1. Introduction
Crystalline bacterial surface layers (called S-layers) are known to be one of the most common cell surface structures in archaea and bacteria [
1,
2,
3,
4,
5]. S-layers are monomolecular arrays of a single protein or glycoprotein species (M
w 40 to 200 kDa) and completely cover the archaeal or bacterial cell (
Figure 1). Furthermore, S-layer proteins can be considered one of the most abundant biopolymers on earth since the biomass of prokaryotic organisms exceeds that of eukaryotic organisms [
6]. S-layers exhibit either oblique (p1, p2), square (p4) or hexagonal (p3, p6) lattice symmetry. Accordingly, a unit cell (morphological unit) consists of one, two, four, three, or six identical monomers.
Figure 1 shows a transmission electron microscopy (TEM) image of a bacterial cell with an S-layer with square lattice symmetry. The unit cell dimensions of S-layers range from 3 to 30 nm, while the thickness is between 5 and 10 nm (up to 70 nm in archaea). Due to their crystalline nature, S-layers are porous protein networks (30–70% porosity) with pores of uniform size (2–8 nm) and morphology [
7,
8].
The widespread occurrence and high physiological cost of forming S-layer proteins raises the question of the selection advantage of S-layer carrying organisms in their natural and often highly competitive habitats. This is because approximately 500,000 S-layer monomers are required to completely cover a rod-shaped bacterial cell. Assuming a generation time of 20 min, this means more than 400 copies of a single protein or glycoprotein species are synthesized per second [
10]. In this context, it is interesting to recall that under optimal growth conditions for bacteria (in continuous laboratory cultures), S-layer deficient mutants, or variants possessing S-layers composed of (glyco)protein subunits with lower molecular mass, often outgrow of wild-type strains. Since the S-layer covers the entire cell surface, it has been inferred that many biological functions may depend on the completeness in the coverage and on the repetition of the ultrastructure and chemical groups on the surface down to the sub nanometer range [
4]. Furthermore, the surfaces of many S-layer carrying bacteria show excellent antifouling properties. It may serve as model system for the development of ecologically benign and long-lasting marine antifouling coatings in the maritime sector [
11]. This surface property was first observed in TEM images of freeze-etched and metal-shadowed samples where intact cells were vitrified (30,000 K/s) [
2]. Even when cells were harvested from complex environments or growth media containing a variety of macromolecular components, the S-layer lattices were never covered by adsorbed molecules. In this context, it should be borne in mind that the self-cleaning ability of biological surfaces depends not only on the wettability but also on the water structure close to the substrate. It can be speculated that glycosylation of the S-layer may even enhance this effect [
12]. Detailed studies on molecular interactions and permeability of S-layers confirmed that the outer S-layer surface is usually charge-neutral, caused by an equal amount of carboxyl- and amino groups, which prevents non-specific binding of molecules and blocking of pores [
12]. S-layers were adjusted by nature and outperform chemical polymer modification, resulting in amphiphilic marine antifouling/fouling-release coatings by tuning the ratio of hydrophilic and hydrophobic side chains [
13].
In contrast, the inner surface is either net positively or negatively charged, caused by an excess of one of the two groups. In this context, the permeability properties of S-layers should also be considered, which have been intensively studied and characterized over several decades [
8]. Using S-layers from Bacillaceae as a model system, it was clearly shown that the S-layer lattices have well-defined exclusion limits in the range of 30–40 kDa, indicating a limiting pore diameter in the range of 3–4.5 nm [
7]. In another approach, impedance spectroscopy was used to investigate the ion gating of S-layers, e.g., for calcium ions [
14]. The ion current through the pores was driven by an electrical gradient within the pores, most likely caused by the difference between the charge-neutral outer and the negatively charged inner S-layer surface.
In vivo and in vitro studies elucidating the dynamic assembly process of S-layer proteins from Gram-positive bacteria during cell growth revealed that S-layers maintain an equilibrium with lowest free energy during the reassembly process [
4]. Although most of the functions assigned to S-layers are still hypothetical, the supramolecular concept of a closed, isoporous protein lattice must represent a specific adaptation to different ecological and environmental conditions. Synthetic superhydrophobic (SH) surfaces were developed starting in the 1990s and have been the subject of nearly 13,500 publications. The approaches, some of which were very innovative, found no industrial application. The water-repellent properties of SH surfaces fall prey to weak mechanical properties as well as to contamination, which reduces or even destroys the original SH properties [
15]. Consequently, we develop techniques to access the biomechanical properties and link them to integral models.
