1. Introduction
The global abundance of methane gas has led to numerous investigations seeking effective methods for its utilization [
1,
2,
3]. The use of thermal reactions to convert methane into products with high carbon contents appears to be a promising approach [
4,
5]. Among the various methane conversion reactions, direct methane dehydroaromatization (MDA) has attracted significant attention in the fields of chemical engineering and material design because it can produce high-value aromatic compounds without producing oxidized carbon gases, such as carbon dioxide and carbon monoxide [
6,
7]. According to the bifunctional reaction mechanism, methane gas diffuses into the micropores of Mo-loaded zeolites and is converted to ethylene at the Mo sites before undergoing aromatization in the confined microporous channels. However, the mechanistic pathway of methane conversion is still under debate [
8,
9,
10,
11]. Al-ZSM-5 is a suitable candidate for use as an MDA catalyst because of its strong acidity, thermal stability, and the fact that its average pore size (~0.55 nm) is comparable to the size of aromatic compounds [
12]. Many studies employing Mo-loaded Al-ZSM-5 catalysts have reported high aromatic yields, with methane conversions close to the thermodynamic equilibrium [
4,
12,
13]. However, a gradual decrease after 2–3 h in the methane conversion and aromatic yield was also observed, which was attributed to the deactivation of Al-ZSM-5 as a result of coke formation in the micropores [
14].
As Al-ZSM-5 serves as an anchoring site for Mo species and provides microporous channels in which aromatization occurs, both features depend closely on its physiochemical properties [
9,
12]. Optimizing the design of Al-ZSM-5 may, therefore, provide an opportunity to solve the deactivation problem, and research to this end has been conducted. Optimization of the lengths of the microporous channels, for example, has been extensively researched [
15,
16,
17,
18]. Various structures (mesoporous, hollow, and hierarchical) prepared by templating methods [
15,
16,
19], post-treatment by dealumination [
20] and desilication [
21] have been studied in the context of the MDA reaction. Control of the acid strength of Al-ZSM-5 [
13,
22] and the distribution of Al [
23] in the crystal have also been shown to be effective in improving the catalytic performance of the MDA reaction. The results obtained by tuning these properties indicate that optimizing the design of Al-ZSM-5 is a key factor in achieving commercially attractive MDA reaction processes.
The core–shell zeolite structure allows for significant control over both, the acid-site distribution and the diffusion-channel length, making it suitable for use in MDA [
24]. Core–shell zeolites consisting of an Al-ZSM-5 core and a silicalite-1 shell have been prepared to eliminate the presence of external active sites, and precise control of the silicalite-1 layer thickness has been achieved by controlling certain synthesis parameters such as the amount of silica nutrients and the growth time [
23,
25]. The core–shell catalyst was found to exhibit improved catalytic stability; it was also found that the shell thickness plays a crucial role in determining the MDA reaction performance. A core–shell zeolite consisting of Al-ZSM-5 grown on the core of silicalite-1 has been previously reported [
26,
27]. This structure was found to induce a high rate of benzene formation and a reduction in coke formation owing to the effective loading of Mo and rapid diffusion of reaction species through the thin Al-ZSM-5 layer [
26]. Our research group recently reported the growth of Al-ZSM-5 (<100 nm) on 30–40 nm silicalite-1 crystals, leading to the formation of monodisperse core–shell zeolites [
28]. This approach allows for precise control over the Al-ZSM-5 shell layer via the seeded-growth process, and could also be used to tailor its design for use as a catalyst in the MDA reaction by the optimizing channel length and acid-site distribution. However, Al-ZSM-5 has a slightly different unit cell size compared to silicalite-1 because of the framework distortion which occurs due to Al–O–Si bonding. Furthermore, the solubility of silica and the corresponding nucleation/growth kinetics depend on the concentration of Al species in the sol used in the synthesis (hereafter referred to as the synthesis sol) [
29]. Such features may cause complications in the preparation of core–shell zeolites consisting of silicalite-1 cores and Al-ZSM-5 shells. To the best of our knowledge, there have been no reports on the microstructural evolution (revealing the Al distribution and the presence of Si–OH defects) during the growth of Al-ZSM-5 on silicalite-1 seeds.
