
LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target

Abstract
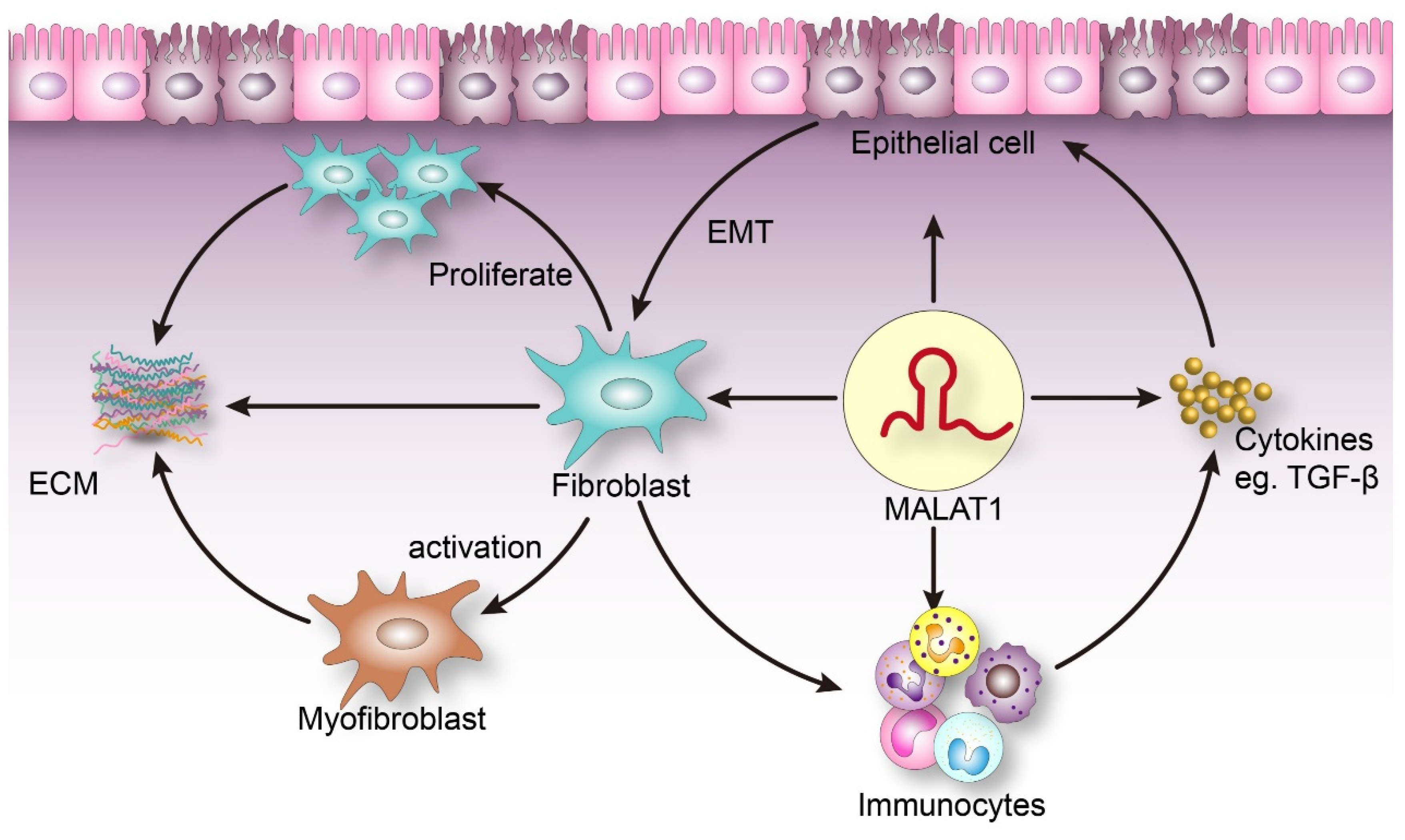
:1. Introduction
2. Association between MALAT1 and the Pathogenesis of Fibrosis
2.1. MALAT1 Can Stimulate Fibroblast Proliferation and Affect ECM Balance
2.2. The Role of MALAT1 in EMT
2.3. MALAT1 Is Involved in Inflammation
2.4. MALAT1 Interacts with TGF-β to Promote the Development of Fibrosis
3. MALAT1 and Fibrotic Disease
3.1. MALAT1 and Liver Fibrosis
3.2. MALAT1 and Cardiac Fibrosis
3.3. MALAT1 and Renal Fibrosis
3.4. MALAT1 and Pulmonary Fibrosis
4. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Birbrair, A.; Zhang, T.; Files, D.C.; Mannava, S.; Smith, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res. Ther. 2014, 5, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fani, F.; Regolisti, G.; Delsante, M.; Cantaluppi, V.; Castellano, G.; Gesualdo, L.; Villa, G.; Fiaccadori, E. Recent advances in the pathogenetic mechanisms of sepsis-associated acute kidney injury. J. Nephrol. 2018, 31, 351–359. [Google Scholar] [CrossRef]
- Fischereder, M.; Schroppel, B. The role of chemokines in acute renal allograft rejection and chronic allograft injury. Front. Biosci. 2009, 14, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- Gomez, H.; Kellum, J.A.; Ronco, C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat. Rev. Nephrol. 2017, 13, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusano, K.F.; Pola, R.; Murayama, T.; Curry, C.; Kawamoto, A.; Iwakura, A.; Shintani, S.; Ii, M.; Asai, J.; Tkebuchava, T.; et al. Sonic hedgehog myocardial gene therapy: Tissue repair through transient reconstitution of embryonic signaling. Nat. Med. 2005, 11, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.; Collett, J.A.; Gunst, S.J.; Basile, D.P. Th17 cells contribute to pulmonary fibrosis and inflammation during chronic kidney disease progression after acute ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R265–R273. [Google Scholar] [CrossRef] [PubMed]
- Popovic, B.; Sutic, I.; Skocibusic, N.; Ljubotina, A.; Diminic-Lisica, I.; Bukmir, L. Cholestasis and Inflammation of the Pancreas in Family Medicine. Acta Med. Croatica 2015, 69, 319–326. [Google Scholar]
- Cao, L.; Nicosia, J.; Larouche, J.; Zhang, Y.; Bachman, H.; Brown, A.C.; Holmgren, L.; Barker, T.H. Detection of an Integrin-Binding Mechanoswitch within Fibronectin during Tissue Formation and Fibrosis. ACS Nano 2017, 11, 7110–7117. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, P.; Jayakumar, C.; Ramesh, G. Proximal tubule-specific overexpression of netrin-1 suppresses acute kidney injury-induced interstitial fibrosis and glomerulosclerosis through suppression of IL-6/STAT3 signaling. Am. J. Physiol. Renal Physiol. 2013, 304, F1054–F1065. [Google Scholar] [CrossRef] [Green Version]
- Tewes, S.; Gueler, F.; Chen, R.; Gutberlet, M.; Jang, M.S.; Meier, M.; Mengel, M.; Hartung, D.; Wacker, F.; Rong, S.; et al. Functional MRI for characterization of renal perfusion impairment and edema formation due to acute kidney injury in different mouse strains. PLoS ONE 2017, 12, e0173248. [Google Scholar] [CrossRef]
- Xiao, Y.; Yang, N.; Zhang, Q.; Wang, Y.; Yang, S.; Liu, Z. Pentraxin 3 inhibits acute renal injury-induced interstitial fibrosis through suppression of IL-6/Stat3 pathway. Inflammation 2014, 37, 1895–1901. [Google Scholar] [CrossRef]
- Espindola, M.S.; Habiel, D.M.; Narayanan, R.; Jones, I.; Coelho, A.L.; Murray, L.A.; Jiang, D.; Noble, P.W.; Hogaboam, C.M. Targeting of TAM Receptors Ameliorates Fibrotic Mechanisms in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1443–1456. [Google Scholar] [CrossRef]