Considering the combination of antifouling properties [
16,
17] and the transport of nutrients and metabolites through the pores of the S-layer, the central question of how bacteria and archaea can overcome the diffusion limit (based on Brownian motion) to bring nutrients into the cell and remove waste products from the cell is still open. This question was raised more than thirty years ago by E. M. Purcell in a pioneering lecture in which he studied life at low Reynolds numbers, particularly the locomotion of flagellum-driven bacterial cells [
18]. For such small objects, the viscosity of water dominates their life. Since bacterial cells have an extremely low Reynolds number R, typically of 10
−4, they entrain the dense environment, which decreases only slowly. The viscous forces dominate, and inertia is entirely irrelevant. To overcome the local diffusion-limited transport of nutrients into the cell and waste away from its surface, flagellated prokaryotes swim certain distances before stopping or changing their direction. This is the only way for the bacterial cell to find a difference in the environment or induce local turbulence and, in this way, take the opportunity to increase the exchange rate. Since bacteria are not constantly moving and are sedentary to some extent, the turbulences generated by the S-layer could be an evolutionary solution to overcome the diffusion limit in nutrient supply, especially for non-mobile cells bound to surfaces or trapped in a biofilm.
Based on preliminary drag experiments with an optical trap, which indicated that micrometer-sized S-layer coated microbeads have a slightly higher flow resistance than blank beads (data not shown), we decided to calculate the water flow across S-layers in silico and to try to develop a theoretical model that will allow us to better understand the S-layer–water interface. As a model system, we used high-resolution cryo-TEM tomography data of the S-layer protein SbpA from
Lysinibacillus sphaericus ATCC 4525 (identical to
Lysinibacillus sphaercius CCM2177, see [
19]) [
9,
20].
The combination of SAXS data and cryo-EM data is completely new in the field and will later serve as a basis for more detailed investigations. To the best of our knowledge, no previous study has combined two orthogonal techniques, here SAXS (reciprocal space) and cryo-EM tomography (real space), using a linear programming technique. By this approach, we create a high-resolution model and consider the biophysics of the S-layer. We solve the Navier–Stokes and Poisson equations and determine the size of a simulation box required to examine the impacts of the S-layer on the water matrix.
3. Results and Discussion
To our knowledge, this is the first time, in the literature, that cryo-EM and small-angle X-ray scattering datasets have been combined to obtain a high-resolution model of the SbpA S-layer protein. The cryo-EM data were taken from a previous publication [
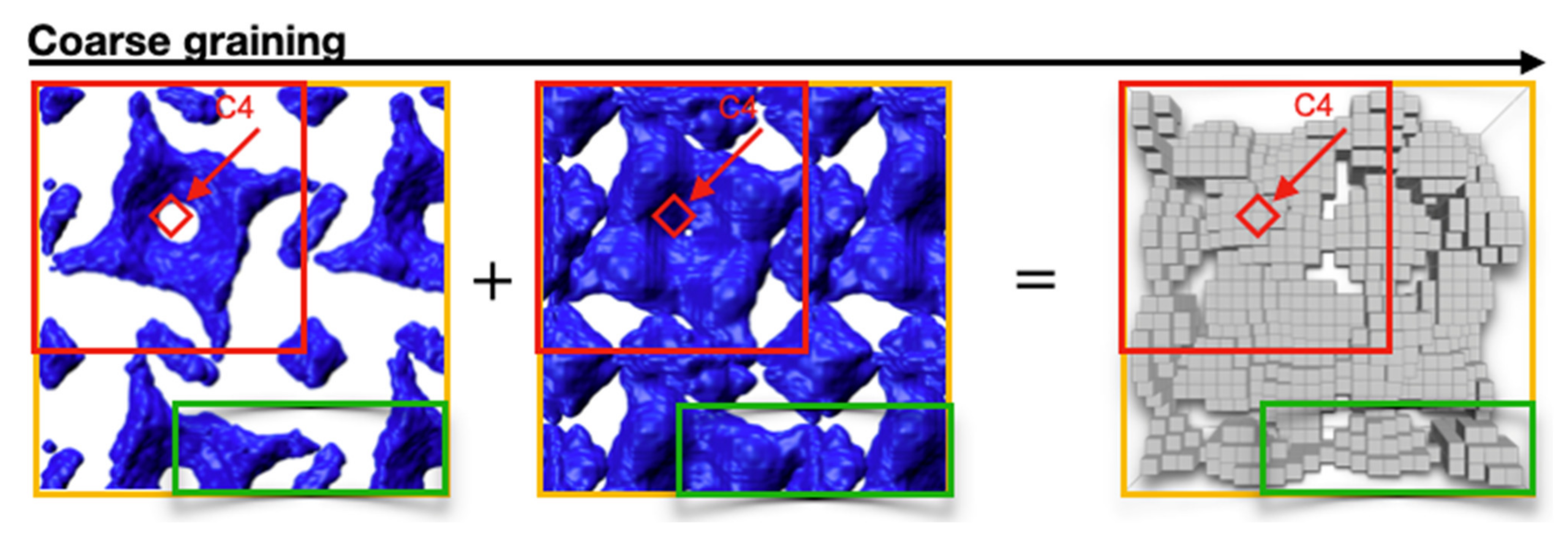
20]. The morphological unit of SbpA with its p4 lattice symmetry was assembled in silico in
