
Phase Properties of Different HfO2 Polymorphs: A DFT-Based Study





Abstract
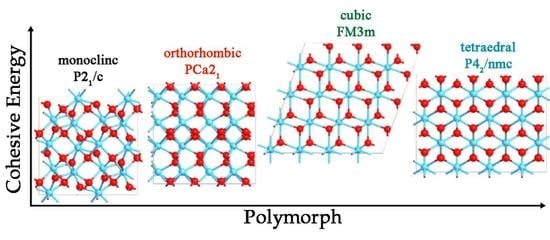
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Structure Analysis
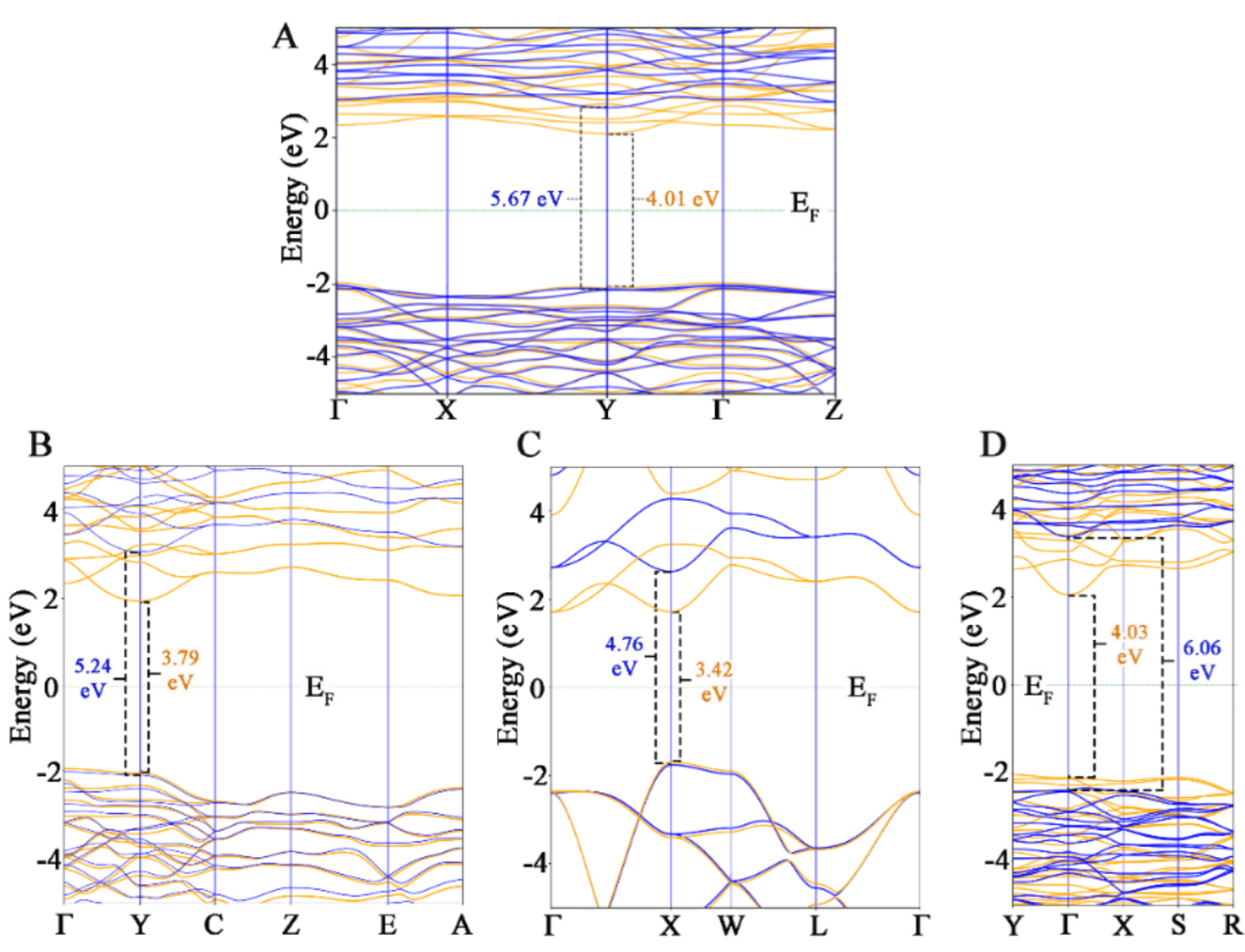
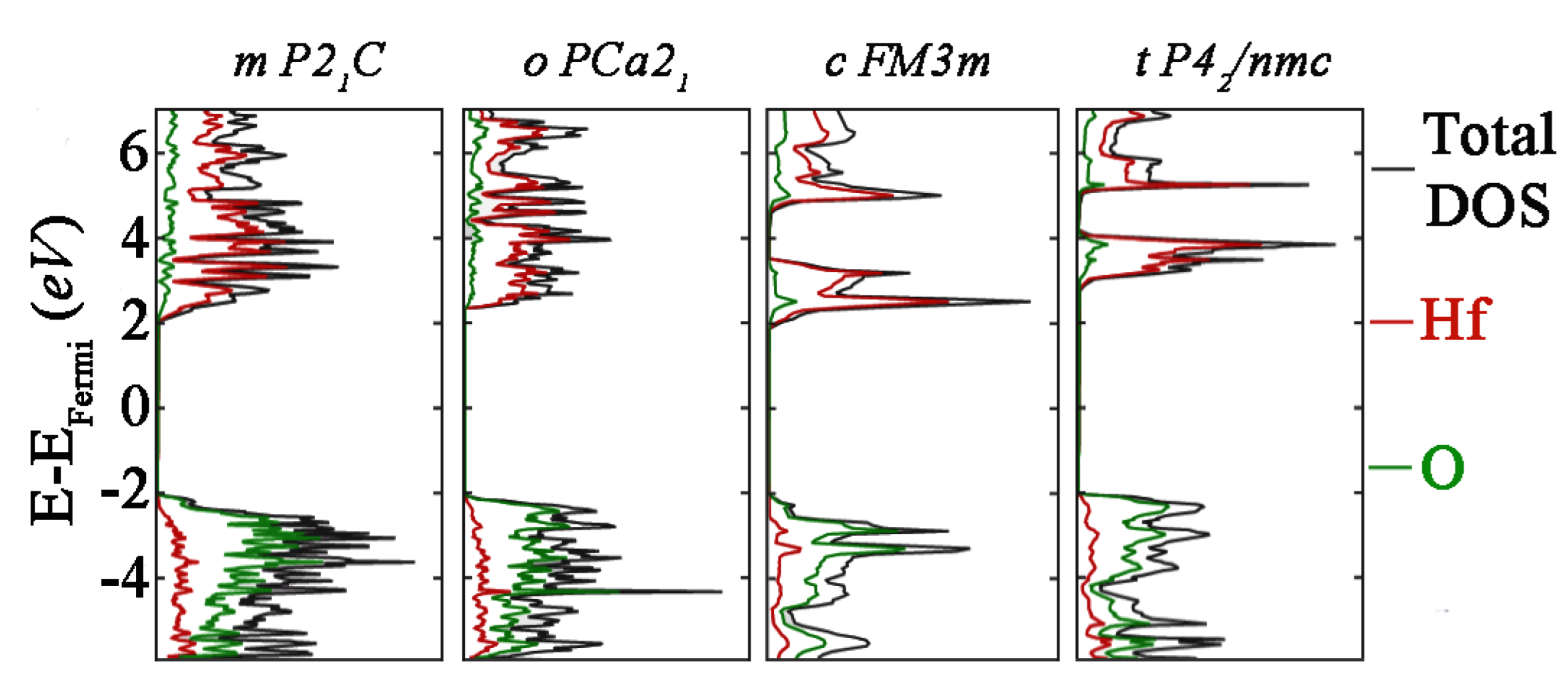
3.2. Electrical Analysis
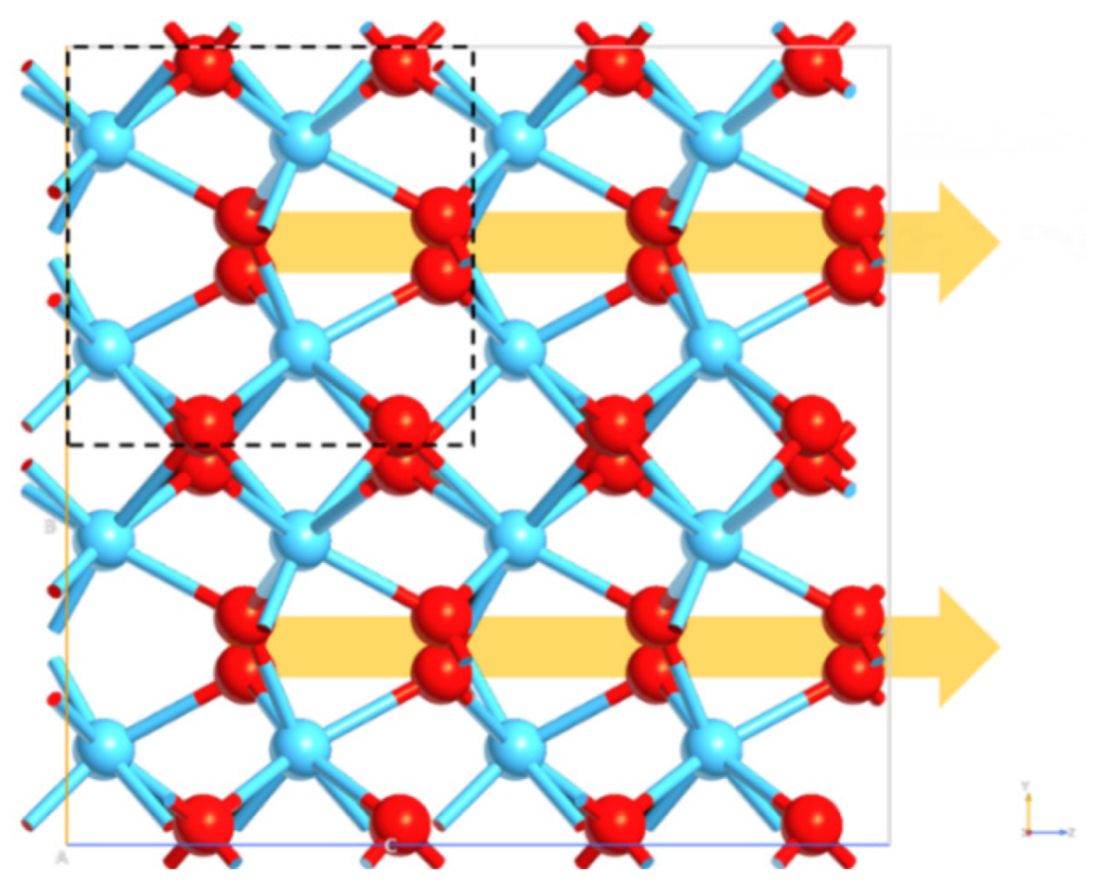
3.3. Oxygen Vacancies Effects
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilk, G.D.; Wallace, R.M.; Anthony, J.M. High-κ gate dielectrics: Current status and materials properties considerations. J. Appl. Phys. 2001, 89, 5243. [Google Scholar] [CrossRef]
- Robertson, J. High dielectric constant oxides. EPJ Appl. Phys. 2004, 28, 265–291. [Google Scholar] [CrossRef] [Green Version]
- Buiu, O.; Lu, Y.; Mitrovic, I.Z.; Hall, S.; Chalker, P.; Potter, R.J. Spectroellipsometric assessment of HfO2 thin films. Thin Solid Films 2006, 2, 623–626. [Google Scholar] [CrossRef]
- Jones, M.N.; Kwon, Y.W.; Norton, D.P. Dielectric constant and current transport for HfO2 thin films on ITO. Appl. Phys. A Mater. Sci. Process. 2005, 81, 285–288. [Google Scholar] [CrossRef]
- Schlom, D.G.; Haeni, J.H. A thermodynamic approach to selecting alternative gate dielectrics. MRS Bull. 2002, 27, 198–204. [Google Scholar] [CrossRef]
- Clima, S.; Wouters, D.J.; Adelmann, C.; Schenk, T.; Schroeder, U.; Jurczak, M.; Pourtois, G. Identification of the ferroelectric switching process and dopant-dependent switching properties in orthorhombic HfO2: A first principles insight. Appl. Phys. Lett. 2014, 104, 092906. [Google Scholar] [CrossRef]
- Sang, X.; Grimley, E.D.; Schenk, T.; Schroeder, U.; Lebeau, J.M. On the structural origins of ferroelectricity in HfO2 thin films. Appl. Phys. Lett. 2015, 106, 162905. [Google Scholar] [CrossRef]