- Lemmer, A.; VanWagner, L.B.; Ganger, D. Assessment of Advanced Liver Fibrosis and the Risk for Hepatic Decompensation in Patients With Congestive Hepatopathy. Hepatology 2018, 68, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Jiang, S.; Yang, Y. Database Selection and Heterogeneity-More Details, More Credibility. JAMA Oncol. 2018, 4, 1295. [Google Scholar] [CrossRef]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef]
- Hocher, B.; Adamski, J. Metabolomics for clinical use and research in chronic kidney disease. Nat. Rev. Nephrol. 2017, 13, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Q.; Cao, G.; Chen, H.; Liu, D.; Su, W.; Yu, X.Y.; Vaziri, N.D.; Liu, X.H.; Bai, X.; Zhang, L.; et al. Gene and protein expressions and metabolomics exhibit activated redox signaling and wnt/beta-catenin pathway are associated with metabolite dysfunction in patients with chronic kidney disease. Redox Biol. 2017, 12, 505–521. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Wang, H.L.; Cheng, X.L.; Wei, F.; Bai, X.; Lin, R.C.; Vaziri, N.D. Metabolomics analysis reveals the association between lipid abnormalities and oxidative stress, inflammation, fibrosis, and Nrf2 dysfunction in aristolochic acid-induced nephropathy. Sci. Rep. 2015, 5, 12936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; He, Y.; Li, J. MicroRNA-21: A central regulator of fibrotic diseases via various targets. Curr. Pharm. Des. 2015, 21, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Tsitsiou, E.; Herrick, S.E.; Lindsay, M.A. MicroRNAs and the regulation of fibrosis. FEBS J. 2010, 277, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, F. Long noncoding RNA: A new contributor and potential therapeutic target in fibrosis. Epigenomics 2017, 9, 1233–1241. [Google Scholar] [CrossRef]
- Jiang, X.; Lei, R.; Ning, Q. Circulating long noncoding RNAs as novel biomarkers of human diseases. Biomark. Med. 2016, 10, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Guo, L.; Li, Y.; Zhou, Q.; Li, Z. MALAT1 is an oncogenic long non-coding RNA associated with tumor invasion in non-small cell lung cancer regulated by DNA methylation. Int. J. Clin. Exp. Pathol. 2015, 8, 15903–15910. [Google Scholar]
- Wang, W.; Zhu, Y.; Li, S.; Chen, X.; Jiang, G.; Shen, Z.; Qiao, Y.; Wang, L.; Zheng, P.; Zhang, Y. Long noncoding RNA MALAT1 promotes malignant development of esophageal squamous cell carcinoma by targeting beta-catenin via Ezh2. Oncotarget 2016, 7, 25668–25682. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, R.; Mayeda, A.; Yoshida, M.; Nakagawa, S. MALAT1 long non-coding RNA in cancer. Biochim. Biophys. Acta 2016, 1859, 192–199. [Google Scholar] [CrossRef]
- Zhang, X.; Hamblin, M.H.; Yin, K.J. The long noncoding RNA Malat1: Its physiological and pathophysiological functions. RNA Biol. 2017, 14, 1705–1714. [Google Scholar] [CrossRef]
- Tripathi, V.; Shen, Z.; Chakraborty, A.; Giri, S.; Freier, S.M.; Wu, X.; Zhang, Y.; Gorospe, M.; Prasanth, S.G.; Lal, A.; et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013, 9, e1003368. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Chen, G.; He, R.; Yang, L. LncRNA MALAT1 promotes osteoarthritis by modulating miR-150-5p/AKT3 axis. Cell Biosci. 2019, 9, 54. [Google Scholar] [CrossRef]
- Liu, B.; Qiang, L.; Wang, G.D.; Duan, Q.; Liu, J. LncRNA MALAT1 facilities high glucose induced endothelial to mesenchymal transition and fibrosis via targeting miR-145/ZEB2 axis. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3478–3486. [Google Scholar] [PubMed]