Figure 1. Thus, the blue colored hull consists of 16 protein monomers. The four-fold axes are marked by red upright squares. In addition to the pair densities within the unit cell, pair densities from neighboring unit cells are also included. The green rectangle marks the simulation box for the Navier–Stokes and Poisson equations, which are solved at the end of this paper.
The core model in
Figure 2a was discriminated from the hull model in
Figure 2b. In the core model, all potential scattering sites were placed within the hull of the cryo-EM data, while in the hull model, they were placed on the hull of the cryo-EM data. Pair densities were computed thereof and are indicated in the inserts by full and dashed blue lines.
At ease, the form factor,
P(Q), was computed. It is the Fourier transform,
of the individual pair density distributions:
It was computed and is shown by the solid and dashed blue lines in
Figure 2. Both deviated significantly from the experimental scattering data,
I(Q). Standard small-angle X-ray scattering approaches [
21,
28] relate these discrepancies between the two curves to the apparent structure factor of the SbpA self-assembly:
This approach has already been used in earlier work [
21]. There, the small-angle X-ray scattering signal was also divided into two factors. One corresponded to the form factor, while the second factor corresponded to the structure factor of the primitive unit cell. Although this approach is justifiable, we can show that it is no longer necessary from now on [
21]. The small-angle X-ray scattering signal can be recovered by the Fourier transform of a pair density distribution from the S-layer model. Pair densities calculated from cryo-EM data were used as initial configuration, and hypothetical pair densities were calculated by a linear programming algorithm:
Parameter α = 1, and Equation (2) was optimized by the use of the L
2 Norm. The constants
c1 and
c2 were introduced due to experimental uncertainties. The results obtained by applying the linear programming algorithm were remarkable. In
Figure 2, computed pair densities and the corresponding hypothetical scattering intensities are given by red lines. By the use of the linear programming approach, hypothetical scattering intensities matched the experimental data perfectly. The chosen unit cell already had enough information about the periodicity of self-assembled array to explain the scattering pattern. The computed pair density shows small protrusions at identical positions independent of the pair density distributions. These protrusions are interpreted as high populations of relative distances within the primitive p4 unit cell.
It is not shown here, but increasing the number of unit cells would not affect the quality of the fit. It is obvious because, for larger distances, the calculated pair densities scale with the power of 2 of the relative distances, and consequently, the Fourier transform falls to zero:
Since the core and hull models give similar results, both were combined, and a final coarse-grained box model was designed, which is introduced in
Figure 3. The core and hull models were superimposed. The resulting model was rasterized, leaving only volumes that enclosed potential scattering sites. This approach reduced the resolution to approximately 1 nm, which is the approximate side length of an individual grain.
