
Molecular Mechanism Study on Stereo-Selectivity of α or β Hydroxysteroid Dehydrogenases




Abstract
:1. Introduction
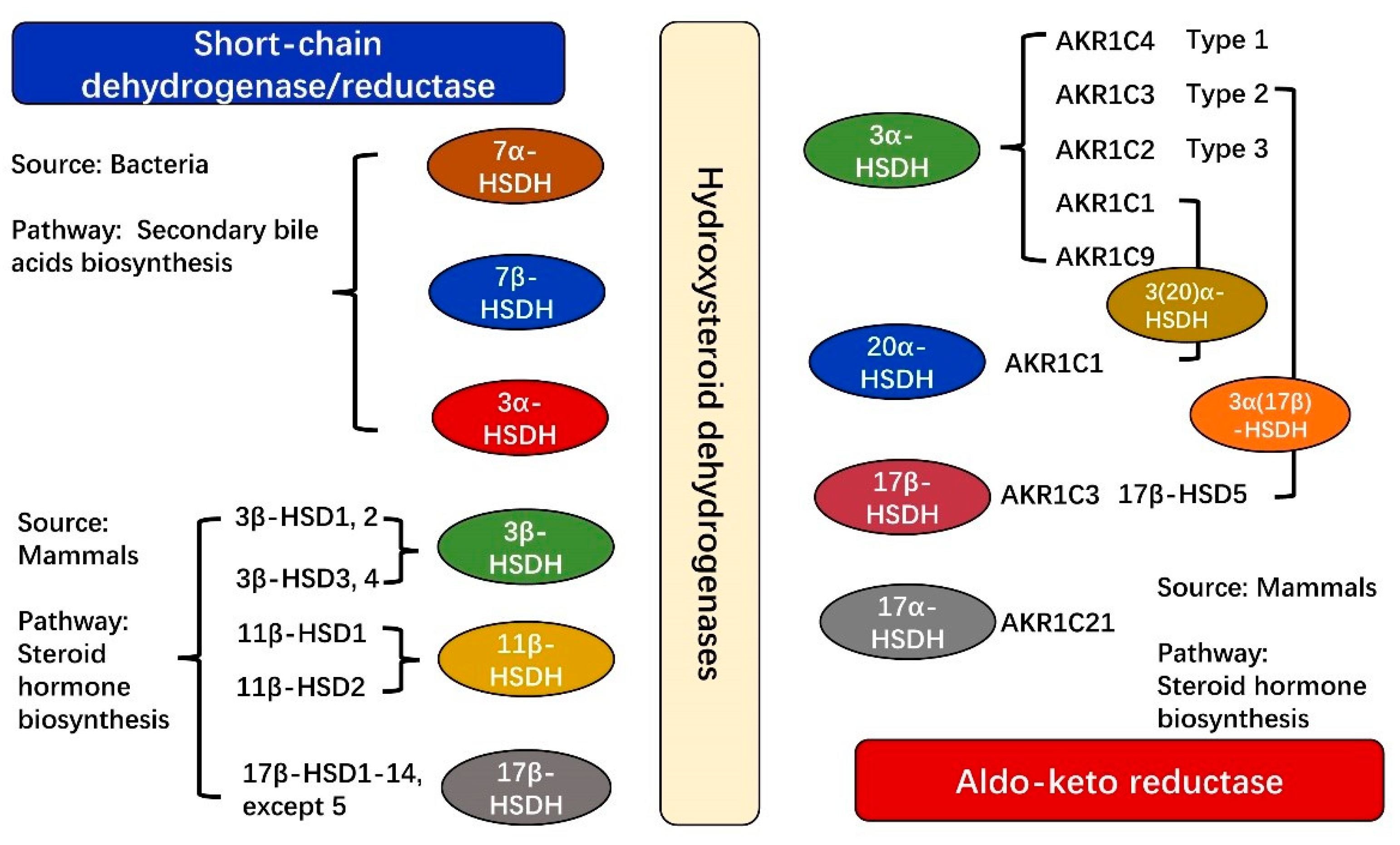
2. Classification and Functions of HSDHs
2.1. Characteristics and Functions of HSDHs from SDRs
2.1.1. HSDHs in Primary or Secondary Bile Acid Biosynthesis
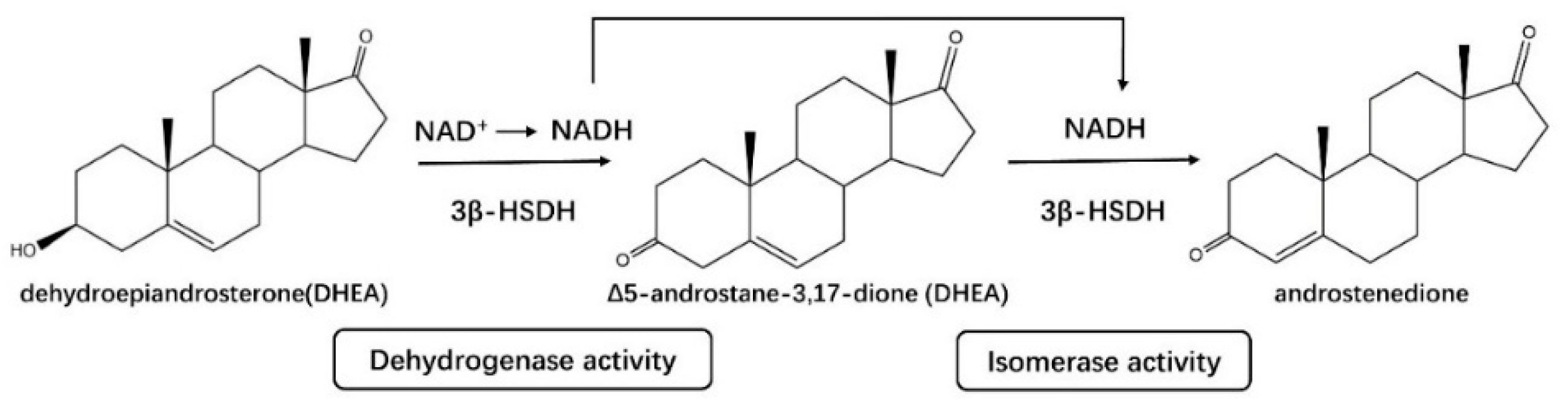
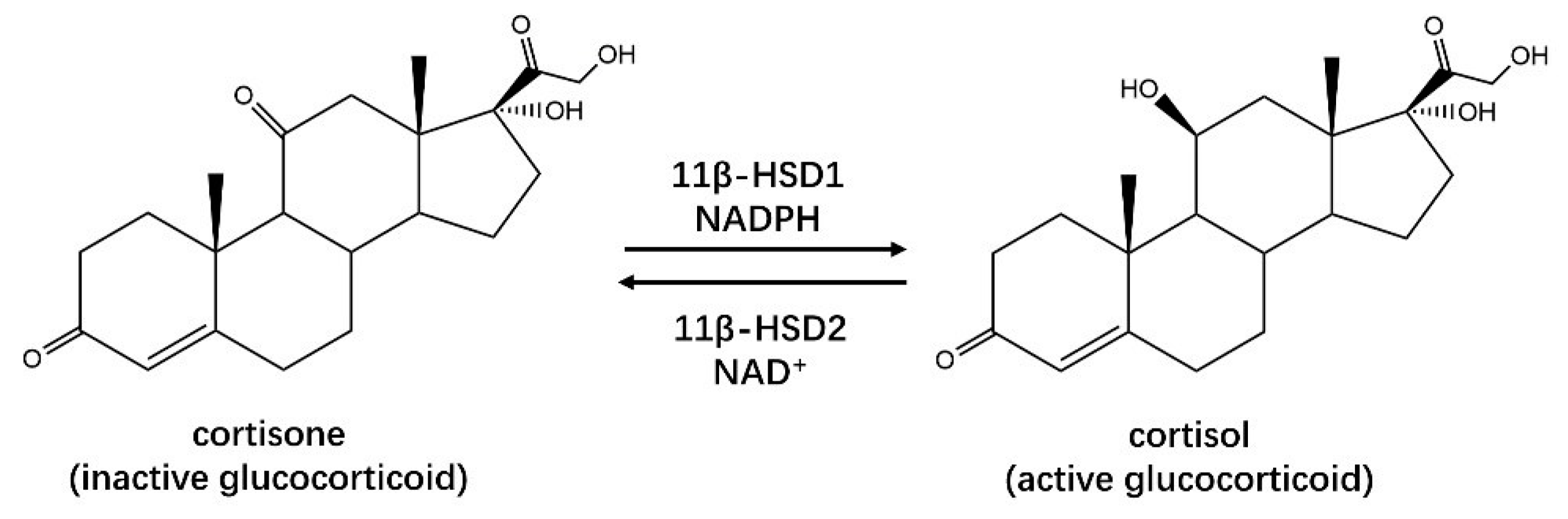
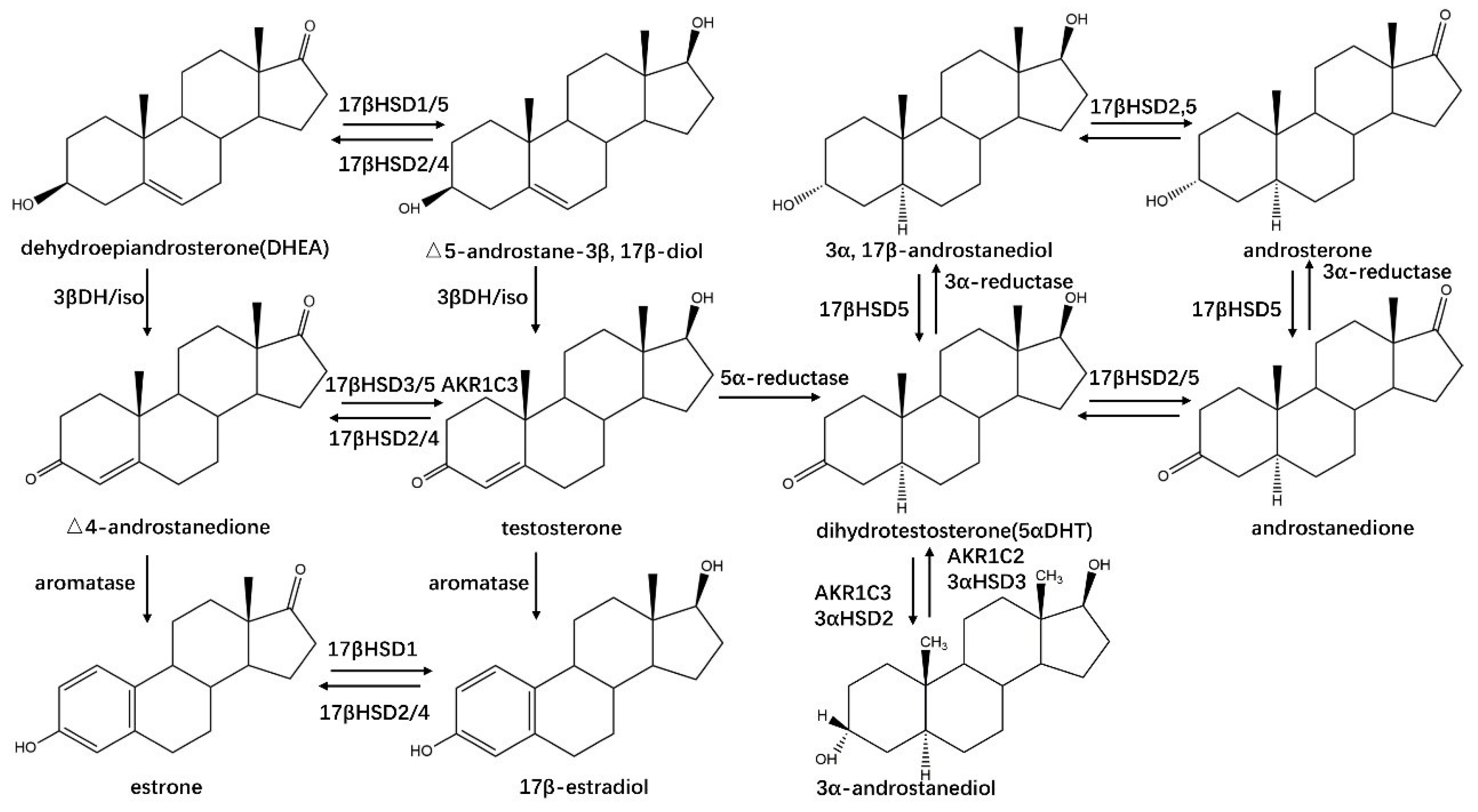
2.1.2. HSDHs in Steroid Hormone Biosynthesis
2.2. Characteristics and Functions of HSDHs from AKRs
3. Coenzyme Binding Modes of HSDHs
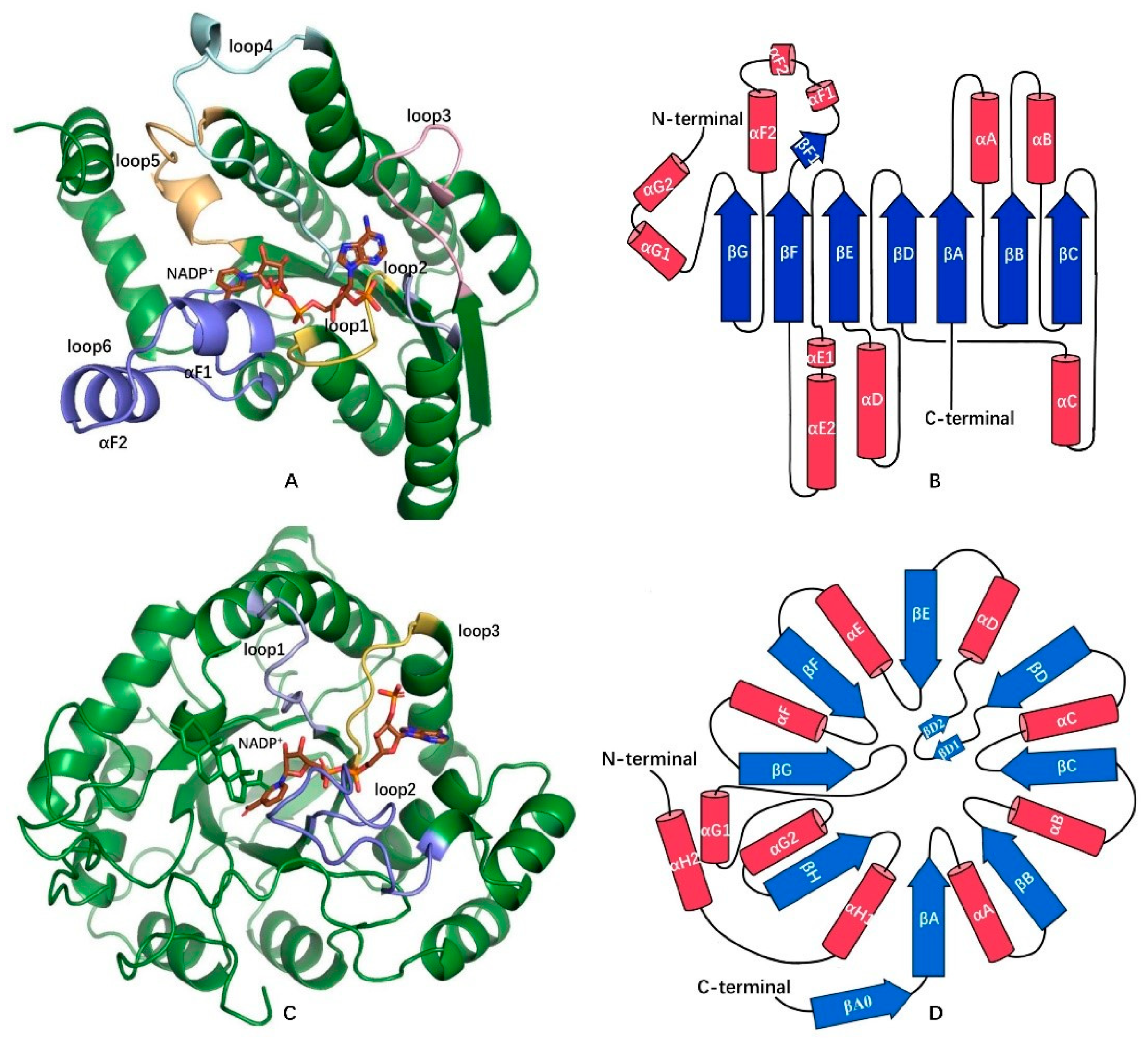
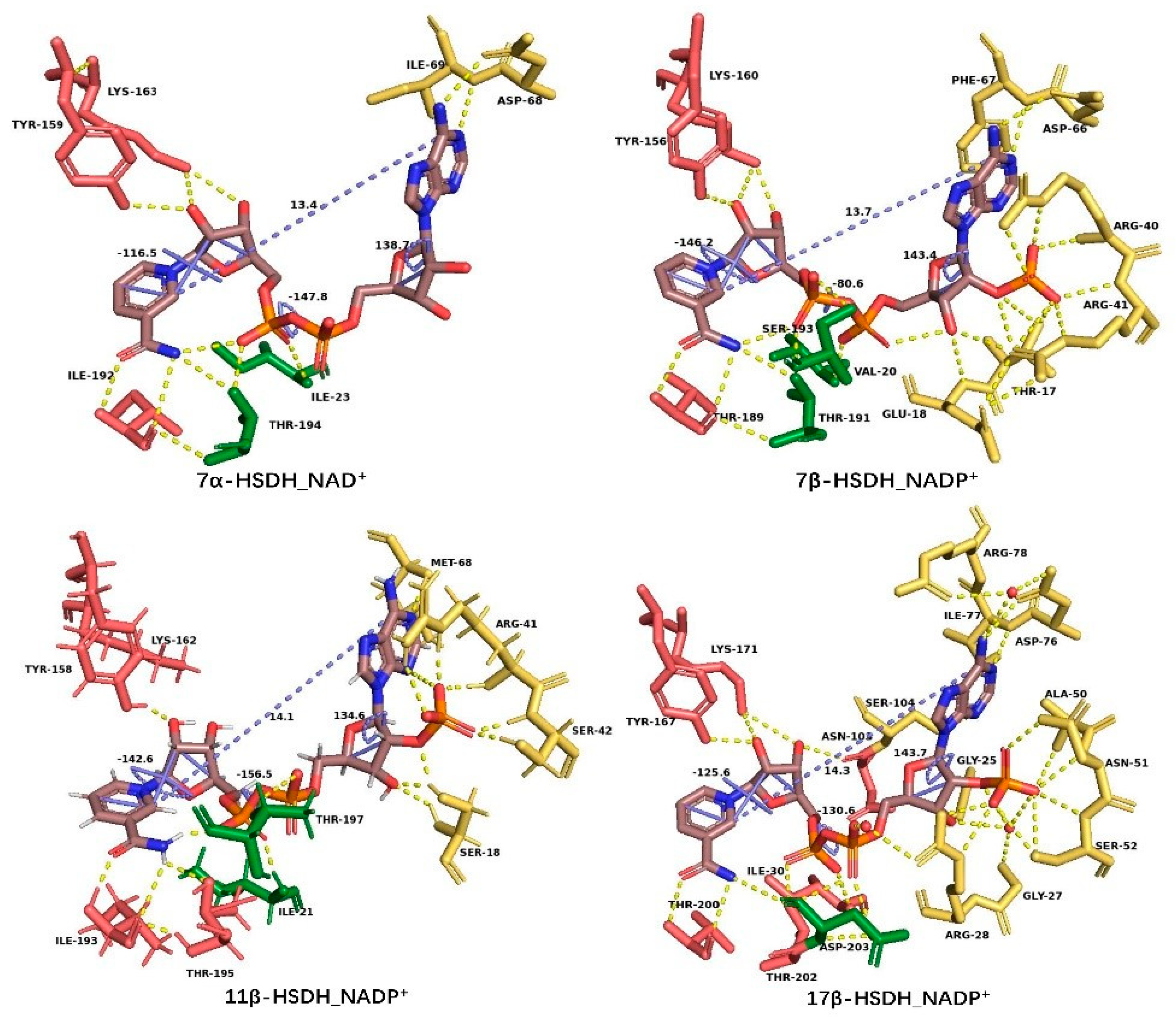
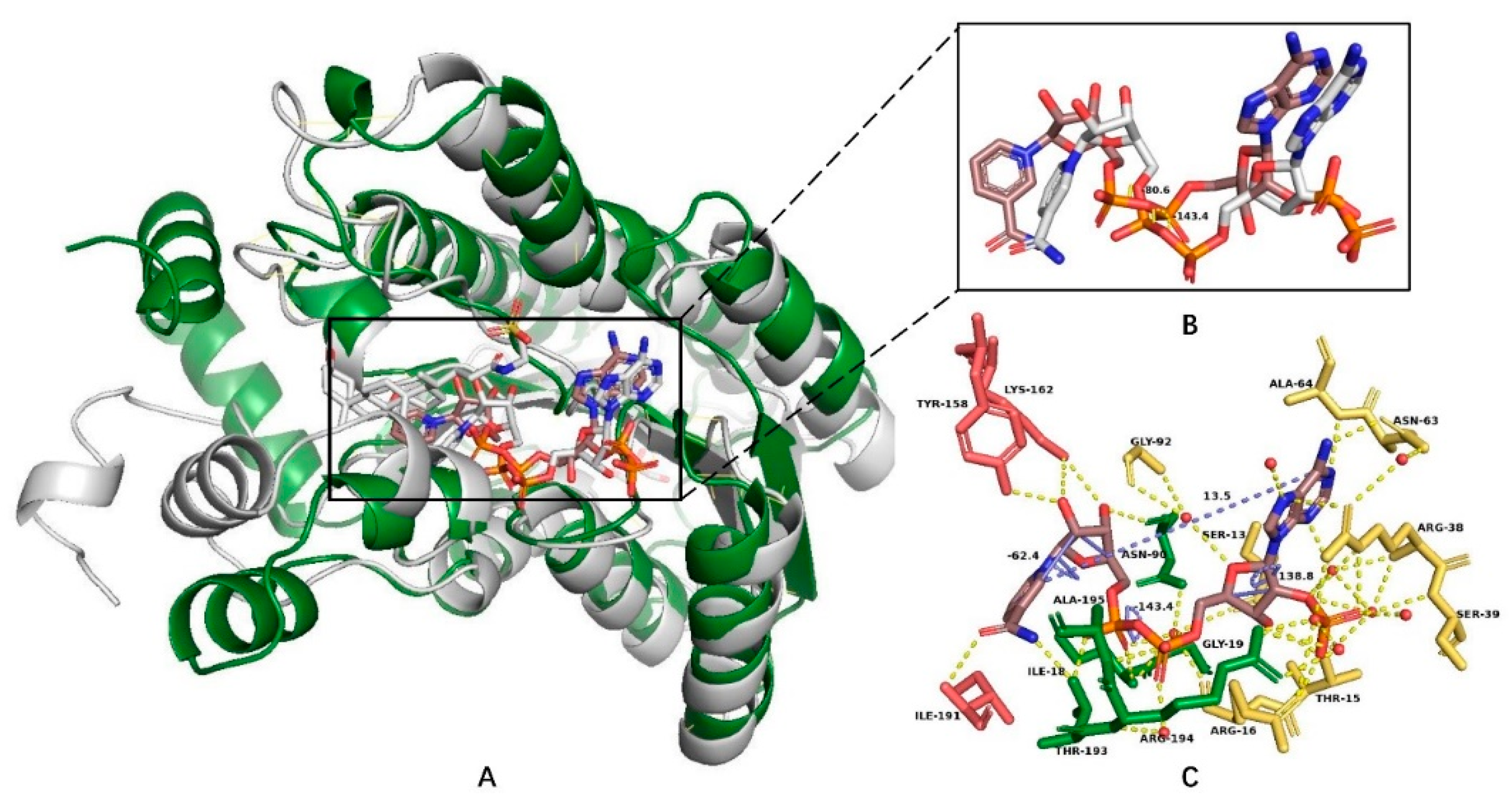
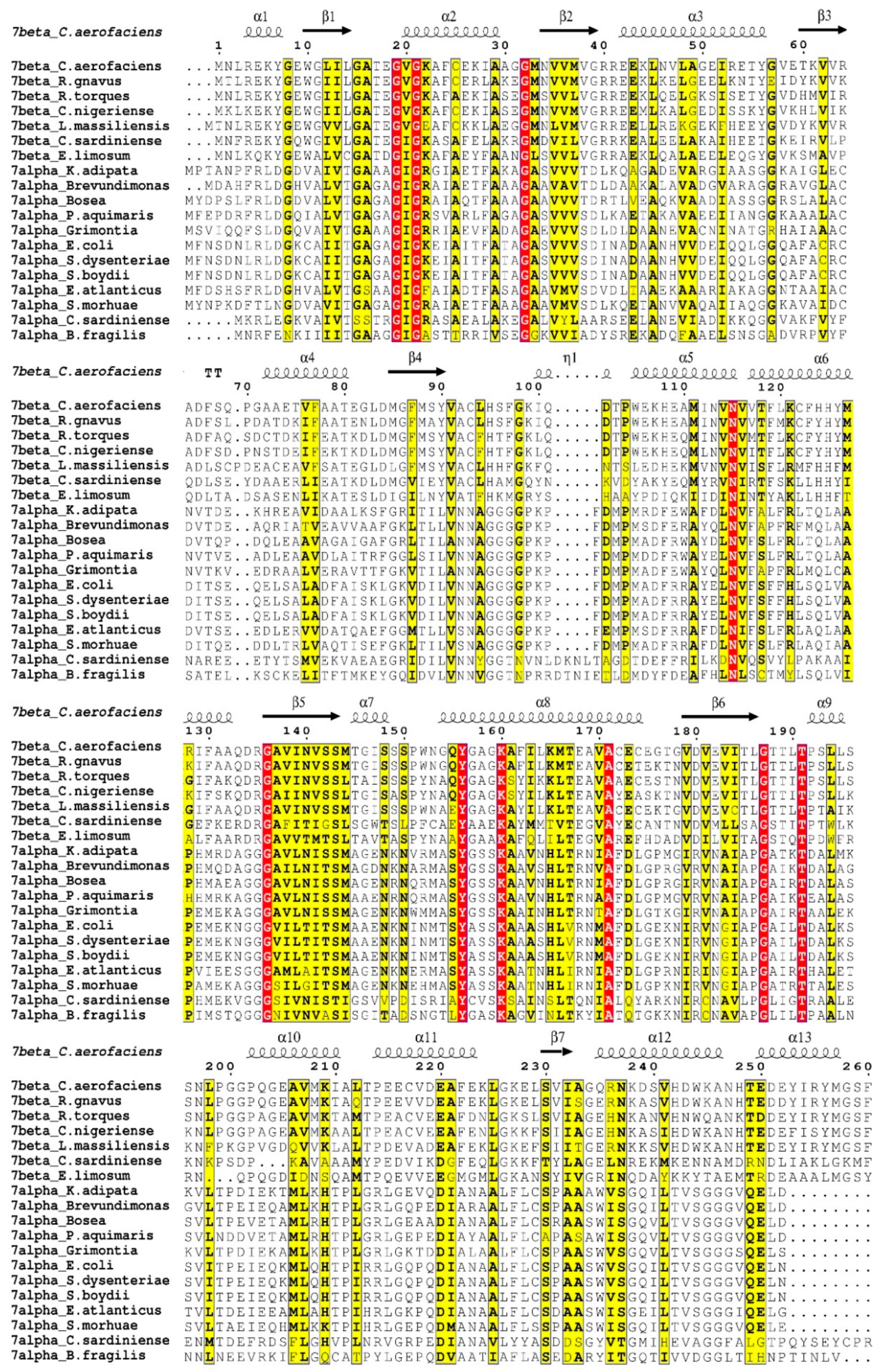
3.1. Coenzyme Binding Modes of the HSDHs from SDRs
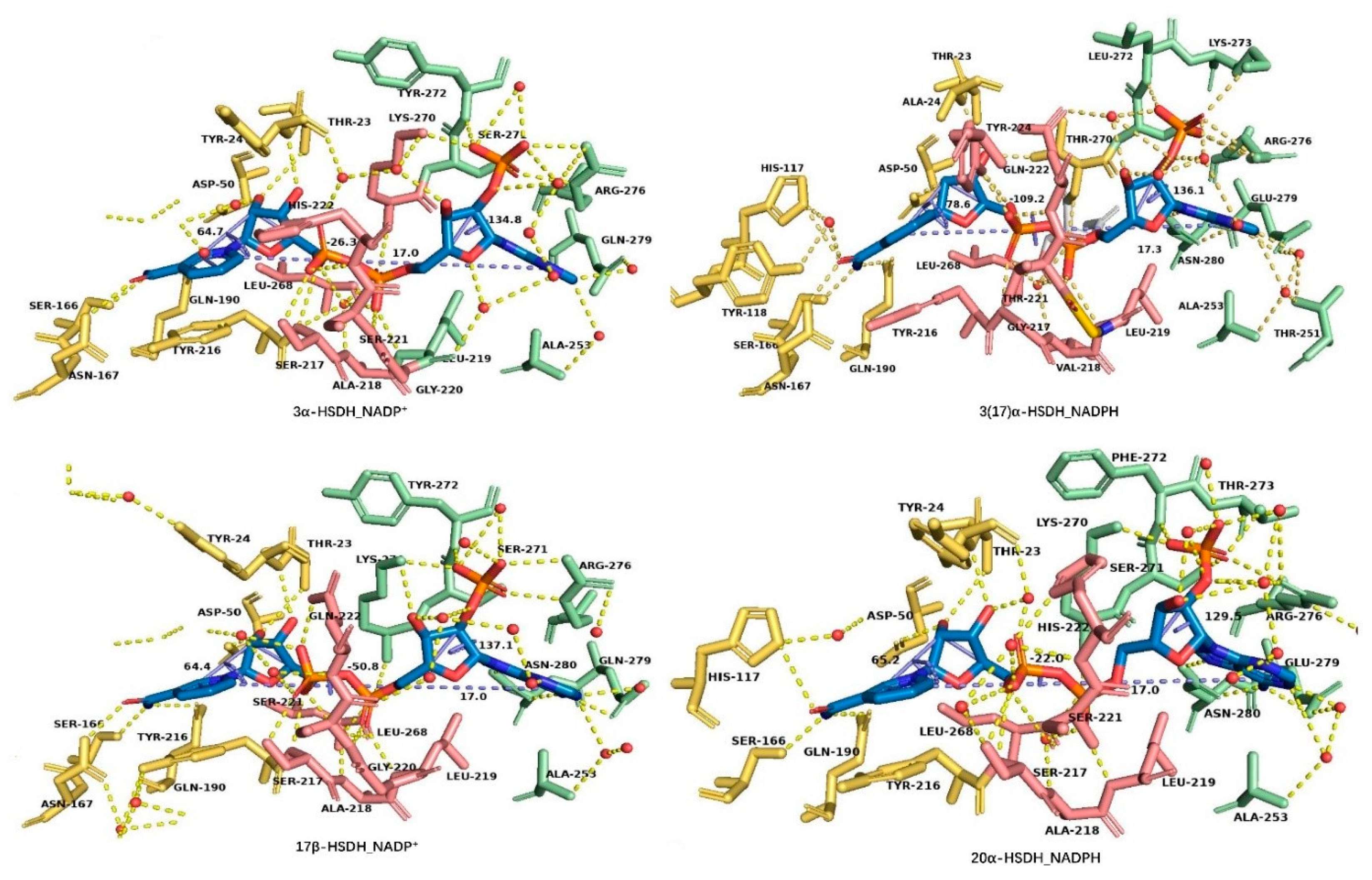
3.2. NAD(H)/NADP(H) Binding Modes of the HSDHs from AKRs
4. Catalytic Mechanism of HSDHs
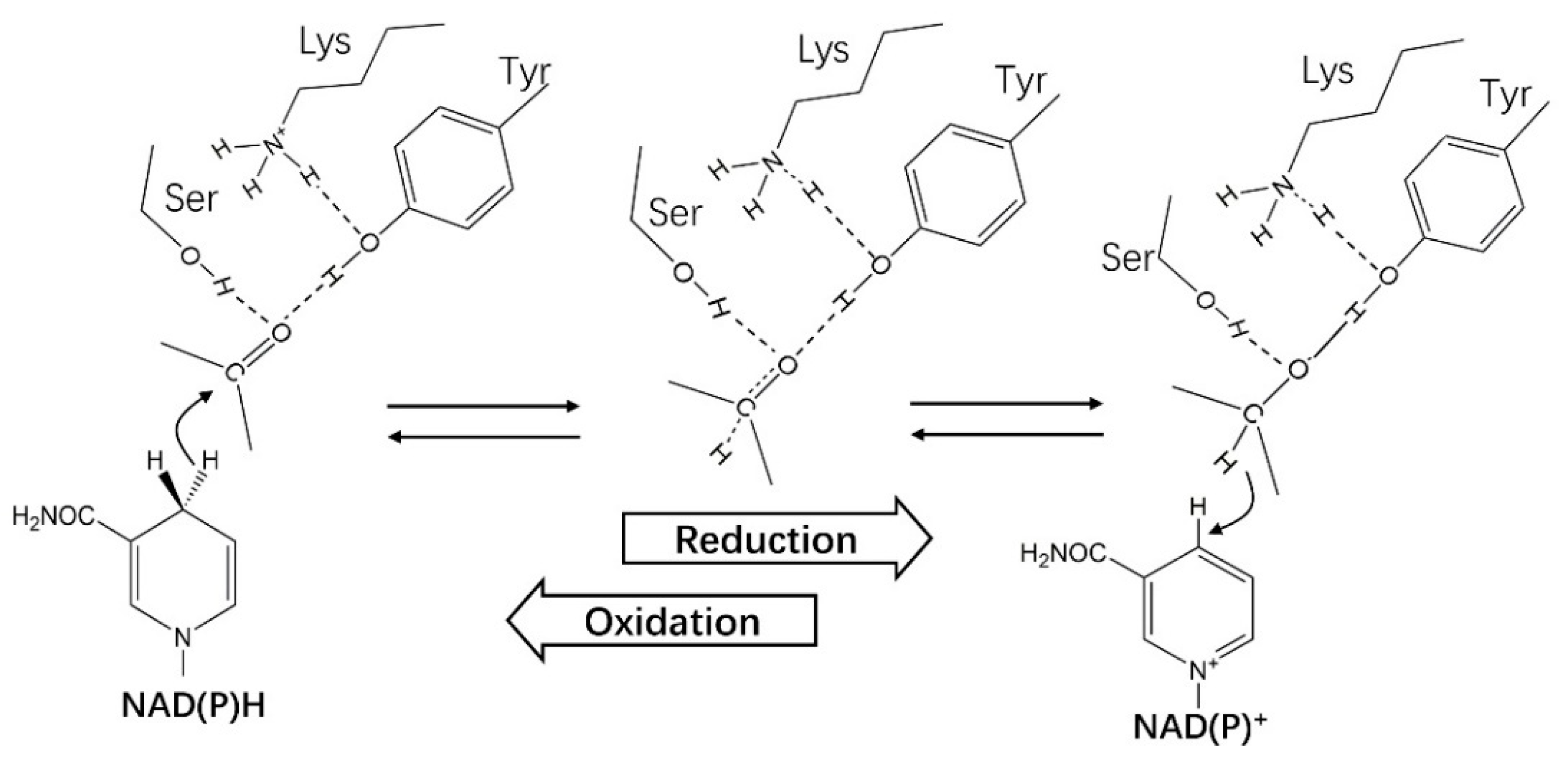
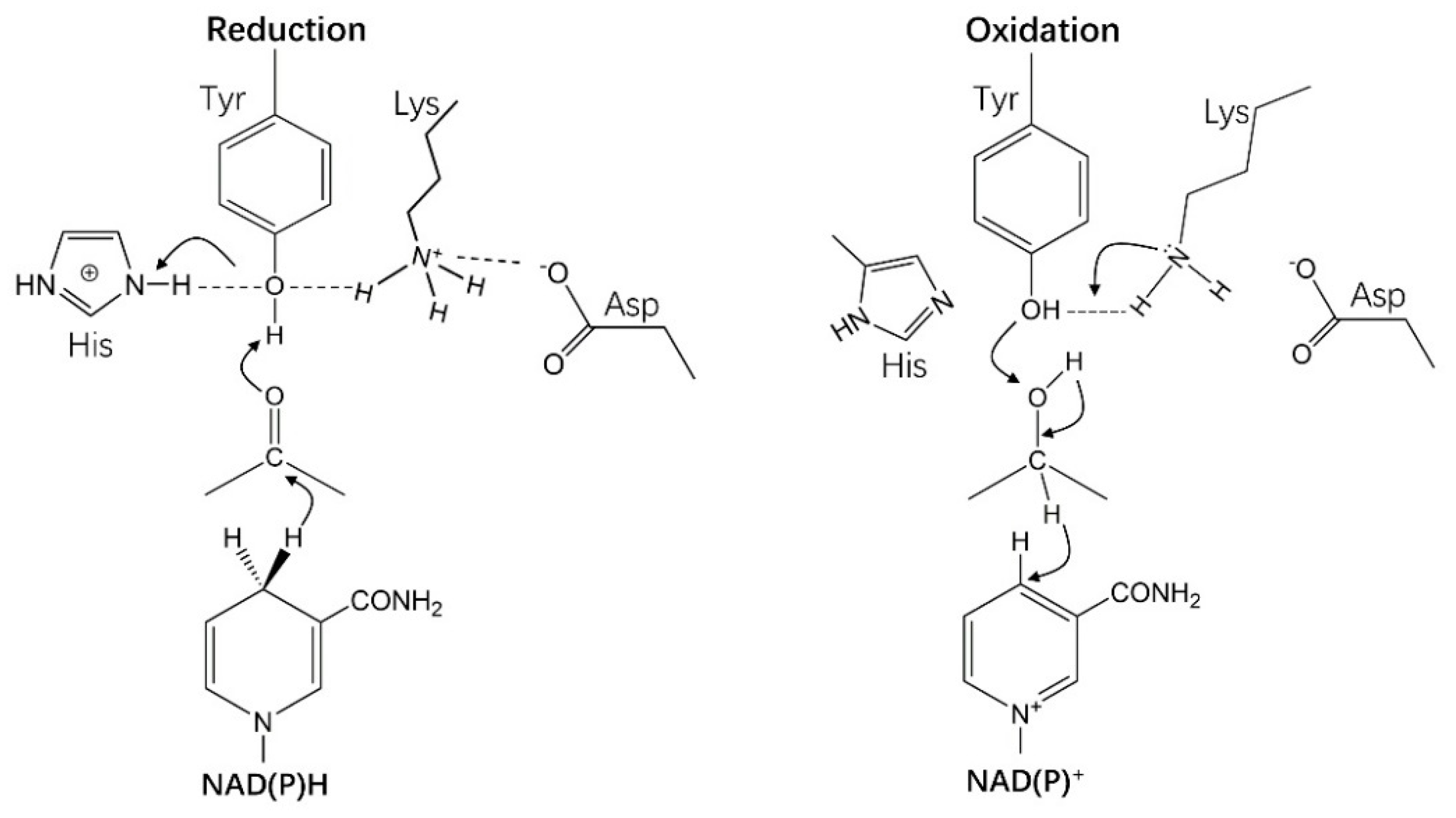
4.1. Catalytic Mechanism of HSDHs from SDRs
4.2. Catalytic Mechanism of HSDHs from AKRs
5. Stereospecificity Study of HSDHs through Molecular Docking
5.1. The Methods of Molecular Docking
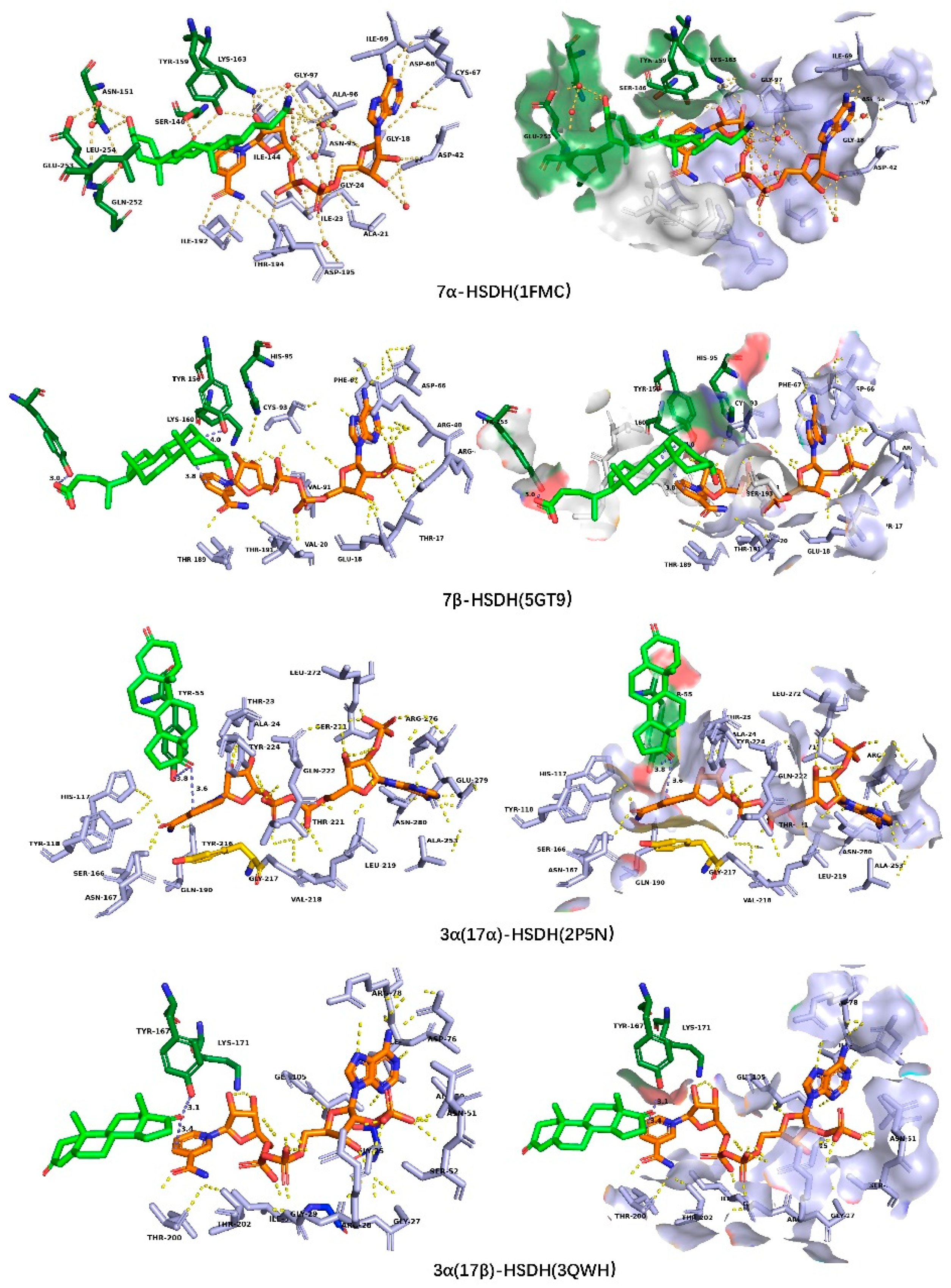
5.2. Results and Discussion of Molecular Docking
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kisiela, M.; Skarka, A.; Ebert, B.; Maser, E. Hydroxysteroid dehydrogenases (HSDs) in bacteria—A bioinformatic perspective. J. Steroid Biochem. Mol. Biol. 2012, 129, 31–46. [Google Scholar] [CrossRef]
- Penning, T.M. Human hydroxysteroid dehydrogenases and pre-receptor regulation: Insights into inhibitor design and evaluation. J. Steroid Biochem. Mol. Biol. 2011, 125, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, T.; Higashi, H.; Kanatani, A.; Lin, X.S.; Nagai, H.; Oyama, H.; Kurazono, K.; Tsuru, D. Cloning and sequencing of the 7 alpha-hydroxysteroid dehydrogenase gene from Escherichia coli HB101 and characterization of the expressed enzyme. J. Bacteriol. 1991, 173, 2173–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, S.; Masuda, N. Characterization of NADP-dependent 7 beta-hydroxysteroid dehydrogenases from Peptostreptococcus productus and Eubacterium aerofaciens. Appl. Environ. Microbiol. 1982, 43, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Tannin, G.M.; Agarwal, A.K.; Monder, C.; New, M.I.; White, P.C. The human gene for 11 beta-hydroxysteroid dehydrogenase. Structure, tissue distribution, and chromosomal localization. J. Biol. Chem. 1991, 266, 16653–16658. [Google Scholar] [CrossRef]
- Jörnvall, H.; Höög, J.-O.; Persson, B. SDR and MDR: Completed genome sequences show these protein families to be large, of old origin, and of complex nature. FEBS Lett. 1999, 445, 261–264. [Google Scholar] [CrossRef]
- Jörnvall, H.; Persson, B.; Krook, M.; Atrian, S.; Gonzalez-Duarte, R.; Jeffery, J.; Ghosh, D. Short-chain dehydrogenases/reductases (SDR). Biochemistry 1995, 34, 6003–6013. [Google Scholar] [CrossRef]
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Jez, J.M.; Bennett, M.J.; Schlegel, B.P.; Lewis, M.; Penning, T.M. Comparative anatomy of the aldo–keto reductase superfamily. Biochem. J. 1997, 326, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Möbus, E.; Maser, E. Molecular Cloning, Overexpression, and Characterization of Steroid-inducible 3α-Hydroxysteroid Dehydrogenase/Carbonyl Reductase from Comamonas testosteroni. J. Biol. Chem. 1998, 273, 30888–30896. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Ueda, S.; Sugiyama, M.; Imamura, S. Cloning and expression of a Pseudomonas 3α-hydroxy steroid dehydrogenase-encoding gene in Escherichia coli. Gene 1993, 130, 137–140. [Google Scholar] [CrossRef]
- Bennett, M.J.; Schlegel, B.P.; Jez, J.M.; Penning, T.M.; Lewis, M. Structure of 3α-Hydroxysteroid/Dihydrodiol Dehydrogenase Complexed with NADP+. Biochemistry 1996, 35, 10702–10711. [Google Scholar] [CrossRef]
- Deyashiki, Y.; Ogasawara, A.; Nakayama, T.; Nakanishi, M.; Miyabe, Y.; Sato, K.; Hara, A. Molecular cloning of two human liver 3 α-hydroxysteroid/dihydrodiol dehydrogenase isoenzymes that are identical with chlordecone reductase and bile-acid binder. Biochem. J. 1994, 299, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. The PyMol Molecular Graphics System. Proteins 2002, 30, 442–454. [Google Scholar]