Motivated by this, zeolites of various sizes were prepared through the growth of Al-ZSM-5 on silicalite-1 nanocrystals, and the Si/Al ratio (SAR) in the synthesis sol was varied. The microstructural properties of the resultant crystals were characterized by X-ray diffraction (XRD), Ar isotherm analysis, field emission–scanning electron microscopy (FE-SEM), and field-emission–transmission electron microscopy (FE-TEM). Interestingly, no significant changes were observed in the MDA reactions when different crystal sizes were used, regardless of the SAR. To investigate these results, the microscopic and catalytic properties of the prepared zeolites were characterized by energy dispersive spectroscopy (EDS), elemental analysis (EA), infrared spectroscopy (IR), and ammonia temperature-programmed desorption (NH3-TPD). Interesting features of core–shell zeolites were observed using these methods, including the unique Al distribution in the crystals and the presence of Si–OH nests depending on the crystal size. Based on these data, the growth kinetics of the silicalite-1 core/Al-ZSM-5 shell zeolite was deduced, and future directions for designing effective MDA catalysts were proposed.
2. Experimental
2.1. Materials
Tetraethyl orthosilicate (TEOS, 98%), silicic acid (99.9%), tetrapropylammonium hydroxide (TPAOH, 1M), and ammonium heptamolybdate ((NH4)6Mo7O24·4H2O) were purchased from Sigma-Aldrich. Aluminum isopropoxide (>98.0%) and sodium hydroxide beads (NaOH, 98.0%) were purchased from SAMCHUN. The deionized (DI) water used for all procedures and analytical measurements was purified using an aqua MAX-Ultra 370 Series purification system (18.2 Ω; YOUNGIN CHROMAS, Anyang, Korea). All chemicals were used as received without further purification.
2.2. Design Strategy of Core–Shell Zeolites
The procedure for the preparation of monodisperse core–shell zeolites (<100 nm) grown from silicalite-1 was described in our previous report [
30]. Only a slight improvement in the catalytic stability was exhibited using the core–shell nanocrystal catalysts in MDA, prompting the need for further microstructural engineering of the catalyst in terms of epitaxial growth and acid-site properties. Core–shell zeolites with various SARs and crystal sizes were prepared from silicalite-1 seed crystals, as shown in
Figure 1. The target crystal sizes were 80–100 nm and 600–800 nm, and the target SARs of the corresponding crystals were 35 and 140. The morphologies of the crystals and the growth kinetics which affect the distribution of Al species in the crystal were investigated, and the catalytic stability and hydrocarbon selectivity were studied in detail.
2.3. Synthesis of Silicalite-1 Nanocrystals
Silicalite-1 nanocrystals were synthesized using a two-stage varying-temperature synthesis process with a sol composition of 114 H2O:2.49 TPAOH:0.8 NaOH:10 SiO2. Approximately 8.93 g of 1.0 M TPAOH solution was mixed with 0.16 g of deionized (DI) water, and 0.127 g of sodium hydroxide was added to this solution. After stirring at 30 °C for 0.5 h, 2.5 g of silicic acid was added as the silica source to the reaction mixture. The mixture was then vigorously stirred at 30 °C for 24 h, and hydrothermal treatment was performed at 100 °C for 2 days after reaction at 60 °C for 6 days in a convection oven. Afterward, the white precipitate in the reaction mixture was washed three times with DI water by centrifugation at 13,000 rpm. The resulting product was then redispersed in DI water by sonication to prevent aggregation of the silicalite-1 seed crystals.
2.4. Synthesis of Conventional Al-ZSM-5
Conventional micron-sized ZSM-5 crystals were synthesized using precursor sols with 4050 H2O:4.5 TPAOH:20 SiO2:x Al2O3:2x NaOH, (where x = 0.29, 0.25, 0.15, 0.07, and 0). The mixture was stirred at 30 °C for 24 h. After filtering the precursor sol with a syringe filter of 0.45 μm (polypropylene), the mixture was transferred into a Teflon-lined stainless-steel autoclave, and a hydrothermal treatment was performed at 150 °C for 3 days. The product was washed with DI water by centrifugation at 13,000 rpm and dried overnight at 60 °C in a convection oven.
