur. Phys. J. B 2013, 86, 361. [Google Scholar] [CrossRef] [Green Version]
- Solana-Madruga, E.; Aguilar-Maldonado, C.; Ritter, C.; Huvé, M.; Mentré, O.; Attfield, J.P.; Arévalo-López, Á.M. Complex magnetism in Ni3TeO6-type Co3TeO6 and high-pressure polymorphs of Mn3−xCoxTeO6 solid solutions. Chem. Commun. 2021, 57, 2511–2514. [Google Scholar] [CrossRef]
- Arévalo-López, Á.M.; Solana-Madruga, E.; Aguilar-Maldonado, C.; Ritter, C.; Mentré, O.; Attfield, J.P. Magnetic frustration in the high-pressure Mn2MnTeO6 (Mn3TeO6-II) double perovskite. Chem. Commun. 2019, 55, 14470–14473. [Google Scholar] [CrossRef]
- Kim, J.W.; Artyukhin, S.; Mun, E.D.; Jaime, M.; Harrison, N.; Hansen, A.; Yang, J.J.; Oh, Y.S.; Vanderbilt, D.; Zapf, V.S.; et al. Successive Magnetic-Field-Induced Transitions and Colossal Magnetoelectric Effect in Ni3TeO6. Phys. Rev. Lett. 2015, 115, 137201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ra, H.-S.; Ok, K.M.; Halasyamani, P.S. Combining Second-Order Jahn−Teller Distorted Cations to Create Highly Efficient SHG Materials: Synthesis, Characterization, and NLO Properties of BaTeM2O9 (M = Mo6+ or W6+). J. Am. Chem. Soc. 2003, 125, 7764–7765. [Google Scholar] [CrossRef] [PubMed]
- Halasyamani, P.S.; Zhang, W. Viewpoint: Inorganic Materials for UV and Deep-UV Nonlinear-Optical Applications. Inorg. Chem. 2017, 56, 12077–12085. [Google Scholar] [CrossRef] [PubMed]
- Höss, P.; Schleid, T. Sc2Te5O13 und Sc2TeO6: Die ersten Oxotellurate des Scandiums. Z. Anorg. Allg. Chem. 2007, 633, 1391–1396. [Google Scholar] [CrossRef]
- Meier, S.F.; Schleid, T. Oxotellurate(IV) der Lanthanide: II. Die isotype Reihe M2Te5O13 (M = Dy − Lu) / Oxotellurates(IV) of Lanthanides: II. The Isotypic Series M2Te5O13 (M = Dy − Lu). Z. Für. Nat. B Chem. Sci. 2005, 60, 720–726. [Google Scholar] [CrossRef]
- Höss, P.; Osvet, A.; Meister, F.; Batentschuk, M.; Winnacker, A.; Schleid, T. Synthesis, crystal structures and luminescence properties of the Eu3+-doped yttrium oxotellurates(IV) Y2Te4O11 and Y2Te5O13. J. Solid State Chem. 2008, 181, 2783–2788. [Google Scholar] [CrossRef]
- Song, S.Y.; Lee, D.W.; Ok, K.M. Rich Structural Chemistry in Scandium Selenium/Tellurium Oxides: Mixed-Valent Selenite–Selenates, Sc2(SeO3)2(SeO4) and Sc2(TeO3)(SeO3)(SeO4), and Ternary Tellurite, Sc2(TeO3)3. Inorg. Chem. 2014, 53, 7040–7046. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Xu, X.; Mao, J.-G. A Series of New Ternary and Quaternary Compounds in the LiI–GaIII–TeIV–O System. Inorg. Chem. 2010, 49, 11573–11580. [Google Scholar] [CrossRef] [PubMed]
- Höss, P.; Meier, S.F.; Schleid, T. Lu2Te3O9: The First Example of the Triclinic C-Type Lanthanoid(III) Oxotellurates(IV) with the Composition M2Te3O9. Z. Anorg. Allg. Chem. 2013, 639, 2548–2553. [Google Scholar] [CrossRef]
- Meier, S.F.; Höss, P.; Schleid, T. Dy2Te3O9: Der erste Vertreter von Lanthanoid(III)-Oxotelluraten der Zusammensetzung M2Te3O9. Z. Anorg. Allg. Chem. 2009, 635, 768–775. [Google Scholar] [CrossRef]
- Gates-Rector, S.; Blanton, T. The Powder Diffraction File: A quality materials characterization database. Powder Diffr. 2019, 34, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Blasse, G. Lanthanide tellurates Ln6TeO12. J. Inorg. Nucl. Chem. 1969, 31, 3335–3336. [Google Scholar] [CrossRef]
- Walker, D. Lubrication, gasketing, and precision in multianvil experiments. Am. Mineral. 1991, 76, 1092–1100. [Google Scholar]