- Bashton, M.; Chothia, C. The geometry of domain combination in proteins 1 1Edited by J. Thornton. J. Mol. Biol. 2002, 315, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Hyndman, D.; Bauman, D.R.; Heredia, V.V.; Penning, T.M. The aldo-keto reductase superfamily homepage. Chem. Interact. 2003, 144, 621–631. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, L.; Barnard, L.; Baranowski, E.S.; Gilligan, L.C.; Taylor, A.E.; Arlt, W.; Shackleton, C.H.; Storbeck, K.-H. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2019, 194, 105439. [Google Scholar] [CrossRef] [PubMed]
- Higaki, Y.; Kamiya, T.; Usami, N.; Shintani, S.; Shiraishi, H.; Ishikura, S.; Yamamoto, I.; Hara, A. Molecular Characterization of Two Monkey Dihydrodiol Dehydrogenases. Drug Metab. Pharmacokinet. 2002, 17, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Matsunaga, T.; Arai, Y.; Ikari, A.; Tajima, K.; El-Kabbani, O.; Yamano, S.; Hara, A.; Kitade, Y. Cloning and Characterization of Four Rabbit Aldo-Keto Reductases Featuring Broad Substrate Specificity for Xenobiotic and Endogenous Carbonyl Compounds: Relationship with Multiple Forms of Drug Ketone Reductases. Drug Metab. Dispos. 2014, 42, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Persson, B.; Kallberg, Y. Classification and nomenclature of the superfamily of short-chain dehydrogenases/reductases (SDRs). Chem. Interact. 2013, 202, 111–115. [Google Scholar] [CrossRef]
- Maser, E. Xenobiotic carbonyl reduction and physiological steroid oxidoreduction. Biochem. Pharmacol. 1995, 49, 421–440. [Google Scholar] [CrossRef]
- Kallberg, Y.; Oppermann, U.; Persson, B. Classification of the short-chain dehydrogenase/reductase superfamily using hidden Markov models. FEBS J. 2010, 277, 2375–2386. [Google Scholar] [CrossRef]
- Roth, S.; Stockinger, P.; Steff, J.; Steimle, S.; Sautner, V.; Tittmann, K.; Pleiss, J.; Müller, M. Crossing the Border: From Keto- to Imine Reduction in Short-Chain Dehydrogenases/Reductases. ChemBioChem 2020, 21, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.R.; Kaserer, T.; Schuster, D.; Odermatt, A. Virtual screening applications in short-chain dehydrogenase/reductase research. J. Steroid Biochem. Mol. Biol. 2017, 171, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Maser, E. Carbonyl Reductases and Pluripotent Hydroxysteroid Dehydrogenases of the Short-chain Dehydrogenase/reductase Superfamily. Drug Metab. Rev. 2007, 39, 87–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lukacik, P.; Kavanagh, K.L.; Oppermann, U. SDR-type human hydroxysteroid dehydrogenases involved in steroid hormone activation. Mol. Cell. Endocrinol. 2007, 265, 71–76. [Google Scholar] [CrossRef]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef]
- Sivamaruthi, B.S.; Fern, L.A.; Ismail, D.S.N.R.P.H.; Chaiyasut, C. The influence of probiotics on bile acids in diseases and aging. Biomed. Pharmacother. 2020, 128, 110310. [Google Scholar] [CrossRef]
- Oppermann, U.C.T.; Maser, E. Characterization of a 3alpha-Hydroxysteroid Dehydrogenase/Carbonyl Reductase from the Gram-Negative Bacterium Comamonas testosteroni. JBIC J. Biol. Inorg. Chem. 1996, 241, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, S. Comparative analysis of Corynebacterium glutamicum genomes: A new perspective for the industrial production of amino acids. BMC Genom. 2017, 18, 940. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Aigner, A.; Schmid, R.D. Identification, cloning, heterologous expression, and characterization of a NADPH-dependent 7β-hydroxysteroid dehydrogenase from Collinsella aerofaciens. Appl. Microbiol. Biotechnol. 2010, 90, 127–135. [Google Scholar] [CrossRef]
- Chen, X.; Cui, Y.; Feng, J.; Wang, Y.; Liu, X.; Wu, Q.; Zhu, D.; Ma, Y. Flavin Oxidoreductase-Mediated Regeneration of Nicotinamide Adenine Dinucleotide with Dioxygen and Catalytic Amount of Flavin Mononucleotide for One-Pot Multi-Enzymatic Preparation of Ursodeoxycholic Acid. Adv. Synth. Catal. 2019, 361, 2497–2504. [Google Scholar] [CrossRef] [Green Version]
- He, X.-L.; Wang, L.-T.; Gu, X.-Z.; Xiao, J.-X.; Qiu, W.-W. A facile synthesis of ursodeoxycholic acid and obeticholic acid from cholic acid. Steroids 2018, 140, 173–178. [Google Scholar] [CrossRef]