2.5. Synthesis of Al-ZSM-5 Nanocrystals
Nano-sized ZSM-5 crystals were synthesized using a seeded growth method with silicalite-1 nanocrystals as seeds. Typically, the precursor sols with 4050 H2O:4.5 TPAOH:20 SiO2:x Al2O3:2x NaOH (where x = 0.63, 0.29, 0.25, 0.14, 0.07, and 0) were stirred at 30 °C for 24 h. After filtering the precursor sol with a 0.45 µm (polypropylene) syringe filter, a predetermined amount of silicalite-1 nanocrystals was added to the precursor sols. The mixture was then transferred into a Teflon-lined stainless-steel autoclave, and the hydrothermal treatment was carried out at 150 °C for 1 d. After the cleaning procedure by centrifugation at 13,000 rpm, the products were dried overnight in a convection oven at 60 °C. Subsequently, the synthesized crystals were calcined at 550 °C for 6 h at a ramp rate of 1 °C/min, to remove the organic template.
2.6. Ion Exchange
The as-synthesized crystal samples were ion-exchanged in an aqueous NH4NO3 solution. Subsequently, 1 g of zeolite was added to 25 mL of NH4NO3 solution (0.5 M), and the mixture was then stored at 60 °C for 3 h and stirred. After the ion-exchange process, the samples were washed with DI water. This step was repeated three times, and the final samples were dried overnight at 60 °C. The dried samples were calcined at 500 °C for 3 h at a ramp rate of 1 °C/min.
2.7. Preparation of the Mo/HZSM-5 Catalysts
Mo/HZSM-5 catalysts were prepared using a conventional wet impregnation method. The nano-seeded HZSM-5 zeolites and commercial HZSM-5 zeolites (SiO2/Al2O3 = 60, Zeolyst) were obtained by the calcination of the NH4-ZSM-5 zeolites at 500 °C for 5 h in air to convert NH4+ to H+ before impregnation. Ammonium heptamolybdate ((NH4)6Mo7O24·4H2O, Sigma-Aldrich) was used as the Mo precursor; a Mo concentration of 4 wt.% (4 Mo/HZSM-5) was used in the catalysts. After Mo impregnation, the impregnated powder was dried at 80 °C for 12 h, followed by calcination at 500 °C for 5 h in air. The catalysts were pressed to form pellets, which were then crushed and sieved to obtain a particle size of 212–500 µm (35–70 mesh) for using in catalytic reactions.
2.8. MDA Reaction
The MDA reactions were conducted with 0.3 g of the catalysts in a fixed bed tubular quartz reactor (inner diameter of 10 mm) at atmospheric pressure and 700 °C. The catalyst bed was made stationary using a porous quartz wool. A uniform temperature profile along the catalyst bed was achieved using three different heating zones with an aluminum heating block inserted into the void space between the furnace and the tubular reactor. The catalyst bed temperature was monitored using three K-type thermocouples in direct contact with the aluminum block. Before conducting the MDA reaction, the catalyst samples were pretreated in the presence of He to remove water by increasing the temperature from 25 °C to 700 °C. A feed gas mixture of CH
4/Ar (90% CH
4 and 10% Ar, 10 mL/min) was introduced into the reactor with Ar serving as the internal standard. Calibration of the gas chromatography (GC) was carried out using a standard gas mixture consisting of linear hydrocarbons (CH
4, C
2H
4, C
2H
6, C
3H
8, C
4H
10, C
5H
12, and C
6H
14) and aromatic hydrocarbons (benzene, toluene, xylene (m/p mixture), 1,3,5-trimethylbenzene, 1,2,4-trimethylbenzene, and naphthalene), as shown in
Figure S1a,b. As shown in
Figure S1c, hydrocarbons higher than C5 were not detected in the chromatograph of the reaction product.