- Gong, Y.; Zhu, Y.; Zhu, B.; Si, X.; Heng, D.; Tang, Y.; Sun, X.; Lin, L. LncRNA MALAT1 is up-regulated in diabetic gastroparesis and involved in high-glucose-induced cellular processes in human gastric smooth muscle cells. Biochem. Biophys. Res. Commun. 2018, 496, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, T.; Liang, X.; Shu, S.; Xiang, X.; Zhang, W.; Guo, T.; Xie, W.; Deng, W.; Tang, X. lncRNA MALAT1 mediated high glucose-induced HK-2 cell epithelial-to-mesenchymal transition and injury. J. Physiol. Biochem. 2019, 75, 443–452. [Google Scholar] [CrossRef]
- Marlar, S.; Abdellatef, S.A.; Nakanishi, J. Reduced adhesive ligand density in engineered extracellular matrices induces an epithelial-mesenchymal-like transition. Acta Biomater. 2016, 39, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Syn, W.K.; Karaca, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Kalluri, R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 2004, 82, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, Z.; Li, H.; Fan, J.; Yang, S.; Chen, C.; Wang, D.W. MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell 2017, 16, 387–400. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, Y.; Tang, Y.; Li, Q. MALAT1 Modulates TGF-beta1-Induced Endothelial-to-Mesenchymal Transition through Downregulation of miR-145. Cell. Physiol. Biochem. 2017, 42, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yao, H.; Li, M.; Li, H.; Wang, F. Long Non-Coding RNA MALAT1 Mediates Transforming Growth Factor Beta1-Induced Epithelial-Mesenchymal Transition of Retinal Pigment Epithelial Cells. PLoS ONE 2016, 11, e0152687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Ou, C.; Liu, J.; Chen, C.; Zhou, Q.; Yang, S.; Li, G.; Wang, G.; Song, J.; Li, Z.; et al. YAP1-induced MALAT1 promotes epithelial-mesenchymal transition and angiogenesis by sponging miR-126-5p in colorectal cancer. Oncogene 2019, 38, 2627–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, W.; Li, L.; Shi, Y.; Bu, X.; Xia, Y.; Wang, J.; Djangmah, H.S.; Liu, X.; You, Y.; Xu, B. Long non-coding RNA MALAT1 acts as a competing endogenous RNA to promote malignant melanoma growth and metastasis by sponging miR-22. Oncotarget 2016, 7, 63901–63912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014, 5, e1506. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, J.M.; Thum, T. Long noncoding RNAs in kidney and cardiovascular diseases. Nat. Rev. Nephrol. 2016, 12, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, X.; Liu, K.; Hamblin, M.H.; Yin, K.J. Long Noncoding RNA Malat1 Regulates Cerebrovascular Pathologies in Ischemic Stroke. J. Neurosci. 2017, 37, 1797–1806. [Google Scholar] [CrossRef]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell. Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Schroen, B.; Voigt, A.; Haas, J.; Kuehl, U.; Lassner, D.; Skurk, C.; Escher, F.; Wang, X.; Kratzer, A.; et al. Long noncoding RNA MALAT1-derived mascRNA is involved in cardiovascular innate immunity. J. Mol. Cell. Biol. 2016, 8, 178–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gast, M.; Rauch, B.H.; Nakagawa, S.; Haghikia, A.; Jasina, A.; Haas, J.; Nath, N.; Jensen, L.; Stroux, A.; Bohm, A.; et al. Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE-/-mice. Cardiovasc Res. 2019, 115, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Su, Z.; Song, D.; Mao, Y.; Mao, X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett. 2016, 590, 2884–2895. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Anzano, M.A.; Lamb, L.C.; Smith, J.M.; Frolik, C.A.; Marquardt, H.; Todaro, G.J.; Sporn, M.B. Isolation from murine sarcoma cells of novel transforming growth factors potentiated by EGF. Nature 1982, 295, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-beta Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, N.; Factor, V.; Nagy, P.; Kopp, J.; Kondaiah, P.; Wakefield, L.; Roberts, A.B.; Sporn, M.B.; Thorgeirsson, S.S. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. USA 1995, 92, 2572–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Chung, A.C.; Huang, X.R.; Dong, Y.; Yu, X.; Lan, H.Y. Identification of novel long noncoding RNAs associated with TGF-beta/SMAD3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol. 2014, 184, 409–417. [Google Scholar] [CrossRef]
- Zhang, J.; Han, C.; Song, K.; Chen, W.; Ungerleider, N.; Yao, L.; Ma, W.; Wu, T. The long-noncoding RNA MALAT1 regulates TGF-beta/SMAD signaling through formation of a lncRNA-protein complex with SMADs, SETD2 and PPM1A in hepatic cells. PLoS ONE 2020, 15, e0228160. [Google Scholar]
- Hu, T.J.; Huang, H.B.; Shen, H.B.; Chen, W.; Yang, Z.H. Role of long non-coding RNA MALAT1 in chronic obstructive pulmonary disease. Exp. Ther. Med. 2020, 20, 2691–2697. [Google Scholar] [PubMed]
- Zhang, S.S.; Gong, Z.J.; Xiong, W.; Wang, X.; Min, Q.; Luo, C.D.; Ling, T.Y. A rat model of oral submucous fibrosis induced by bleomycin. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2016, 122, 216–223. [Google Scholar] [CrossRef]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, N.; Zhao, S.X.; Kong, L.B.; Du, J.H.; Ren, W.G.; Han, F.; Zhang, Q.S.; Li, W.C.; Cui, P.; Wang, R.Q.; et al. LncRNA-ATB/microRNA-200a/beta-catenin regulatory axis involved in the progression of HCV-related hepatic fibrosis. Gene 2017, 618, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Li, Y.M.; Yin, S.; Xu, M.J.; Feng, D.; Zhou, Z.; Zang, M.; Mukhopadhyay, P.; Varga, Z.V.; Pacher, P.; et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 2017, 66, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, G.; De Chiara, F.; Bottcher, K.; Levi, A.; Dhar, D.; Longato, L.; Mazza, G.; Zhang, Z.; Marrali, M.; Fernandez-Iglesias, A.; et al. The adenosine monophosphate-activated protein kinase-vacuolar adenosine triphosphatase-pH axis: A key regulator of the profibrogenic phenotype of human hepatic stellate cells. Hepatology 2018, 68, 1140–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, N.C.; Iredale, J.P. Liver fibrosis: Cellular mechanisms of progression and resolution. Clin. Sci. 2007, 112, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.W.; Hien, T.T.; Lim, S.C.; Jun, D.W.; Choi, H.S.; Yoon, J.H.; Cho, I.J.; Kang, K.W. Pin1 induction in the fibrotic liver and its roles in TGF-beta1 expression and SMAD2/3 phosphorylation. J. Hepatol. 2014, 60, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, X.; Zhou, Q.; Huang, C.; Meng, X.; Xu, F.; Li, J. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol. Appl. Pharmacol. 2015, 289, 163–176. [Google Scholar] [CrossRef]
- Sun, L.; Fan, Z.; Chen, J.; Tian, W.; Li, M.; Xu, H.; Wu, X.; Shao, J.; Bian, Y.; Fang, M.; et al. Transcriptional repression of SIRT1 by protein inhibitor of activated STAT 4 (PIAS4) in hepatic stellate cells contributes to liver fibrosis. Sci. Rep. 2016, 6, 28432. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ghosh, A.K.; Chu, H.; Fang, F.; Hinchcliff, M.E.; Wang, J.; Marangoni, R.G.; Varga, J. The Histone Deacetylase Sirtuin 1 Is Reduced in Systemic Sclerosis and Abrogates Fibrotic Responses by Targeting Transforming Growth Factor beta Signaling. Arthritis Rheumatol. 2015, 67, 1323–1334. [Google Scholar] [CrossRef]