Next, the grey line in the upper panel in
Figure 4c indicates the pair density of the grey box model. This pair density was chosen as an initial pair density distribution. Again, we applied the linear programming algorithm of Equation (7) to compute the best hypothetical scattering function. This is the dashed grey line in the lower panel of
Figure 4c. The mismatch between the pair density calculated from the box model and the pair density that provided the best hypothetical scattering function was balanced by introducing weights. Each box was given a weight between 0 and 1. Here, again, we have applied a linear programming algorithm:
The weights were continued until the pair densities computed from the weighted box model matched the pair densities calculated from the experimental scattering data (
Figure 4c, upper and lower panel). Boxes that have received a rating of less than 0.3 are not shown in the colored box model. While light red boxes weigh 0.3, boxes in darker red have been colored because their weighting is closer to 1. The consequences can be interpreted in two ways: The boxes could be volumes of high electronic contrast, indicating the electronic morphology of the unit cell. We emphasize the following interpretation: darker boxes are entities within the SbpA layer that do not move, and thus their probability of contributing to the scattering signal is higher than the lighter boxes, which had a lower probability of scattering coherently within the S-layer. The dark boxes are then the pillars spanning the S-layer. The lighter boxes on the other side comprise scattering sites that are weaklier bound within the SbpA lattice. They could move, they could open or close the S-layer, and they could form selective pores.
We chose the idea that darker volumes represent regions of lower mobility within the S-layer. These darker volumes act like pillars and span the SbpA self-assembly as described above. We apply a simple Navier–Stokes computation to calculate the flow profile that the S-layer exerts on the fluid around it. We will investigate the impact of the S-layer on the water matrix at a later stage in a 3D lattice Boltzmann molecular dynamics simulation. Instead, we explored the question of whether, at low Reynolds numbers, a particular distribution of rigid regions in the S-layer could influence the flow of the fluid above. We simplified our approach and argued that the flow over a unit cell is unidirectional due to the high viscosity, and any lateral contributions within one unit cell are small.
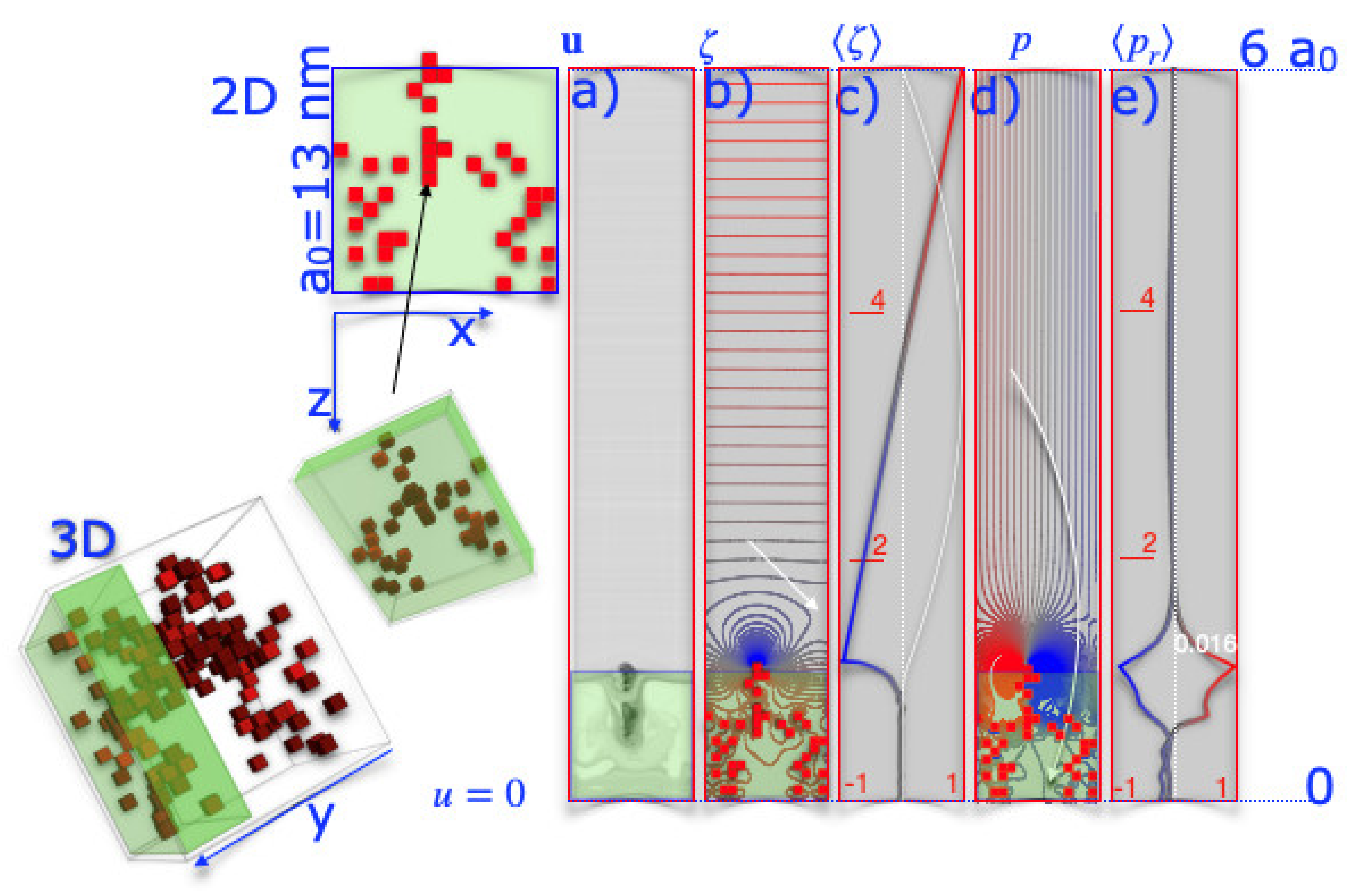
This raises the question of what the overall flow field looks like when the rigid volumes act as obstacles. The graphs in