- Simard, J.; Ricketts, M.-L.; Gingras, S.; Soucy, P.; Feltus, F.A.; Melner, M.H. Molecular Biology of the 3β-Hydroxysteroid Dehydrogenase/Δ5-Δ4 Isomerase Gene Family. Endocr. Rev. 2005, 26, 525–582. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.L.; Mack, V.L.; Sun, J.; Terrell, J.R.; Bucholtz, K.M. The functions of key residues in the inhibitor, substrate and cofactor sites of human 3β-hydroxysteroid dehydrogenase type 1 are validated by mutagenesis. J. Steroid Biochem. Mol. Biol. 2010, 120, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.L.; Duax, W.L.; Addlagatta, A.; Brandt, S.; Fuller, R.R.; Norris, W. Structure/Function Relationships Responsible for Coenzyme Specificity and the Isomerase Activity of Human Type 1 3β-Hydroxysteroid Dehydrogenase/Isomerase. J. Biol. Chem. 2003, 278, 35483–35490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.L.; Bose, H.S. Regulation of human 3-beta-hydroxysteroid dehydrogenase type-2 (3βHSD2) by molecular chaperones and the mitochondrial environment affects steroidogenesis. J. Steroid Biochem. Mol. Biol. 2015, 151, 74–84. [Google Scholar] [CrossRef]
- Pletnev, V.Z.; Thomas, J.L.; Rhaney, F.L.; Holt, L.S.; Scaccia, L.A.; Umland, T.C.; Duax, W.L. Rational proteomics V: Structure-based mutagenesis has revealed key residues responsible for substrate recognition and catalysis by the dehydrogenase and isomerase activities in human 3β-hydroxysteroid dehydrogenase/isomerase type 1. J. Steroid Biochem. Mol. Biol. 2006, 101, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettel, D.; Sharifi, N. HSD3B1 status as a biomarker of androgen deprivation resistance and implications for prostate cancer. Nat. Rev. Urol. 2017, 15, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Yue, W.; Wang, J.-P. Estrogen metabolites and breast cancer. Steroids 2015, 99, 61–66. [Google Scholar] [CrossRef]
- Duarsa, G.W.K.; Sari, Y.A.; Oka, A.A.G.; Santosa, K.B.; Yudiana, I.W.; Tirtayasa, P.M.W.; Pramana, I.B.P.; Kloping, Y.P. Serum testosterone and prostate-specific antigen levels are major risk factors for prostatic volume increase among benign prostatic hyperplasia patients. Asian J. Urol. 2020, 1–9. [Google Scholar] [CrossRef]
- Damiani, F.; Makieva, S.; Rinaldi, S.F.; Hua, L.; Marcolongo, P.; Petraglia, F.; Norman, J.E. 11β-hydroxysteroid dehydrogenase type 1 and pregnancy: Role in the timing of labour onset and in myometrial contraction. Mol. Cell. Endocrinol. 2017, 447, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Dammann, C.; Stapelfeld, C.; Maser, E. Expression and activity of the cortisol-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 is tissue and species-specific. Chem. Interact. 2019, 303, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Valeur, E.; Christmann-Franck, S.; Lepifre, F.; Carniato, D.; Cravo, D.; Charon, C.; Bozec, S.; Musil, D.; Hillertz, P.; Doare, L.; et al. Structure-based design of 7-azaindole-pyrrolidine amides as inhibitors of 11β-hydroxysteroid dehydrogenase type I. Bioorgan. Med. Chem. Lett. 2012, 22, 5909–5914. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, F.W.; Dossetter, A.G.; Scott, J.S.; Robb, G.R.; Boyd, S.; Groombridge, S.D.; Kemmitt, P.D.; Sjögren, T.; Gutierrez, P.M.; Deschoolmeester, J.; et al. Optimization of Brain Penetrant 11β-Hydroxysteroid Dehydrogenase Type I Inhibitors and in Vivo Testing in Diet-Induced Obese Mice. J. Med. Chem. 2014, 57, 970–986. [Google Scholar] [CrossRef]
- Siu, M.; Johnson, T.O.; Wang, Y.; Nair, S.K.; Taylor, W.D.; Cripps, S.J.; Matthews, J.J.; Edwards, M.P.; Pauly, T.A.; Ermolieff, J.; et al. N-(Pyridin-2-yl) arylsulfonamide inhibitors of 11β-hydroxysteroid dehydrogenase type 1: Discovery of PF-915275. Bioorgan. Med. Chem. Lett. 2009, 19, 3493–3497. [Google Scholar] [CrossRef]
- Sandeep, T.C.; Walker, B.R. Pathophysiology of modulation of local glucocorticoid levels by 11β-hydroxysteroid dehydrogenases. Trends Endocrinol. Metab. 2001, 12, 446–453. [Google Scholar] [CrossRef]
- Zhu, Q.; Ge, F.; Dong, Y.; Sun, W.; Wang, Z.; Shan, Y.; Chen, R.; Sun, J.; Ge, R.-S. Comparison of flavonoids and isoflavonoids to inhibit rat and human 11β-hydroxysteroid dehydrogenase 1 and 2. Steroids 2018, 132, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E. Evolution of 11β-hydroxysteroid dehydrogenase-type 1 and 11β-hydroxysteroid dehydrogenase-type 3. FEBS Lett. 2010, 584, 2279–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mindnich, R.; Möller, G.; Adamski, J. The role of 17 beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2004, 218, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Moeller, G.; Adamski, J. Multifunctionality of human 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2006, 248, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Moeller, G.; Adamski, J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2009, 301, 7–19. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: Protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef]