- Walker, D.; Carpenter, M.A.; Hitch, C.M. Some simplifications to multianvil devices for high pressure experiments. Am. Mineral. 1990, 75, 1020–1028. [Google Scholar]
- Huppertz, H.; Heymann, G.; Schwarz, U.; Schwarz, M.R. High-Pressure Methods in Solid-State Chemistry. In Handbook of Solid State Chemistry; Dronskowski, R., Kikkawa, S., Stein, A., Eds.; Wiley-VCH: Weinheim, Germany, 2017; Volume 2, pp. 23–48. [Google Scholar]
- Huppertz, H. Multianvil high-pressure/high-temperature synthesis in solid state chemistry. Z. Kristallogr. 2004, 219, 330–338. [Google Scholar] [CrossRef]
- Thompson, P.; Cox, D.E.; Hastings, J.B. Rietveld refinement of Debye-Scherrer synchrotron X-ray data from Al2O3. J. Appl. Crystallogr. 1987, 20, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Young, R.A.; Desai, P. Crystallite Size and Microstrain Indicators in Rietveld Refinement. Arch. Nauki. Mater. 1989, 10, 71–90. [Google Scholar]
- APEX3 (v. 2017.3-0), CELL_NOW (v. 2008/4), SAINT (v. 8.38A), TWINABS (v. 2012/1), and SADABS (v. 2016/2); Bruker AXS GmbH: Karlsruhe, Germany, 2016.
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. ShelXL—Crystal Structure Refinement—Multi-CPU Version; 2017/1; University of Göttingen: Göttingen, Germany, 2017. [Google Scholar]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Le Page, Y. MISSYM1.1—A flexible new release. J. Appl. Crystallogr. 1988, 21, 983–984. [Google Scholar] [CrossRef]
- Spek, A. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Efremov, V.A.; Tyulin, A.V.; Trunov, V.K.; Kudin, O.V.; Yanovskij, V.K.; Voronkova, V.I. Crystal structure of monoclinic Y2WO6 and Yb2WO6. Kristallografiya 1984, 28, 904–909. [Google Scholar]
- Becker, R.; Johnsson, M.; Berger, H. A new synthetic cobalt tellurate: Co3TeO6. Acta Crystallogr. C 2006, 62, i67–i69. [Google Scholar] [CrossRef]
- Geller, S.; Romo, P.; Remeika, J.P. Refinement of the structure of scandium sesquioxide. Z. Kristallogr. 1967, 124, 136–142. [Google Scholar] [CrossRef]
- Prewitt, C.T.; Downs, R.T. High-pressure crystal chemistry. Rev. Mineral. Geochem. 1998, 37, 283–317. [Google Scholar]
- Hoppe, R.; Voigt, S.; Glaum, H.; Kissel, J.; Müller, H.P.; Bernet, K. A new route to charge distributions in ionic solids. J. Less-Common Met. 1989, 156, 105–122. [Google Scholar] [CrossRef]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. B 1985, 41, 244–247. [Google Scholar] [CrossRef] [Green Version]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. B 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Hübenthal, R. MAPLE; v.4; Universität Gießen: Gießen, Germany, 1993. [Google Scholar]
- Nespolo, M.; Guillot, B. CHARDI2015: Charge distribution analysis of non-molecular structures. J. Appl. Crystallogr. 2016, 49, 317–321. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | HP-Sc2TeO6 |
| Molar mass, g∙mol−1 | 313.52 |
| Crystal system | monoclinic |
| Space group | P2/c |
| Cell formula units | 4 |
| Powder diffractometer | STOE STADI P |
| Radiation | Mo-Kα1 (λ = 70.93 pm) |
| Powder data: | |
| a, pm | 729.224(7) |
| b, pm | 512.576(5) |
| c, pm | 1095.04(2) |
| β, deg | 103.898(1) |
| V, Å3 | 397.323(7) |
| Single-crystal diffractometer | Bruker D8 Quest |
| Radiation | Mo-Kα (λ = 71.073 pm) |
| Single-crystal data: | |
| a, pm | 729.43(3) |
| b, pm | 512.52(2) |