- Yu, F.; Lu, Z.; Cai, J.; Huang, K.; Chen, B.; Li, G.; Dong, P.; Zheng, J. MALAT1 functions as a competing endogenous RNA to mediate Rac1 expression by sequestering miR-101b in liver fibrosis. Cell Cycle 2015, 14, 3885–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Witek, R.P.; Yang, L.; Omenetti, A.; Syn, W.K.; Moylan, C.A.; Jung, Y.; Karaca, G.F.; Teaberry, V.S.; Pereira, T.A.; et al. Activation of Rac1 promotes hedgehog-mediated acquisition of the myofibroblastic phenotype in rat and human hepatic stellate cells. Hepatology 2010, 52, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Dai, X.; Chen, C.; Xue, J.; Xiao, T.; Mostofa, G.; Wang, D.; Chen, X.; Xu, H.; Sun, Q.; Li, J.; et al. Exosomal MALAT1 derived from hepatic cells is involved in the activation of hepatic stellate cells via miRNA-26b in fibrosis induced by arsenite. Toxicol. Lett. 2019, 316, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, R.; Huang, Z.; Gurley, E.C.; Wang, X.; Wang, J.; He, H.; Yang, H.; Lai, G.; Zhang, L.; et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans. Hepatology 2018, 68, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Zhu, W.; Wang, Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.; Lai, G.; et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology 2019, 70, 1317–1335. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Leti, F.; Legendre, C.; Still, C.D.; Chu, X.; Petrick, A.; Gerhard, G.S.; DiStefano, J.K. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl. Res. 2017, 190, 25–39.e21. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Flichman, D.; Garaycoechea, M.E.; San Martino, J.; Castano, G.O.; Pirola, C.J. Metastasis-associated lung adenocarcinoma transcript 1 as a common molecular driver in the pathogenesis of nonalcoholic steatohepatitis and chronic immune-mediated liver damage. Hepatol. Commun. 2018, 2, 654–665. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Tyagi, S.C.; Cleutjens, J.P. Collagen network of the myocardium: Function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 1994, 26, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, L.; Song, J.; Wang, Z.; Huang, X.; Guo, Z.; Chen, F.; Zhao, X. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J. Cell. Physiol. 2019, 234, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, W.; Xu, R.; Nie, Y.; Cao, X.; Meng, J.; Xu, X.; Hu, S.; Zheng, Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med. 2012, 16, 2150–2160. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, J.; Tan, W.L.W.; Jiang, Y.; Wang, S.; Li, Q.; Yu, X.; Tan, J.; Liu, S.; Zhang, P.; et al. Extracellular vesicles from human embryonic stem cell-derived cardiovascular progenitor cells promote cardiac infarct healing through reducing cardiomyocyte death and promoting angiogenesis. Cell Death Dis. 2020, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28. [Google Scholar] [CrossRef]
- Adeghate, E.; Singh, J. Structural changes in the myocardium during diabetes-induced cardiomyopathy. Heart Fail. Rev. 2014, 19, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gu, H.; Xu, W.; Zhou, X. Down-regulation of lncRNA MALAT1 reduces cardiomyocyte apoptosis and improves left ventricular function in diabetic rats. Int. J. Cardiol. 2016, 203, 214–216. [Google Scholar] [CrossRef]
- Zhang, M.; Gu, H.; Chen, J.; Zhou, X. Involvement of long noncoding RNA MALAT1 in the pathogenesis of diabetic cardiomyopathy. Int. J. Cardiol. 2016, 202, 753–755. [Google Scholar] [CrossRef]