- Gangloff, A.; Shi, R.; Nahoum, V.; Lin, S. Pseudo-symmetry of C19-steroids, alternative binding orientations and multispecificity in human estrogenic 17β-hydroxysteroid dehydrogenase. FASEB J. 2002, 17, 274–276. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Calvo, E.-L.; Yang, C.-Q.; Liu, J.; Sang, X.-Y.; Lin, S.-X. Transcriptome of 17β-hydroxysteroid dehydrogenase type 2 plays both hormone-dependent and hormone-independent roles in MCF-7 breast cancer cells. J. Steroid Biochem. Mol. Biol. 2019, 195, 105471. [Google Scholar] [CrossRef]
- Abdelsamie, A.S.; Salah, M.; Siebenbürger, L.; Hamed, M.M.; Börger, C.; Van Koppen, C.J.; Frotscher, M.; Hartmann, R.W. Development of potential preclinical candidates with promising in vitro ADME profile for the inhibition of type 1 and type 2 17β-Hydroxysteroid dehydrogenases: Design, synthesis, and biological evaluation. Eur. J. Med. Chem. 2019, 178, 93–107. [Google Scholar] [CrossRef]
- Ning, X.; Yang, Y.; Deng, H.; Zhang, Q.; Huang, Y.; Su, Z.; Fu, Y.; Xiang, Q.; Zhang, S. Development of 17β-hydroxysteroid dehydrogenase type 3 as a target in hormone-dependent prostate cancer therapy. Steroids 2017, 121, 10–16. [Google Scholar] [CrossRef]
- Mindnich, R.D.; Penning, T.M. Aldo-keto reductase (AKR) superfamily: Genomics and annotation. Hum. Genom. 2009, 3, 362–370. [Google Scholar] [CrossRef]
- Jin, Y.; Penning, T.M. Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 263–292. [Google Scholar] [CrossRef]
- Rižner, T.L.; Penning, T.M. Role of aldo–keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Rižner, T.L.; Penning, T.M. Aldo-keto reductase 1C3—Assessment as a new target for the treatment of endometriosis. Pharmacol. Res. 2020, 152, 104446. [Google Scholar] [CrossRef]
- Penning, T.M.; Jin, Y.; Heredia, V.V.; Lewis, M. Structure–function relationships in 3α-hydroxysteroid dehydrogenases: A comparison of the rat and human isoforms. J. Steroid Biochem. Mol. Biol. 2003, 85, 247–255. [Google Scholar] [CrossRef]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.-F.; Lin, H.-K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1‒AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar] [CrossRef]
- Wan, R.; Kong, X.; Yang, Y.; Tao, S.; Chen, Y.; Teichmann, A.T.; Wieland, F.H. Role of human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C3) in the extrahepatic metabolism of the steroidal aromatase inactivator Formestane. J. Steroid Biochem. Mol. Biol. 2020, 198, 105527. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol. Cell. Endocrinol. 2019, 489, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-K.; Steckelbroeck, S.; Fung, K.-M.; Jones, A.N.; Penning, T.M. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3α-hydroxysteroid dehydrogenase/type 5 17β-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids 2004, 69, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Lin, H.-K.; Rogers, K.A.; Brame, L.S.; Yeh, M.M.; Yang, Q.; Fung, K.-M. Expression of aldo-keto reductase family 1 member C3 (AKR1C3) in neuroendocrine tumors & adenocarcinomas of pancreas, gastrointestinal tract, and lung. Int. J. Clin. Exp. Pathol. 2013, 6, 2419–2429. [Google Scholar]
- Šmuc, T.; Rižner, T.L. Aberrant pre-receptor regulation of estrogen and progesterone action in endometrial cancer. Mol. Cell. Endocrinol. 2009, 301, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Rižner, T.L.; Lin, H.K.; Penning, T.M. Role of human type 3 3α-hydroxysteroid dehydrogenase (AKR1C2) in androgen metabolism of prostate cancer cells. Chem. Interact. 2003, 144, 401–409. [Google Scholar] [CrossRef]
- El-Kabbani, O.; Dhagat, U.; Hara, A. Inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1). J. Steroid Biochem. Mol. Biol. 2011, 125, 105–111. [Google Scholar] [CrossRef]
- El-Kabbani, O.; Scammells, P.J.; Day, T.; Dhagat, U.; Endo, S.; Matsunaga, T.; Soda, M.; Hara, A. Structure-based optimization and biological evaluation of human 20α-hydroxysteroid dehydrogenase (AKR1C1) salicylic acid-based inhibitors. Eur. J. Med. Chem. 2010, 45, 5309–5317. [Google Scholar] [CrossRef]