The reactor effluent was transferred via heated transfer lines to an Agilent 7890B GC system (Agilent, Santa Clara, USA) equipped with two capillary columns: an HP-PLOT/Q column (30.0 m × 0.32 mm × 0.5 µm; Agilent, Santa Clara, CA, USA) connected to a thermal conductivity detector to separate and analyze the permanent gases and a GS-GasPro PLOT column (60.0 m × 0.32 mm; Agilent, Santa Clara, CA, USA) connected to a flame ionization detector to separate and analyze the hydrocarbons. Ar, serving as the internal standard, was used to analyze the CH
4 conversion, product yield, and selectivity. The methane conversion efficiency of the catalysts was evaluated in terms of the number of moles of methane reacted per mole of methane fed into the system. The product selectivity was calculated on a carbon atom basis considering all carbon-containing products using Equation (1).
where
i is the carbon-containing species and
n is the number of carbon atoms in species
i. The evolution of the transient products throughout the course of the reaction was analyzed using an online MKS Cirrus II Quadrupole MS system (Micromeritics, Norcross, GA, USA) connected to the outlet of the GC.
2.9. Characterization
The XRD patterns of the samples were recorded using a New DB-Adavance (Cu Kα radiation) diffractometer (Bruker-AXS, Madison, WI, USA). The product FE-SEM images were obtained by SIGMA (Carl Zeiss, Jena, Germany). FE-TEM and EDS mapping images were obtained using a JEM-F200 microscope (JEOL, Akishima, Japan). FE-TEM specimens were prepared by dispersing zeolite crystals in ethanol. A small droplet of dispersion was coated on a TEM grid and dried at 30 °C for 1 day. The Ar adsorption and desorption isotherms of all the samples were obtained at −196 °C using an ASAP 2020 system (Micromeritics, Norcross, GA, USA). The catalyst surface area was calculated using the Brunauer–Emmett–Teller (BET) equation. The average pore diameter of each catalyst was calculated using the Barrett–Joyner–Halenda desorption method. The total pore volume was determined at a relative pressure (P/P0) of 0.99. The conventional t-plot method was used to extract the micropore volume and external surface area from the Ar adsorption data. Before the measurements, the samples were degassed under vacuum at 300 °C for 12 h. The in situ Fourier transform infrared (FT-IR) spectroscopy analysis of the samples was conducted on a Nicolet iS10 spectrometer (Thermo Fisher Scientific, Somerset, NJ, USA). Self-supported pellets with a density of 14.9 mg/cm2 were prepared at a pressure of 1000 kPa and placed in the IR cell. The prepared pellets were pre-activated at 400 °C for 2 h under vacuum (~10−5 Torr). All spectra were recorded within the spectral range of 4000–6000 cm−1 with a resolution of 4 cm−1. The SAR of the zeolite samples was determined using 5100 ICP-OES (Agilent, Santa Clara, CA, USA). The following conditions were used: plasma power = 1500 W, plasma gas flow = 10 L/min, auxiliary gas flow = 0.2 L/min, and nebulizer gas flow = 0.70 L/min. Each sample was completely dissolved in an acid mixture (8 mL HNO3, 1 mL HF, 1 mL H2O2) diluted with up to 50 mL of DI water. The wavelengths of the spectrometric lines used for the analysis were 251.611 nm (Si) and 396.152 nm (Al).
Temperature-programmed desorption of ammonia (NH3-TPD) was conducted to investigate the acidic properties of the catalysts. Prior to the analyses, 100 mg of the catalyst was treated at 500 °C for 1 h in He to remove the adsorbed water and organic species. After saturation with a 15 vol% NH3/He gas mixture at 100 °C for 30 min, the catalysts were degassed with He for 1 h. NH3 was then desorbed over the temperature range of 100–500 °C at a heating rate of 5 °C/min under a He flow of 50 mL/min. The desorbed NH3 was monitored using an AutoChem II 2920 (Micromeritics, Norcross, GA, USA).
3. Result and Discussion
The preparation of monodisperse core–shell zeolite (<100 nm) grown from silicalite-1 was reported in our previous report [
30]. Only a slight improvement in the catalytic stability was obtained using the core–shell nanocrystal catalysts in MDA, prompting the need for further microstructural engineering of the catalyst in terms of epitaxial growth and acid-site properties. Zeolites with various SARs and crystal sizes were prepared from silicalite-1 seed crystals, as shown in
Figure 1. The crystal sizes of zeolites grown on silicalite-1 were designed to be 80–100 nm and 600–800 nm, and the SAR of the corresponding crystals was tuned to 35 and 140. The morphologies of the crystals and growth kinetics affecting the distribution of Al species in the crystal were investigated, and the catalyst stability and hydrocarbon selectivity were thoroughly studied.