- Kita, K.; Kyuno, K.; Toriumi, A. Permittivity increase of yttrium-doped HfO2 through structural phase transformation. Appl. Phys. Lett. 2005, 86, 102906. [Google Scholar] [CrossRef]
- Heo, S.; Tahir, D.; Chung, J.G.; Lee, J.C.; Kim, K.; Lee, J.; Lee, H.I.; Park, G.S.; Oh, S.K.; Kang, H.J.; et al. Band alignment of atomic layer deposited (HfZrO4)1−x(SiO2)x gate dielectrics on Si (100). Appl. Phys. Lett. 2015, 107, 182101. [Google Scholar] [CrossRef]
- Zhao, X.; Vanderbilt, D. First-principles study of structural, vibrational, and lattice dielectric properties of hafnium oxide. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 65, 233106. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, Y.; Feng, J.; Ye, C.; Wang, B.Y.; Wang, H.B.; Li, Q.; Jiang, Y.; Huang, A.P.; Xiao, Z.S. Structure and electrical properties of HfO2 high-k films prepared by pulsed laser deposition on Si (100). Appl. Phys. A Mater. Sci. Process 2008, 93, 681–684. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Aryasetiawan, F.; Lichtenstein, A.I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: The LDA + U method. J. Phys. Condens. Matter 1997, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Gomez-Abal, R.I.; Rinke, P.; Scheffler, M. First-principles modeling of localized d states with the GW@LDA + U approach. Phys. Rev. B 2010, 82, 045108. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA + U Framework. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 195107. [Google Scholar] [CrossRef] [Green Version]
- Plata, J.J.; Márquez, A.M.; Sanz, J.F. Communication: Improving the density functional theory U description of CeO2 by including the contribution of the O 2p electrons. J. Chem. Phys. 2012, 136, 041101. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.J.; Page, A.J. The Hubbard-U correction and optical properties of d0 metal oxide photocatalysts. J. Chem. Phys. 2020, 153, 224116. [Google Scholar] [CrossRef]