- Lima, M.A.; Silva, S.V.; Jaeger, R.G.; Freitas, V.M. Progesterone decreases ovarian cancer cells migration and invasion. Steroids 2020, 161, 108680. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, G.C.; Tosto, V.; Tsibizova, V. Progesterone: History, facts, and artifacts. Best Pr. Res. Clin. Obstet. Gynaecol. 2020, 69, 2–12. [Google Scholar] [CrossRef]
- Brožič, P.; Cesar, J.; Kovač, A.; Davies, M.; Johnson, A.; Fishwick, C.; Rižner, T.L.; Gobec, S. Derivatives of pyrimidine, phthalimide and anthranilic acid as inhibitors of human hydroxysteroid dehydrogenase AKR1C1. Chem. Interact. 2009, 178, 158–164. [Google Scholar] [CrossRef]
- Beranič, N.; Gobec, S.; Rižner, T.L. Progestins as inhibitors of the human 20-ketosteroid reductases, AKR1C1 and AKR1C3. Chem. Interact. 2011, 191, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Vidal, L.S.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta BBA Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Huang, R.; Wei, X.; Zhu, Z.; Zhang, Y.-H.P. Protein engineering of oxidoreductases utilizing nicotinamide-based coenzymes, with applications in synthetic biology. Synth. Syst. Biotechnol. 2017, 2, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Iyanagi, T. Molecular mechanism of metabolic NAD(P)H-dependent electron-transfer systems: The role of redox cofactors. Biochim. Biophys. Acta BBA Bioenerg. 2019, 1860, 233–258. [Google Scholar] [CrossRef]
- Filling, C.; Berndt, K.D.; Benach, J.; Knapp, S.; Prozorovski, T.; Nordling, E.; Ladenstein, R.; Jörnvall, H.; Oppermann, U. Critical Residues for Structure and Catalysis in Short-chain Dehydrogenases/Reductases. J. Biol. Chem. 2002, 277, 25677–25684. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.L.; Jornvall, H.; Persson, B.; Oppermann, U. Medium- and short-chain dehydrogenase/reductase gene and protein families. Cell. Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, C.; Oerum, S.; Bray, J.; Kavanagh, K.L.; Shafqat, N.; Yue, W.; Oppermann, U. Towards a systematic analysis of human short-chain dehydrogenases/reductases (SDR): Ligand identification and structure–activity relationships. Chem. Interact. 2015, 234, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.E.; Marsden, B.D.; Oppermann, U. The human short-chain dehydrogenase/reductase (SDR) superfamily: A bioinformatics summary. Chem. Interact 2009, 178, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Filling, C.; Wu, X.; Shafqat, N.; Hult, M.; Mårtensson, E.; Shafqat, J.; Oppermann, U.C. Subcellular targeting analysis of SDR-type hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2001, 171, 99–101. [Google Scholar] [CrossRef]
- Duax, W.L.; Ghosh, D.; Pletnev, V. Steroid dehydrogenase structures, mechanism of action, and disease. Vitam. Horm. 2000, 58, 121–148. [Google Scholar] [CrossRef]
- Grant, G.A. Contrasting catalytic and allosteric mechanisms for phosphoglycerate dehydrogenases. Arch. Biochem. Biophys. 2012, 519, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, S.; Zhang, C.; Zhu, X.; Hammad, M.A.; Zhang, X.; Christian, M.; Zhang, H.; Liu, P. Hydroxysteroid dehydrogenase family proteins on lipid droplets through bacteria, C. elegans, and mammals. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2018, 1863, 881–894. [Google Scholar] [CrossRef]
- Sherbet, D.P.; Papari-Zareei, M.; Khan, N.; Sharma, K.K.; Brandmaier, A.; Rambally, S.; Chattopadhyay, A.; Andersson, S.; Agarwal, A.K.; Auchus, R.J. Cofactors, redox state, and directional preferences of hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2007, L, 83–88. [Google Scholar] [CrossRef]
- Wushur, I.; Sylte, I.; Winberg, J.-O. The catalytic reaction mechanism of drosophilid alcohol dehydrogenases. Perspect. Sci. 2015, 4, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Nonaka, T.; Tanabe, T.; Yoshimoto, T.; Tsuru, D.; Mitsui, Y. Crystal Structures of the Binary and Ternary Complexes of 7α-Hydroxysteroid Dehydrogenase from Escherichia coli. Biochemistry 1996, 35, 7715–7730. [Google Scholar] [CrossRef]
- Wang, R.; Wu, J.; Jin, D.K.; Chen, Y.; Lv, Z.; Chen, Q.; Miao, Q.; Huo, X.; Wang, F. Structure of NADP+-bound 7β-hydroxysteroid dehydrogenase reveals two cofactor-binding modes. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 246–252. [Google Scholar] [CrossRef]