Figure 5 show the velocity, vorticity, and pressure of the fluid in a slit. The boundary conditions are periodic in the x-direction. The S-layer is located at the bottom edge, while the top edge remains untouched. Non-slipping boundary conditions are introduced at z = 0 and z = 6
a0. There, the velocity is kept equal to zero. The flow field, as shown in
Figure 5a, is hardly disturbed. It was to be expected, as the viscosity was set high. The obstacles presented by the rigid regions strongly disturb the flow field only in their vicinity. In the S-layer, we find curved trajectories, which can be interpreted as an indication that the periodic structure of the S-layer imprints a flow field that triggers transport through the S-layer.
Again, the flow field is hardly disturbed near the S-layer, and even at distances of less than one lattice constant, it is laminar. From the trajectories, we then calculated the vorticity, which is more structured. A high vorticity is found near the surface of the S-layer (blue, red), and due to the periodicity of the S-layer, the vorticity is structured next to it. Near the SbpA S-layer surface, where the vorticity (blue) is high, the trajectories of molecules suspended in the liquid can be affected, leading to their out-of-plane transport. In the present calculation, the SbpA S-layer induced vorticity profiles of 10–20 nm deep into the liquid. In regions to the left and right of this, the vorticity was close to zero.
The trajectories of proteins or nanoparticles (
Figure 5, white arrow) would not be affected there. According to the pore size, particles with spatial dimensions smaller than
a0 would rest above the pore and could thus stay long enough near the pores to pass the S-layer lattice towards the cell. On the other hand, it could be seen as an indication of a Lotus leaf effect, as particles larger than the repeating distance of the S-layer would be transported parallel to the S-layer [
29]. On average, the vorticity was very different near the S-layer compared to a blank surface. Pressure is a variable that changes the most when the nanostructured surface is compared to a blank surface. Areas with lower pressure are marked in blue, and areas with higher pressure are marked in red. The pressure differences are small in total. These low-pressure differences could have translated into lower and higher resistance probabilities of water near the protein stabilizing the protein conformation. Could it be that the protein is more flexible in regions with lower pressure and therefore changes its conformation so molecules and particles can pass the S-layer?