Figure 2 shows the FE-SEM, FE-TEM, and EDS results of core-shell Al-ZSM-5 crystals. The samples were designated as 80 nm_SAR 35, 600 nm_SAR 35, 100 nm_SAR 140, and 800 nm_SAR 140. The first number describes the crystal size, and the second number denotes the SAR of the synthesis sol. The crystallization reaction was conducted at 150 °C for 24 h. As shown in
Figure 2a, the 100 nm_SAR 140 sample shows ~100 nm crystal size and rough, irregular shape by FE-SEM. Spherical protrusions were observed by FE-TEM, and a single crystalline structure was confirmed by electron diffraction (ED). The EDS results of the sample display uniform distribution of Si throughout the crystal; the distribution of Al in the crystal was not distinct because of the small crystal size. These data indicate that aggregate growth occurred on silicalite-1 crystal, whereas the densification stage, which results in a smooth surface, was not reached because of the small crystal size. In contrast, a well-faceted coffin-shaped crystal was observed for the 800 nm_SAR 140 sample, as shown in the FE-SEM image in
Figure 2b. The corresponding ED pattern revealed the single-crystalline nature of the zeolite, and the EDS mapping indicated that both Si and Al were uniformly distributed in the zeolite. Thus, the 800 nm_SAR 140 sample evolved to the densification stage of crystal growth. The 80 nm_SAR 35 sample exhibited irregular spherical morphologies, as observed by FE-SEM, and ellipsoidal morphologies, as observed by FE-TEM (
Figure 2c). It had a smaller crystal size than that of 100 nm_SAR 140 under the same seed/nutrient gel condition because of the difficulty in crystal growth in low SAR, as discussed in our previous report [
30]. A single crystalline structure was revealed by ED for 80 nm_SAR_35, whereas the distribution of Al in this sample could not be observed by EDS mapping, possibly due to its small crystal size. Similar to 100 nm_SAR 140, the crystal of 80 nm_SAR_35 was mainly composed of small crystallites by aggregative growth without proceeding to the densification stage. Conversely, the 600 nm_SAR 35 sample exhibited a rough external surface despite its well-developed coffin shape, as shown in the FE-SEM and FE-TEM images in
Figure 2d. The ED pattern revealed that the 600 nm_SAR 35 sample was single crystalline, and the EDS mapping showed that Si and Al were well-dispersed throughout the entire crystal. Thus, this crystal proceeded to the densification stage after the early stage of crystal growth, similar to 800 nm_SAR 140. The results in
Figure 2 indicate that the large crystals (800 nm_SAR 140 and 600 nm_SAR 35) were formed through a two-step growth process, consisting of early aggregative growth on silicalite-1 seeds and subsequent densification by the addition of monomeric and oligomeric silica/alumina species [
31,
32,
33].
The MDA reaction was then conducted using the Mo-impregnated Al-ZSM-5, where Mo-loaded zeolites were designated as “crystal size_Mo_SAR ratio.” For instance, 80 nm of Al-ZSM-5 with a SAR of 35 was represented as 80 nm_Mo_SAR 35.
Figure 3 shows the methane conversion efficiency of the zeolites as a function of the time-on-stream (TOS) characterized by GC-MS at atmospheric pressure and 700 °C. As shown in
Figure 3, after 1 h of induction, the conversion in the presence of each zeolite sample reached 12% of the equilibrium conversion for all the samples and then gradually decreased because of catalyst deactivation. The 80 nm_Mo SAR 35 and 100 nm_Mo SAR 140 samples showed slightly higher methane conversion than the large zeolite samples. However, this difference was marginal. In addition, the methane conversion and deactivation efficiencies of the zeolite samples were not significantly different from those of the conventional zeolite (SAR = 120) (
Figure S2). Therefore, although the Al-ZSM-5 samples functioned as catalysts for the MDA reaction, they did not show substantial improvement in the methane conversion efficiency and catalyst stability compared to the conventional zeolite sample.
Figure 4 shows the hydrocarbon selectivity of the MDA reaction in the presence of the zeolite samples.