- Lou, D.; Wang, B.; Tan, J.; Zhu, L.; Cen, X.; Ji, Q.; Wang, Y. The three-dimensional structure of Clostridium absonum 7α-hydroxysteroid dehydrogenase: New insights into the conserved arginines for NADP(H) recognition. Sci. Rep. 2016, 6, 22885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhu, D.-W.; Hu, X.-J.; Zhou, M.; Shang, P.; Lin, S.-X. Human 3-alpha hydroxysteroid dehydrogenase type 3 (3α-HSD3): The V54L mutation restricting the steroid alternative binding and enhancing the 20α-HSD activity. J. Steroid Biochem. Mol. Biol. 2014, 141, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.-F.; Legrand, P.; Cantin, L.; Labrie, F.; Luu-The, V.; Breton, R. Loop Relaxation, A Mechanism that Explains the Reduced Specificity of Rabbit 20α-Hydroxysteroid Dehydrogenase, A Member of the Aldo-Keto Reductase Superfamily. J. Mol. Biol. 2004, 339, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ratnam, K.; Penning, T.M. Mutation of Nicotinamide Pocket Residues in Rat Liver 3α-Hydroxysteroid Dehydrogenase Reveals Different Modes of Cofactor Binding. Biochemistry 2000, 39, 102–109. [Google Scholar] [CrossRef]
- Wierenga, R. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett. 2001, 492, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Banfield, M.J.; Salvucci, M.E.; Baker, E.N.; Smith, C.A. Crystal structure of the NADP(H)-dependent ketose reductase from Bemisia argentifolii at 2.3 Å resolution. J. Mol. Biol. 2001, 306, 239–250. [Google Scholar] [CrossRef]
- Jin, Y.; Penning, T.M. Molecular docking simulations of steroid substrates into human cytosolic hydroxysteroid dehydrogenases (AKR1C1 and AKR1C2): Insights into positional and stereochemical preferences. Steroids 2006, 71, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Bennett, M.J.; Smith-Hoog, S.; Schlegel, B.P.; Jez, J.M.; Lewis, M. Structure and function of 3α-hydroxysteroid dehydrogenase. Steroids 1997, 62, 101–111. [Google Scholar] [CrossRef]
- Khan, M.S.; Qais, F.A.; Rehman, T.; Ismail, M.H.; Alokail, M.S.; Altwaijry, N.; Alafaleq, N.O.; Alajmi, M.F.; Gaber, N.S.; Alqhatani, R. Mechanistic inhibition of non-enzymatic glycation and aldose reductase activity by naringenin: Binding, enzyme kinetics and molecular docking analysis. Int. J. Biol. Macromol. 2020, 159, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.C.; Jin, Y.; Penning, T.M. Elucidation of a Complete Kinetic Mechanism for a Mammalian Hydroxysteroid Dehydrogenase (HSD) and Identification of All Enzyme Forms on the Reaction Coordinate. J. Biol. Chem. 2007, 282, 33484–33493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borhani, D.; Harter, T.; Petrash, J. The crystal structure of the aldose reductase NADPH binary complex. J. Biol. Chem. 1992, 267, 24841–24847. [Google Scholar] [CrossRef]
- Persson, B.; Kallberg, Y.; Oppermann, U.; Jörnvall, H. Coenzyme-based functional assignments of short-chain dehydrogenases/reductases (SDRs). Chem. Interact. 2003, 144, 271–278. [Google Scholar] [CrossRef]
- Ghosh, D.; Wawrzak, Z.; Weeks, C.M.; Duax, W.L.; Erman, M. The refined three-dimensional structure of 3α,20β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure 1994, 2, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Gani, O.A.B.S.M.; Adekoya, O.A.; Giurato, L.; Spyrakis, F.; Cozzini, P.; Guccione, S.; Winberg, J.-O.; Sylte, I. Theoretical Calculations of the Catalytic Triad in Short-Chain Alcohol Dehydrogenases/Reductases. Biophys. J. 2008, 94, 1412–1427. [Google Scholar] [CrossRef] [Green Version]
- Koumanov, A.; Benach, J.; Atrian, S.; Gonzàlez-Duarte, R.; Karshikoff, A.; Ladenstein, R. The catalytic mechanism of Drosophilaalcohol dehydrogenase: Evidence for a proton relay modulated by the coupled ionization of the active site Lysine/Tyrosine pair and a NAD+ ribose OH switch. Proteins Struct. Funct. Bioinform. 2003, 51, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Winberg, J.-O.; Brendskag, M.K.; Sylte, I.; Lindstad, R.I.; McKinley-Mckee, J.S. The catalytic triad in Drosophila alcohol dehydrogenase: pH, temperature and molecular modelling studies. J. Mol. Biol. 1999, 294, 601–616. [Google Scholar] [CrossRef]
- Hovik, R.; Winberg, J.-O.; McKinley-Mckee, J.S. Drosophila melanogaster alcohol dehydrogenase. Insect Biochem. 1984, 14, 345–351. [Google Scholar] [CrossRef]