- Electronic structures and optical properties of monoclinic ZrO2 studied by first-principles local density approximation + U approach. J. Adv. Ceram 2017, 6, 43–49. [CrossRef] [Green Version]
- Coury, M.E.A.; Dudarev, S.L.; Foulkes, W.M.C.; Horsfield, A.P.; Ma, P.W.; Spencer, J.S. Hubbard-like Hamiltonians for interacting electrons in S, P, and D orbitals. Phys. Rev. B 2016, 93, 07510. [Google Scholar] [CrossRef] [Green Version]
- Sai Gautam, G.; Carter, E.A. Evaluating transition metal oxides within DFT-SCAN and SCAN+U frameworks for solar thermochemical applications. Phys. Rev. Mater. 2018, 2, 095401. [Google Scholar] [CrossRef]
- Li, J.; Meng, S.; Li, L.; Lu, H.; Tohyama, T. First-principles generalized gradient approximation (GGA) + Ud + Up studies of electronic structures and optical properties in cubic HfO2. Comput. Mater. Sci. 2014, 81, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An integrated platform of electronic and atomic-scale modelling tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Setten, M.J.; Giantomassi, M.; Bousquet, E.; Verstraete, M.J.; Hamann, D.R.; Gonze, X.; Rignanese, G.M. The PseudoDojo: Training and grading a 85 element optimized norm-conserving pseudopotential table. Comput. Phys. Commun. 2018, 226, 39. [Google Scholar] [CrossRef] [Green Version]
- King-Smith, R.D.; Vanderbilt, D. Theory of polarization of crystalline solids. Phys. Rev. B 1993, 47, 1651. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.T.; Chen, Y.W.; Liu, C.W. Strain effect on the stability in ferroelectric HfO2 simulated by first-principles calculations. J. Phys. D Appl. Phys. 2020, 53, 23LT01. [Google Scholar] [CrossRef]