- Che, H.; Wang, Y.; Li, H.; Li, Y.; Sahil, A.; Lv, J.; Liu, Y.; Yang, Z.; Dong, R.; Xue, H.; et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-beta1/SMADs signaling in diabetic cardiomyopathy. FASEB J. 2020, 34, 5282–5298. [Google Scholar] [CrossRef]
- Liu, J.; Xu, L.; Zhan, X. LncRNA MALAT1 regulates diabetic cardiac fibroblasts through the Hippo/YAP signaling pathway. Biochem. Cell Biol. 2020, 98, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, C.; Li, J.; Che, J.; Yang, X.; Xian, Y.; Li, X.; Cao, C. Long non-coding RNA MALAT1 promotes cardiac remodeling in hypertensive rats by inhibiting the transcription of MyoD. Aging 2019, 11, 8792–8809. [Google Scholar] [CrossRef]
- Murry, C.E.; Kay, M.A.; Bartosek, T.; Hauschka, S.D.; Schwartz, S.M. Muscle differentiation during repair of myocardial necrosis in rats via gene transfer with MyoD. J. Clin. Investig. 1996, 98, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.; Hermans-Beijnsberger, S.; Beqqali, A.; Bitsch, N.; Nakagawa, S.; Prasanth, K.V.; de Windt, L.J.; van Oort, R.J.; Heymans, S.; Schroen, B. Long Non-Coding RNA Malat-1 Is Dispensable during Pressure Overload-Induced Cardiac Remodeling and Failure in Mice. PLoS ONE 2016, 11, e0150236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Kaissling, B.; Lehir, M.; Kriz, W. Renal epithelial injury and fibrosis. Biochim. Biophys. Acta 2013, 1832, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chen, J.K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFbeta-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zeng, L.; Cao, C.; Lu, C.; Lian, W.; Han, J.; Zhang, X.; Zhang, J.; Tang, T.; Li, M. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp. Cell Res. 2017, 350, 327–335. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, D.Y.; Sha, W.G.; Shen, L.; Lu, G.Y. Long non-coding RNA MALAT1 interacts with transcription factor Foxo1 to regulate SIRT1 transcription in high glucose-induced HK-2cells injury. Biochem. Biophys. Res. Commun. 2018, 503, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, R.; Li, X.; Fan, M.; Lin, J.; Zhen, J.; Chen, L.; Lv, Z. LncRNA MALAT1 is dysregulated in diabetic nephropathy and involved in high glucose-induced podocyte injury via its interplay with beta-catenin. J. Cell. Mol. Med. 2017, 21, 2732–2747. [Google Scholar] [CrossRef]
- Monga, S.P. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015, 148, 1294–1310. [Google Scholar] [CrossRef] [Green Version]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanthakumar, C.B.; Hatley, R.J.; Lemma, S.; Gauldie, J.; Marshall, R.P.; Macdonald, S.J. Dissecting fibrosis: Therapeutic insights from the small-molecule toolbox. Nat. Rev. Drug Discov. 2015, 14, 693–720. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, B.; Chen, Z.; He, Y.; Du, Y.; Liu, Y.; Chen, X. m(6)A-induced lncRNA MALAT1 aggravates renal fibrogenesis in obstructive nephropathy through the miR-145/FAK pathway. Aging 2020, 12, 5280–5299. [Google Scholar] [CrossRef]
- Li, H.; He, L.; Lou, W.; Wang, H.; Li, Y. Metastasis-related lung adenocarcinoma transcript 1 (MALAT1) promotes hypoxia-induced transdifferentiation of renal tubular mesothelial cells. Chin. J. Cell. Mol. Immunol. 2018, 11, 1007–1008. (In Chinese) [Google Scholar]
- Kolling, M.; Genschel, C.; Kaucsar, T.; Hubner, A.; Rong, S.; Schmitt, R.; Sorensen-Zender, I.; Haddad, G.; Kistler, A.; Seeger, H.; et al. Hypoxia-induced long non-coding RNA Malat1 is dispensable for renal ischemia/reperfusion-injury. Sci. Rep. 2018, 8, 3438. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundarakrishnan, A.; Chen, Y.; Black, L.D.; Aldridge, B.B.; Kaplan, D.L. Engineered cell and tissue models of pulmonary fibrosis. Adv. Drug Deliv. Rev. 2018, 129, 78–94. [Google Scholar] [CrossRef]