Figure S2 shows the hydrocarbon selectivity of the conventional zeolite sample (SAR = 120). As shown in
Figure 4 and
Figure S2, at a TOS of 1 h, the zeolites showed significantly different naphthalene and ethylene selectivities depending on their morphology. This difference diminished after 2 h of TOS because of catalyst deactivation. At a TOS of 1 h, the Al-ZSM-5 zeolites showed a naphthalene selectivity of less than 20% (
Figure 4a–d), whereas the conventional zeolite showed a naphthalene selectivity of approximately 30%. The ethylene selectivity of the core–shell zeolites was 10% or higher, while that of the conventional zeolite was less than 5% (
Figure S2c). This difference in the naphthalene and ethylene selectivities of the zeolite samples and conventional zeolite samples can be attributed to the differences in their channel lengths and external surface areas. The low naphthalene selectivity of the zeolite samples indicates that the benzene produced in the zeolite channel was effectively evacuated from the zeolite without excessive aromatization because of the short microporous channels. In contrast, the high ethylene selectivity of the core–shell samples indicates that ethylene was not fully utilized in aromatization. This suggests that ethylene was produced more around the Mo species located on the external surface of the zeolites, which restricted the flow of ethylene into the zeolite microchannels for cyclization.
As shown in
Figure 4a, among all the zeolite samples investigated, the 800 nm_Mo SAR 140 sample exhibited the highest benzene selectivity, followed by the 600 nm_Mo SAR 35 sample (
Figure 4b). This suggests that a certain micropore length scale is required to achieve a high benzene yield, and the sub-100 nm length scale appears to be insufficient for effective cyclization of the C
2 intermediate. In contrast, the benzene selectivity and methane conversion of the samples did not correlate with the target SARs, as shown in
Figure 4a–d. This outcome suggests that the discrepancy originates from microstructural differences in the core–shell zeolites, which affect their catalytic activities. Therefore, further characterization was performed to elucidate the structural evolution of the core–shell zeolites.
Figure 5 shows the XRD pattern and Ar isotherm of the zeolite samples. As can be seen from the XRD pattern (
Figure 5a), all the samples showed pure MFI structures. Stronger XRD signals were observed from samples with large crystals compared with those from samples with smaller sizes and same SAR. Ar isotherm measurements of the samples after calcination are presented in
Figure 5b and
Table 1. A clear uptake of Ar was observed in the low relative pressure region (P/P
0 < 0.01), indicating the presence of micropores in the samples. The two small-sized zeolite samples showed large Ar adsorption in the high-pressure region (P/P
0 > 0.9) because of the voids owing to small particle aggregations. The micropore volume of the 800 nm_SAR 140, 600 nm_SAR 35, and 100 nm_SAR 140 samples was more than 0.17 mL/g, as calculated using the t-plot method (
Table 1). Hence, these samples showed a highly crystalline structure. In contrast, 80 nm_SAR 35 showed a slightly lower micropore volume of 0.14 mL/g, indicating the presence of more non-zeolitic species in the crystal because of low evolution during crystal growth. This is consistent with the microscopic characterization results shown in
Figure 2; there were no unexpected features altering the catalytic properties.
Then, elemental analysis of the samples was conducted by ICP-OES to determine their SARs (
Table 2). Interestingly, the SARs of the sols did not match those of the crystallized samples, and Al incorporation into the zeolite framework depended on the crystal size. For the small zeolites (100 nm_SAR 140 and 80 nm_SAR 35), the SAR in the zeolite sample was higher than that in the synthesis sol. Although the bulk average SAR of 80 nm_SAR 35 was 46.7 (silicalite-1 core and Al-ZSM-5 shell), that of the Al-ZSM-5 shell was calculated to be less than 35. Conversely, for the large core–shell zeolites (800 nm_SAR 140 and 600 nm_SAR 35), the amount of Al incorporated into the framework was lower than that in the original synthesis sol. This result indicates that numerous Al species participated in the early stage of Al-ZSM-5 growth on silicalite-1 seeds, and the contribution of Al species to crystal growth decreased gradually with increasing crystal size. This phenomenon is believed to be associated with the transition of the growth mechanism from the early precursor-particle-based aggregative growth to the densification of aggregates with the progress of crystal growth [
33]. Similar kinetics associated with the contribution of Al species to crystal growth was reported previously [
34]. Thus, it is expected that the Al distribution would have dissimilar crystal sizes, as depicted in
Figure 6. In the case of the small size of Al-ZSM-5, the Al species were concentrated but evenly distributed in the thin Al-ZSM-5 shell layer. On the other hand, a large number of Al sites were present adjacent to the silicalite-1 seed, and the Al distribution was small at the exterior layer for larger crystals (800 nm_SAR 140 and 600 nm_SAR 35). Thus, the crystal-size-dependent variation in Al incorporation into and distribution within the core–shell zeolite may have caused the inconsistencies in the MDA reactions shown in
Figure 3 and
Figure 4. The Al distribution in large crystals is different from the Si/Al distribution in conventional zeolites, which decreases from the interior to the exterior layer due to Al zoning [
35]. The Al distribution observed in core–shell zeolites should be beneficial in MDA catalysis because the low concentration of Al in the outer layer of the crystals helps increase the stability. Previous studies have shown that an Al distribution of this type can help suppress coke formation at the external surface during MDA [
23].