- Li, J.; Lu, H.; Li, Y.; Meng, S.; Zhang, Y. First-principles generalized gradient approximation (GGA)+U studies of electronic structure and optical properties in cubic ZrO2. Solid State Commun. 2015, 211, 38–42. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, U.; Schenk, T.; Shimizu, T.; Funakubo, H.; Sakata, O.; Pohl, D.; Drescher, M.; Adelmann, C.; Materlik, R.; et al. Stabilizing the ferroelectric phase in doped hafnium oxide. J. Appl. Phys. 2015, 118, 072006. [Google Scholar] [CrossRef]
- El-Shanshoury, I.A.; Rudenko, V.A.; Ibrahim, I.A. Polymorphic Behavior of Thin Evaporated Films of Zirconium and Hafnium Oxides. J. Am. Ceram. Soc. 1970, 53, 264–268. [Google Scholar] [CrossRef]
- MacLaren, I.; Ras, T.; MacKenzie, M.; Craven, A.J.; McComb, D.W.; De Gendt, S. Texture, Twinning, and Metastable “Tetragonal” Phase in Ultrathin Films of HfO2 on a Si Substrate. J. Electrochem. Soc. 2009, 156, G103. [Google Scholar] [CrossRef]
- Al-Khatatbeh, Y.; Lee, K.K.M.; Kiefer, B. Phase diagram up to 105 GPa and mechanical strength of HfO2. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 144106. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.C.; Lino, A.A.T.; Scolfaro, L.M.R.; Leite, J.R.; Freire, V.N.; Farias, G.A.; da Silva, E.F., Jr. Band Structure Derived Properties of HfO2 from First Principles Calculations. AIP Conf. Proc. 2005, 772, 189. [Google Scholar]
- Torchio, P.; Gatto, A.; Alvisi, M.; Albrand, G.; Kaiser, N.; Amra, C. High-reflectivity HfO2/SiO2 ultraviolet mirrors. Appl. Opt. 2002, 41, 16. [Google Scholar] [CrossRef] [PubMed]
- Senami, M.; Tsuchida, Y.; Fukushima, A.; Ikeda, Y.; Tachibana, A. Local dielectric property of cubic, tetragonal, and monoclinic hafnium oxides. Jpn. J. Appl. Phys. 2012, 51, 031101. [Google Scholar] [CrossRef]
- Zhao, X.; Vanderbilt, D. Phonons and lattice dielectric properties of zirconia. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 65, 075105. [Google Scholar] [CrossRef] [Green Version]