- Wuxiuer, Y.; Morgunova, E.; Cols, N.; Popov, A.; Karshikoff, A.; Sylte, I.; Gonzàlez-Duarte, R.; Ladenstein, R.; Winberg, J.-O. An intact eight-membered water chain in drosophilid alcohol dehydrogenases is essential for optimal enzyme activity. FEBS J. 2012, 279, 2940–2956. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, T.; Tanaka, N.; Uchikawa, K.; Kabashima, T.; Ito, K.; Nonaka, T.; Mitsui, Y.; Tsuru, M.; Yoshimoto, T. Roles of the Ser146, Tyr159, and Lys163 Residues in the Catalytic Action of 7-Hydroxysteroid Dehydrogenase from Escherichia coli. J. Biochem. 1998, 124, 634–641. [Google Scholar] [CrossRef]
- Penning, T.M.; Drury, J.E. Human aldo–keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch. Biochem. Biophys. 2007, 464, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, T.G.; Green, N.C.; Bhatia, M.B.; El-Kabbani, O. Structure and Mechanism of Aldehyde Reductase. Adv. Exp. Med. Biol. 1995, 372, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bohren, K.M.; Grimshaw, C.E.; Lai, C.J.; Harrison, D.H.; Ringe, D.; Petsko, G.A.; Gabbay, K.H. Tyrosine-48 Is the Proton Donor and Histidine-110 Directs Substrate Stereochemical Selectivity in the Reduction Reaction of Human Aldose Reductase: Enzyme Kinetics and Crystal Structure of the Y48H Mutant Enzyme. Biochemistry 1994, 33, 2021–2032. [Google Scholar] [CrossRef]
- Tarle, I.; Borhani, D.W.; Wilson, D.K.; Quiocho, F.A.; Petrash, J.M. Probing the active site of human aldose reductase. Site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J. Biol. Chem. 1993, 268, 25687–25693. [Google Scholar] [CrossRef]
- Bohren, K.; Grimshaw, C.; Gabbay, K. Catalytic effectiveness of human aldose reductase. Critical role of C-terminal domain. J. Biol. Chem. 1992, 267, 20965–20970. [Google Scholar] [CrossRef]
- Grimshaw, C.E.; Bohren, K.M.; Lai, C.-J.; Gabbay, K.H. Human Aldose Reductase: Rate Constants for a Mechanism Including Interconversion of Ternary Complexes by Recombinant Wild-Type Enzyme. Biochemistry 1995, 34, 14356–14365. [Google Scholar] [CrossRef]
- Schlegel, B.P.; Jez, J.M.; Penning, T.M. Mutagenesis of 3α-Hydroxysteroid Dehydrogenase Reveals a “Push−Pull” Mechanism for Proton Transfer in Aldo−Keto Reductases. Biochemistry 1998, 37, 3538–3548. [Google Scholar] [CrossRef]
- Penning, T. Molecular determinants of steroid recognition and catalysis in aldo-keto reductases. Lessons from 3α-hydroxysteroid dehydrogenase. J. Steroid Biochem. Mol. Biol. 1999, 69, 211–225. [Google Scholar] [CrossRef]
- Eggert, T.; Bakonyi, D.; Hummel, W. Enzymatic routes for the synthesis of ursodeoxycholic acid. J. Biotechnol. 2014, 191, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Arai, H.; Nakamura, Y.; Fukiya, S.; Wada, M.; Yokota, A. Contribution of the 7β-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J. Lipid Res. 2013, 54, 3062–3069. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.-M.; Chen, F.-F.; Li, H.; Li, C.-X.; Xu, J.-H. Continuous Production of Ursodeoxycholic Acid by Using Two Cascade Reactors with Co-immobilized Enzymes. ChemBioChem 2018, 19, 347–353. [Google Scholar] [CrossRef]
- Savino, S.; Ferrandi, E.E.; Forneris, F.; Rovida, S.; Riva, S.; Monti, D.; Mattevi, A. Structural and biochemical insights into 7β-hydroxysteroid dehydrogenase stereoselectivity. Proteins Struct. Funct. Bioinform. 2016, 84, 859–865. [Google Scholar] [CrossRef]
- Nakajima, K.; Hashimoto, T.; Yamada, Y. Two tropinone reductases with different stereospecificities are short-chain dehydrogenases evolved from a common ancestor. Proc. Natl. Acad. Sci. USA 1993, 90, 9591–9595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Yamashita, A.; Akama, H.; Nakatsu, T.; Kato, H.; Hashimoto, T.; Oda, J.; Yamada, Y. Crystal structures of two tropinone reductases: Different reaction stereospecificities in the same protein fold. Proc. Natl. Acad. Sci. USA 1998, 95, 4876–4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnakumar, A.M.; Nocek, B.P.; Clark, D.D.; Ensign, S.A.; Peters, J.W. Structural Basis for Stereoselectivity in the (R)- and (S)-Hydroxypropylthioethanesulfonate Dehydrogenases. Biochemistry 2006, 45, 8831–8840. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, D.A.; Krishnakumar, A.M.; Peters, J.W.; Ensign, S.A. Molecular Basis for Enantioselectivity in the (R)- and (S)-Hydroxypropylthioethanesulfonate Dehydrogenases, a Unique Pair of Stereoselective Short-Chain Dehydrogenases/Reductases Involved in Aliphatic Epoxide Carboxylation. Biochemistry 2010, 49, 3487–3498. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Wu, S.; Xue, H.; Jiang, J. Stereoselective catabolism of compounds by microorganisms: Catabolic pathway, molecular mechanism and potential application. Int. Biodeterior. Biodegrad. 2020, 146, 104822. [Google Scholar] [CrossRef]