- Cao, G.; Zhang, J.; Wang, M.; Song, X.; Liu, W.; Mao, C.; Lv, C. Differential expression of long non-coding RNAs in bleomycin-induced lung fibrosis. Int. J. Mol. Med. 2013, 32, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.; Yu, I.T.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, P.J.; Clevelario, A.L.; Padilha, G.A.; Silva, J.D.; Kitoko, J.Z.; Olsen, P.C.; Capelozzi, V.L.; Rocco, P.R.; Cruz, F.F. Bosutinib Therapy Ameliorates Lung Inflammation and Fibrosis in Experimental Silicosis. Front. Physiol. 2017, 8, 159. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Wu, Q.; Yao, W.; Li, Y.; Liu, Y.; Yuan, J.; Han, R.; Yang, J.; Ji, X.; Ni, C. MiR-503 modulates epithelial-mesenchymal transition in silica-induced pulmonary fibrosis by targeting PI3K p85 and is sponged by lncRNA MALAT1. Sci. Rep. 2017, 7, 11313. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Liu, X.; Li, H.; Ning, H.; Zhou, P.K. PRKCSH Alternative Splicing Involves in Silica-Induced Expression of Epithelial-Mesenchymal Transition Markers and Cell Proliferation. Dose Response 2020, 18, 1559325820923825. [Google Scholar] [CrossRef]
- Cui, H.; Banerjee, S.; Guo, S.; Xie, N.; Ge, J.; Jiang, D.; Zornig, M.; Thannickal, V.J.; Liu, G. Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight 2019, 4, e124522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Chi, Y.; Deng, J.; Liu, Y.; Lu, Y.; Chen, J.; Dong, S. Fine particulate matter 2.5 exerted its toxicological effect by regulating a new layer, long non-coding RNA. Sci. Rep. 2017, 7, 9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Wang, F.; Li, P.; Li, F.S. Integrated Analysis of a Gene Correlation Network Identifies Critical Regulation of Fibrosis by lncRNAs and TFs in Idiopathic Pulmonary Fibrosis. Biomed. Res. Int. 2020, 2020, 6537462. [Google Scholar] [CrossRef] [PubMed]
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.; Moss, M.; Downey, G.P. The fibroproliferative response in acute respiratory distress syndrome: Mechanisms and clinical significance. Eur. Respir. J. 2014, 43, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Bulpa, P.A.; Dive, A.M.; Mertens, L.; Delos, M.A.; Jamart, J.; Evrard, P.A.; Gonzalez, M.R.; Installe, E.J. Combined bronchoalveolar lavage and transbronchial lung biopsy: Safety and yield in ventilated patients. Eur. Respir. J. 2003, 21, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J.; Xie, W.; Li, G.; Yao, L.; Zhang, R.; Xu, B. Overexpression of MALAT1 Relates to Lung Injury through Sponging miR-425 and Promoting Cell Apoptosis during ARDS. Can. Respir. J. 2019, 2019, 1871394. [Google Scholar] [CrossRef]
- Yao, M.Y.; Zhang, W.H.; Ma, W.T.; Liu, Q.H.; Xing, L.H.; Zhao, G.F. Long non-coding RNA MALAT1 exacerbates acute respiratory distress syndrome by upregulating ICAM-1 expression via microRNA-150-5p downregulation. Aging 2020, 12, 6570–6585. [Google Scholar] [CrossRef]
- Dai, L.; Zhang, G.; Cheng, Z.; Wang, X.; Jia, L.; Jing, X.; Wang, H.; Zhang, R.; Liu, M.; Jiang, T.; et al. Knockdown of LncRNA MALAT1 contributes to the suppression of inflammatory responses by up-regulating miR-146a in LPS-induced acute lung injury. Connect. Tissue Res. 2018, 59, 581–592. [Google Scholar] [CrossRef]
- Zhu, W.; Men, X. Negative feedback of NF-kappaB signaling by long noncoding RNA MALAT1 controls lipopolysaccharide-induced inflammation injury in human lung fibroblasts WI-38. J. Cell. Biochem. 2020, 121, 1945–1952. [Google Scholar] [PubMed]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Rat liver tissues of CCl4-induced liver fibrosis. TGF-β1activated HSCs. | SIRT1 | HSCs activation | [69] |