- Richter, C.; Schenk, T.; Park, M.H.; Tscharntke, F.A.; Grimley, E.D.; LeBeau, J.M.; Zhou, C.; Fancher, C.M.; Jones, J.L.; Mikolajick, T.; et al. Si Doped Hafnium Oxide—A “Fragile” Ferroelectric System. Adv. Electron. Mater. 2017, 3, 1700131. [Google Scholar] [CrossRef]
- Huan, T.D.; Sharma, V.; Rossetti, G.A.; Ramprasad, R. Pathways towards ferroelectricity in hafnia. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 90, 064111. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Nukala, P.; Salverda, M.; Matzen, S.; Zhao, H.J.; Momand, J.; Everhardt, A.; Blake, G.R.; Lecoeur, P.; Kooi, B.J.; et al. A rhombohedral ferroelectric phase in epitaxially-strained Hf0.5Zr0.5O2 thin films. Nat. Mater. 2018, 17, 1095–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Chung, C.C.; Schenk, T.; Richter, C.; Hoffmann, M.; Wirth, S.; Jones, J.L.; Mikolajick, T.; Schroeder, U. Origin of Temperature-Dependent Ferroelectricity in Si-Doped HfO2. Adv. Electron. Mater. 2018, 4, 1700489. [Google Scholar] [CrossRef]
- Tao, L.L.; Paudel, T.R.; Kovalev, A.A.; Tsymbal, E.Y. Reversible spin texture in ferroelectric HfO2. Phys. Rev. B 2017, 95, 245141. [Google Scholar] [CrossRef] [Green Version]
- Shein, K.I.; Shein, I.R.; Medvedeva, N.I.; Bamburov, V.G.; Ivanovskii, A.L. Electronic structure of tetragonal thorium silicate in comparison with thorium dioxide. Dokl. Phys. Chem. 2006, 409, 198–201. [Google Scholar] [CrossRef]
- Jaffe, J.E.; Bachorz, R.A.; Gutowski, M. Low-temperature polymorphs of ZrO2 and HfO2: A density-functional theory study. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72, 144107. [Google Scholar] [CrossRef]
- Ondračka, P.; Holec, D.; Nečas, D.; Zajíčková, L. Accurate prediction of band gaps and optical properties of HfO2. J. Phys. D Appl. Phys. 2016, 49, 395301. [Google Scholar] [CrossRef] [Green Version]
- Demkov, A.A. Investigating alternative gate dielectrics: A theoretical approach. Phys. Status Solidi Basic Res. 2001, 1, 57–67. [Google Scholar] [CrossRef]