To further analyze the structure and the corresponding acidity of the prepared zeolites, IR spectroscopy and NH
3-TPD analyses were performed.
Figure 7a shows the IR spectra of the zeolite samples, indicating the presence of different hydroxyl groups in the samples. Commercial Al-ZSM-5 with an SAR of 120 was also characterized for comparison. The zeolites showed three distinct peaks around 3750, 3600, and 3500 cm
−1 (
Figure 7a), corresponding to the external Si-OH group, Brønsted acid sites, and internal Si-OH nests, respectively, [
36]. Highly siliceous MFI zeolites often accompany Si-OH nests [
36,
37]. The commercial Al-ZSM-5 exhibited two distinct bands at 3750 cm
−1 and 3600 cm
−1, and its IR absorption was lower than that of the core–shell zeolite, as shown in
Figure 7a. It can be deduced that the commercial zeolite had a smaller external surface and fewer Si–OH defects than the core–shell zeolite. Three bands with different intestines were observed for the Al-ZSM-5 samples. The 80 nm_SAR 35 sample exhibited two strong bands at ~3750 cm
−1 and 3600 cm
−1 and a weak peak at ~3500 cm
−1, attributed to its large external surface area and low SAR (
Table 2 and
Figure 5b). The 100 nm_SAR 140 sample showed similar features; however, the peaks were weaker than those exhibited by the 80 nm_SAR 35 sample because of its smaller external surface area and higher SAR. An intense band was observed at ~3500 cm
−1 for both, the 800 nm_SAR 140 and 600 nm_SAR 35 samples. The peak at ~3500 cm
−1, corresponding to the Si–OH nests, can be attributed to the increase in the SAR of the sol during Al-ZSM-5 synthesis. However, the 600 nm_SAR 35 sample, which had the same sol composition, exhibited more intense Si–OH nest bands than the 80 nm_SAR 35 sample. The Si–OH nest bands of 600 nm_SAR 35 were stronger than those of the 100 nm_SAR 140 sample, indicating that Al was unevenly distributed and deficient at the external surface of the Al-ZSM-5 layer, as shown in
Table 2 and
Figure 6. The IR absorption was higher for the 800 nm _SAR 140 sample than for the 600 nm _SAR 35 sample because a higher SAR leads to a greater number of Si–OH defects. From the NH
3-TPD patterns (
Figure 7b), it can be observed that all the core–shell samples exhibited typical double peak characteristics with weak acidities at 210 °C and strong acidities at 410 °C. The patterns revealed a clear dependence of the number of acid sites on the SAR of the samples, which is consistent with the elemental analysis (EA) results shown in
Table 2. Among all the samples investigated, the 80 nm_SAR 35 showed the highest weak and strong peak intensities, whereas the 800 nm_SAR 140 showed the lowest strong and weak peak intensities.
In the conversion of core–shell crystals into a catalyst for the MDA reaction, Mo species were introduced by incipient wetting and annealing processes. Mo species possess high dehydrogenation and C-C coupling capabilities and act as metal oxide catalyst in the MDA reaction [
9].