| c, pm | 1095.02(4) |
| β, deg | 103.88(1) |
| V, Å3 | 397.42(3) |
| Calculated density, g∙cm−3 | 5.24 |
| Crystal size, mm3 | 0.02 × 0.02 × 0.02 |
| Temperature, K | 299(2) |
| Absorption coefficient, mm−1 | 10.54 |
| F(000), e | 568 |
| Detector distance, mm | 34 |
| θ range, deg | 2.88–39.42 |
| Range in hkl | −12 ≤ h ≤ 13, ±9, ±19 |
| Total no. reflections | 13924 |
| Data/ref. parameters | 2379/84 |
| Reflections with I > 2σ(I) | 2077 |
| Rint/Rσ | 0.0363, 0.0254 |
| Goodness-of-fit on F2 | 1.055 |
| Absorption correction | multi-scan |
| R1/wR2 for I > 2σ(I) | 0.0191/0.0331 |
| R1/wR2 for all data | 0.0261/0.0344 |
| Extinction coefficient | 0.0026(2) |
| Transmission max./min. | 0.748/0.646 |
| Largest diff. peak/hole/e∙Å−3 | 0.905/−0.962 |
| Atom | Wyck. | x | y | z | SOF | Ueq |
|---|---|---|---|---|---|---|
| Te1 | 4g | 0.22848(2) | 0.26270(2) | 0.61484(2) | 1 | 0.00223(3) |
| Sc1 | 4g | 0.71575(4) | 0.19306(6) | 0.58249(3) | 1 | 0.00365(5) |
| Sc2 | 2f | 1/2 | 0.24868(8) | 1/4 | 1 | 0.00377(6) |
| Sc3 | 2e | 0 | 0.30665(8) | 1/4 | 1 | 0.00442(7) |
| O1 | 4g | 0.0135(2) | 0.6152(2) | 0.3823(2) | 1 | 0.0050(2) |
| O2 | 4g | 0.1418(2) | 0.1363(2) | 0.4484(1) | 1 | 0.0056(2) |
| O3 | 4g | 0.2041(2) | 0.0254(2) | 0.2271(1) | 1 | 0.0044(2) |
| O4 | 4g | 0.3021(2) | 0.5402(2) | 0.2758(1) | 1 | 0.0046(2) |
| O5 | 4g | 0.3462(2) | 0.4355(2) | 0.0674(1) | 1 | 0.0054(2) |
| O6 | 4g | 0.4541(2) | 0.0606(2) | 0.6107(2) | 1 | 0.0050(2) |
| Atom | U11 | U22 | U33 | U23 | U13 | U12 |
|---|---|---|---|---|---|---|
| Te1 | 0.00242(4) | 0.00213(4) | 0.00227(4) | −0.00001(3) | 0.00082(3) | 0.00006(3) |
| Sc1 | 0.0040(1) | 0.0037(1) | 0.0036(1) | 0.00032(8) | 0.00160(8) | 0.00024(8) |
| Sc2 | 0.0041(1) | 0.0030(2) | 0.0041(1) | 0 | 0.0006(1) | 0 |
| Sc3 | 0.0047(2) | 0.0034(2) | 0.0055(2) | 0 | 0.0019(1) | 0 |
| O1 | 0.0031(4) | 0.0062(5) | 0.0060(4) | −0.0009(4) | 0.0017(4) | 0.0006(4) |
| O2 | 0.0076(5) | 0.0054(5) | 0.0030(4) | −0.0018(3) | −0.0004(4) | 0.0007(4) |
| O3 | 0.0059(4) | 0.0031(4) | 0.0043(4) | −0.0013(3) | 0.0013(4) | 0.0006(4) |
| O4 | 0.0068(5) | 0.0043(4) | 0.0027(4) | 0.0010(3) | 0.0012(4) | 0.0017(4) |
| O5 | 0.0061(5) | 0.0049(5) | 0.0051(4) | −0.0013(4) | 0.0010(4) | 0.0015(4) |
| O6 | 0.0029(4) | 0.0044(5) | 0.0081(5) | 0.0013(4) | 0.0023(4) | 0.0014(3) |
| Te1 | Sc1 | Sc2 | Sc3 | O1 | O2 | O3 | O4 | O5 | O6 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ΣV | +5.83 | +3.08 | +2.86 | +2.78 | −2.01 | −1.91 | −1.72 | −1.98 | −2.00 | −1.96 |
| ΣQ | +5.99 | +2.99 | +3.03 | +3.01 | −2.12 | −1.98 | −1.89 | −1.99 | −2.08 | −1.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziegler, R.; Tribus, M.; Hejny, C.; Heymann, G. Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis. Crystals 2021, 11, 1554. https://doi.org/10.3390/cryst11121554
Ziegler R, Tribus M, Hejny C, Heymann G. Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis. Crystals. 2021; 11(12):1554. https://doi.org/10.3390/cryst11121554
Chicago/Turabian StyleZiegler, Raimund, Martina Tribus, Clivia Hejny, and Gunter Heymann. 2021. "Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis" Crystals 11, no. 12: 1554. https://doi.org/10.3390/cryst11121554
APA StyleZiegler, R., Tribus, M., Hejny, C., & Heymann, G. (2021). Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis. Crystals, 11(12), 1554. https://doi.org/10.3390/cryst11121554