| Up | Rat liver tissues of CCl4 and bile duct ligation (BDL) induced liver fibrosis. Activated HSCs and hepatocytes. | miR-101b/Rac1 | HSCs activation and proliferation | [72] |
| Up | Activated HSCs. | CXCL5 | NASH mechanisms of inflammatory chemokines | [80] |
| Up | NAFLD patients. | unknown | NASH | [81] |
| UP | The liver of arsenite induces Mice. Activated HSCs co-culture with arsenite treated L-02 cells. The circulating exosomal of people exposed to arsenite. | miR-26b | HSCs activation | [75] |
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Peri-infarct zone of MI mice hearts. in Ang II-induced CFs. | miR-145 | Cardiac fibrosis after infarction | [88] |
| Up | Infarcted myocardium and cardiomyocytes treated with hCVPC-EVs. | miR-497 | Cardioprotective | [90] |
| UP | High-glucose CFs. DCM mice. | miR-141/NLRP3 | Cardiac fibrosis of DCM | [95] |
| Up | High-glucose CFs. DCM mouse model. | Hippo/YAP CREB | CFs proliferation and invasion Inflammation and collagen accumulation Cardiac fibrosis of DCM | [96] |
| Up | Myocardial tissues and thoracic aortic vascular tissues of SHRs. | MyoD | ASMCs activity Myocardial fibrosis in SHRs | [97] |
| Not change | Pressure overload-induced cardiac remodeling and failure in mice. | Unknown | Unknown | [99] |
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Renal tissues of db/db mice. High glucose-stimulated HK-2 cells | miR-145/ZEB2 | EMT of HK-2 cells Renal fibrosis | [33] |
| Up | Renal fibrosis in patients with obstructive nephropathy. TGF-β1-treated HK2 cells | miR-145/FAK | viability, proliferation and migration of HK2 cells. Renal fibrosis | [109] |
| UP | Renal tissues of unilateral ureteral obstruction mice. Hypoxia-induced HK-2 cells. | miR-100-3p/COL4A1 | EMT of HK-2 cells Renal fibrosis | [110] |
| Up | Hypoxic/ischemic kidney injury in humans, mice, and cells. | Unknown | Unknown | [111] |
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Silica-treated HBE | miR503 PI3K/Akt/mTOR/Snail | Silica-induced pulmonary fibrosis | [118] |
| UP | LPS-treated macrophages | Clec16a | Macrophage activation and bleomycin-induced pulmonary fibrosis | [120] |
| Up | PM2.5-treated HBE | Unknown | Inflammation and EMT process | [121] |
| Up | Peripheral blood of IPF patients | Unknown | Idiopathic pulmonary fibrosis | [123] |
| Up | ARDS patient plasma and PBMCs. | miR-425/PTEN | Cell apoptosis and ARDS | [126] |
| Up | ARDS patients and LPS-treated HPMECs | miR-150-5p/ICAM-1 | Cell apoptosis and lung injury in ARDS | [127] |
| Up | LPS-induced acute lung injury rat model | miR-146a | Inflammatory response | [128] |
| Up | WI-38 cells after treatment by LPS | NF-κB p65 | LPS-induced inflammation injury | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Liu, F.; Cai, Y.; Yang, Y.; Wang, Y. LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target. Crystals 2021, 11, 249. https://doi.org/10.3390/cryst11030249
Li Y, Liu F, Cai Y, Yang Y, Wang Y. LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target. Crystals. 2021; 11(3):249. https://doi.org/10.3390/cryst11030249
Chicago/Turabian StyleLi, Yijie, Fenglin Liu, Yunzhou Cai, Yanqing Yang, and Yuehong Wang. 2021. "LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target" Crystals 11, no. 3: 249. https://doi.org/10.3390/cryst11030249
APA StyleLi, Y., Liu, F., Cai, Y., Yang, Y., & Wang, Y. (2021). LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target. Crystals, 11(3), 249. https://doi.org/10.3390/cryst11030249