- Parales, R.E.; Lee, K.; Resnick, S.M.; Jiang, H.; Lessner, D.J.; Gibson, D.T. Substrate Specificity of Naphthalene Dioxygenase: Effect of Specific Amino Acids at the Active Site of the Enzyme. J. Bacteriol. 2000, 182, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Parales, R.E.; Resnick, S.M.; Yu, C.-L.; Boyd, D.R.; Sharma, N.D.; Gibson, D.T. Regioselectivity and Enantioselectivity of Naphthalene Dioxygenase during Arene cis-Dihydroxylation: Control by Phenylalanine 352 in the α Subunit. J. Bacteriol. 2000, 182, 5495–5504. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, A.; Stojan, J.; Krastanova, I.; Kristan, K.; Švegelj, M.B.; Lamba, D.; Rižner, T.L. Structural basis for inhibition of 17β-hydroxysteroid dehydrogenases by phytoestrogens: The case of fungal 17β-HSDcl. J. Steroid Biochem. Mol. Biol. 2017, 171, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Steckelbroeck, S.; Jin, Y.; Oyesanmi, B.; Kloosterboer, H.J.; Penning, T.M. Tibolone Is Metabolized by the 3α/3β-Hydroxysteroid Dehydrogenase Activities of the Four Human Isozymes of the Aldo-Keto Reductase 1C Subfamily: Inversion of Stereospecificity with a Δ5(10)-3-Ketosteroid. Mol. Pharmacol. 2004, 66, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhagat, U.; Carbone, V.; Chung, R.P.-T.; Schulze-Briese, C.; Endo, S.; Hara, A.; El-Kabbani, O. Structure of 3(17)α-hydroxysteroid dehydrogenase (AKR1C21) holoenzyme from an orthorhombic crystal form: An insight into the bifunctionality of the enzyme. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 825–830. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSDHs | Monomer Size | Coenzymes | Catalytic Reaction | Coenzyme Binding | Active Site | Sequence Identity |
|---|---|---|---|---|---|---|
| Divergent | NAD(H) | Enoyl reductases | GxxxxxSxA | YxxMxxxK | 28–99% | |
| Classical | ~250 aa | NAD(P)(H) | Oxidation/reduction | TGxxxGxG | YxxxK | 8–99% |
| Intermediate | ~250 aa | Drosophila ADH | G/AxxGxxG/A | 27–99% | ||
| Extended | ~350 aa | NAD(P)(H) | Epimerases Dehydratases | TGxxGxxG | 10–99% | |
| Complex | NADP(H) | β-ketoacyl reduction | YxxxN | 20–74% | ||
| Atypical | NmrA-like | Lacks catalytic Tyr |
| HSDHs | Interaction between NAD(P)(H) and HSDHs | Distance/Å | Dihedral Angle/° | ||||
|---|---|---|---|---|---|---|---|
| Adenosine Ring | Pyrophosphate | Nicotinamide Ring | C6A–C2N | A | B | C | |
| 7α–HSDH | Adenine: Asp68, Ile69 | Ile23, Thr194 | Nicotinamide: Ile192, Thr194; Ribose: Tyr159, Lys163 | 13.4 | 138.7 | 47.8 | 116.5 |
| 7β–HSDH | Adenine: Phe67, Asp66; Ribose: Thr17, Glu18, Arg40, Argr41 | Val20, Thr191, Ser193 | Nicotinamide: Thr189, Thr191; Ribose: Tyr156, Lys160 | 13.7 | 143.4 | 80.6 | 146.2 |
| 11β–HSDH | Adenine: Met68; Ribose: Ser18, Arg41, Ser42 | Ile21, Thr197 | Nicotinamide: Ile193, Thr195; Ribose: Tyr158, Lys162 | 14.1 | 134.6 | 156.5 | 142.6 |
| 3α(17β)–HSDH | Adenine: Arg78, Asp76, Ile77, Ser104; Ribose: Gly25, Gly27, Arg28, Ala50, Asn51, Ser52 | Arg28, Ile30, Thr202, Asp203 | Nicotinamide: Thr200, Thr202; Ribose: Asn103, Tyr167, Lys171 | 14.3 | 143.7 | 130.6 | 125.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, M.; Nie, K.; Qin, M.; Xu, H.; Wang, F.; Liu, L. Molecular Mechanism Study on Stereo-Selectivity of α or β Hydroxysteroid Dehydrogenases. Crystals 2021, 11, 224. https://doi.org/10.3390/cryst11030224
Gao M, Nie K, Qin M, Xu H, Wang F, Liu L. Molecular Mechanism Study on Stereo-Selectivity of α or β Hydroxysteroid Dehydrogenases. Crystals. 2021; 11(3):224. https://doi.org/10.3390/cryst11030224
Chicago/Turabian StyleGao, Miaomiao, Kaili Nie, Meng Qin, Haijun Xu, Fang Wang, and Luo Liu. 2021. "Molecular Mechanism Study on Stereo-Selectivity of α or β Hydroxysteroid Dehydrogenases" Crystals 11, no. 3: 224. https://doi.org/10.3390/cryst11030224
APA StyleGao, M., Nie, K., Qin, M., Xu, H., Wang, F., & Liu, L. (2021). Molecular Mechanism Study on Stereo-Selectivity of α or β Hydroxysteroid Dehydrogenases. Crystals, 11(3), 224. https://doi.org/10.3390/cryst11030224