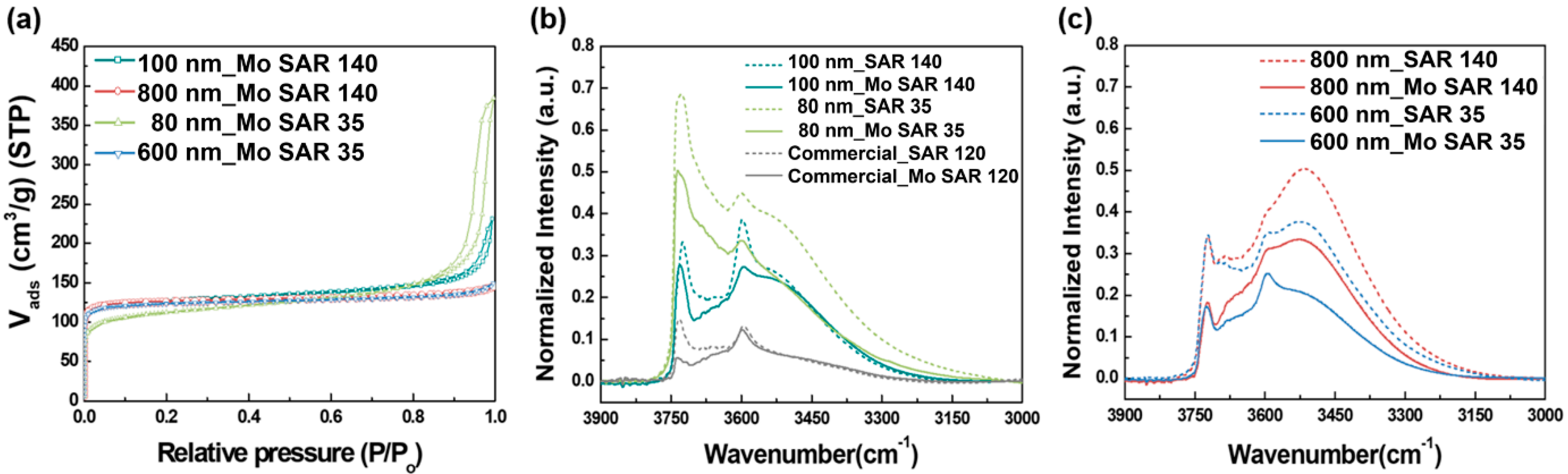
Figure 8 shows the Ar isotherm and IR spectra of the samples after Mo impregnation. After Mo impregnation, the Ar uptake by the samples over the entire P/P
0 range decreased slightly (
Figure 8a), and the micropore volume was also decreased (
Table 2). However, the samples still showed significant Ar uptake in the low-pressure region (P/P
0 < 0.01), indicating that the micropores of the samples were well preserved without pore blockages after Mo impregnation.
Figure 8b,c shows that the IR band intensities decreased over the entire wavenumber range for all the samples, indicating that the Mo species were anchored at every hydroxyl spot in the zeolite samples. Specifically, the two small crystal samples (80 nm_SAR 35 and 100 nm_SAR 140) showed reduced intensities of the IR bands at 3750 and 3600 cm
−1 (
Figure 8b). However, 800 nm_SAR 140 and 600 nm_SAR 35 showed significant decrease in the intensity of the IR band observed at approximately 3500 cm
−1 (
Figure 8c). Hence, most of the Mo species were likely dispersed at the Brønsted acid sites and the external surface for the small core–shell samples. However, the internal Si-OH nests also participated significantly in the dispersion of Mo species in the large Al-ZSM-5 samples. Therefore, the nests could inhibit the incorporation of Mo species into the acid sites of the larger crystals, decreasing the number of active sites for the MDA reaction.
Based on the characterization data shown in
Figure 4,
Figure 5,
Figure 6,
Figure 7 and
Figure 8, it was evident that larger crystals contained fewer Al sites than smaller crystals prepared from the same synthesis sol composition. Thus, epitaxial growth of the core–shell zeolite did not occur, and the microstructure of the core–shell zeolite changed significantly depending on the crystal size. Furthermore, large Si-OH nests in large core–shell zeolites can also decrease the number of active sites for the MDA reaction. These factors might adversely affect the hydrocarbon selectivity and benzene yield for larger crystals, leading to minor changes in the MDA reaction performance, as shown in
Figure 4. Further studies, such as controlling the SAR ratio and removing Si-OH nests, are underway, and the results will be reported in the near future.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}