In a final step, possible electrostatic effects are calculated and outlined in
Figure 6. Periodic boundary conditions were set in the z-direction. At the boundaries at z = 0 and z = 6
a0, the potential was set to zero. Charges randomly decorated the rigid obstacles, and the net charge was balanced to zero. The Poisson equation was solved for the fluid. Differences in dielectricity of the two regions (protein and fluid) or the Poisson–Boltzmann equation were considered. Of course, this is a gross simplification, but the following question is still worth discussing: Does the periodic distribution of charges influence the electric field and affect the water structure? Additionally, how far does it reach in? While the potential and the averaged potential does not change significantly throughout the pore, the electric field does, as shown in
Figure 5. Field lines reaching deep into the fluid are found, which may orient and drag charged nanoparticles. It is surprising that, again, in comparison to the Naiver Stokes equation, the structured surface is, on average, comparable to the unstructured one. However, they deviate significantly at the nanoscale for distances close to the size of the unit cell. At the characteristic distances of the S-layer, the mean electrostatic field is highest at the top of the S-layer and lowest near the pores. This pattern developed even though the charges were randomly distributed. It is the morphology of the S-layer alone that causes differences on small scales. Charges in the liquid would be influenced and, in this way, water would be structured, which happens solely due to the repeating morphology of the S-layer.
4. Summary
Based on preliminary studies and ongoing work, we selected the S-layer protein SbpA from
Lysinibacillus sphaericus ATCC 4525, which is identical to the also often investigated L. sphaericus CCM2177 [
9,
19]. In the presence of calcium ions, SbpA is able to reassemble in suspension, on solid supports (e.g., silicon surfaces, metals, polymers, glass), at the air–water interface, on planar lipid S-layers, on liposomes, nano-capsules, and carbon nanotubes, following a non-classical crystallization pathway [
30,
31,
32,
33,
34,
35]. Furthermore, the formation of an S-layer lattice with p4 lattice symmetry (as in SbpA) was also investigated in a theoretical approach by assuming both unspecific attractive and specific directional bonds between the monomers [
36]. The results were in excellent agreement with the experimental findings, showing, for example, that liquid-like cluster formation precedes crystallization.
In the first part of our work, high-resolution cryo-TEM data were linked with SAXS data. A model was constructed that considered the pair densities of the primitive unit cell and the pair densities coming from the nearest neighbors. The pair densities of this model were calculated and served as initial values for solving linear programming. It must be emphasized that it was possible for the first time in the literature to reconstruct hypothetical scattering data from the primitive unit cell. The approach eliminates the approximation, in which the scattering signal is divided into shape and structure factor. Grooves were found in the pair densities at characteristic intervals. These spacings could be associated with the self-assembled S-layer. Both the core and shell models gave similar results in terms of the best fit and minimized the discrepancies between hypothesized and experimental scattering data. Hull and core model were simplified to a box model.
The resulting box model was refined by choosing the weight assigned to each volume so that the hypothetical and experimental pair densities calculated from it resembled each other. Volumes with high weight were distinguished from those with low weight. Low weight volumes were interpreted as flexible areas in the S-layer lattice, while high weight volumes represented more rigid regions in the S-layer lattice. Navier–Stokes calculations with these volumes showed that these volumes significantly affect the fluid and the electrostatics near the S-layer lattice when electric charges decorated the volumes. The Navier–Stokes equations, as well as the Poisson equation, were solved for a limited region. The flow was assumed to be two-dimensional. The idea was not to give a comprehensive picture, but to push open a door of thought. Both the strength of the turbulences and the pressure near the S-layer surface in the 10 to 50 nm range support our hypothesis that the S-layer increases the exchange rate of metabolites and is responsible for the antifouling properties of the cell surface. The uptake of matter, such as molecules, particles, or any material compounds in this size range, is no longer limited by diffusion. Their removal from the surface leads to a clean S-layer surface. The S-layer thus justifies the high energy consumption in its production in the bacterial cell. In previous work, we sought to predict ab initio structure using reverse-steered molecular dynamics simulations [
21,
37]. The Jarzinsky equation [
38] and later work by Schulten influenced our approach. The disadvantage of our strategy was the absence of a method to link our models to experimental evidence to back up our findings. They were challenged by the crystallographic work of [
39] for Sbsb. In contrast, in this study, we integrate in situ experimental data to construct a coarse model of SbpA. We discuss and establish the concept to deduce this model and sketch an application for which the model may be employed. In subsequent work, the model and application will be refined.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