- Grimley, E.D.; Schenk, T.; Sang, X.; Pešić, M.; Schroeder, U.; Mikolajick, T.; LeBeau, J.M. Structural Changes Underlying Field-Cycling Phenomena in Ferroelectric HfO2 Thin Films. Adv. Electron. Mater. 2016, 2, 1600173. [Google Scholar] [CrossRef]
- Park, M.H.; Kim, H.J.; Kim, Y.J.; Lee, Y.H.; Moon, T.; Kim, K.D.; Hyun, S.D.; Fengler, F.; Schroeder, U.; Hwang, C.S. Effect of Zr Content on the Wake-Up Effect in Hf1-xZrxO2 Films. ACS Appl. Mater. Interfaces 2016, 8, 15466–15475. [Google Scholar] [CrossRef]
- Pešić, M.; Fengler, F.P.G.; Larcher, L.; Padovani, A.; Schenk, T.; Grimley, E.D.; Sang, X.; LeBeau, J.M.; Slesazeck, S.; Schroeder, U.; et al. Physical Mechanisms behind the Field-Cycling Behavior of HfO2-Based Ferroelectric Capacitors. Adv. Funct. Mater. 2016, 26, 4601–4612. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.P.; Stevens, R. Hafnia and hafnia-toughened ceramics. J. Mater. Sci. 1992, 27, 5397–5430. [Google Scholar] [CrossRef]
- Müller, J.; Böscke, T.S.; Schröder, U.; Mueller, S.; Bräuhaus, D.; Böttger, U.; Frey, L.; Mikolajick, T. Ferroelectricity in simple binary ZrO2 and HfO2. Nano Lett. 2012, 12, 4318–4323. [Google Scholar] [CrossRef] [PubMed]
- Kaczkowski, J.; Pugaczowa-Michalska, M.; Płowas’-Korus, I. Comparative density functional studies of pristine and doped bismuth ferrite polymorphs by GGA + U and meta-GGA SCAN + U. Phys. Chem. Chem. Phys. 2021, 23, 8571. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Space Group | Calculated (PBE-GGA) | Calculated (PBE-GGA + U) | Experiments |
|---|---|---|---|---|
| Monoclinic | P21/c | a = 5.068 b = 5.135 c = 5.292 | a = 5.208 b = 5.325 c = 5.424 | a = 5.07 [28] b = 5.14 c = 5.29 |
| Orthorhombic | Pca21 | a = 5.231 b = 5.008 c = 5.052 | a = 5.411 b = 5.220 c = 5.248 | a = 5.23 [29] b = 5.00 c = 5.05 |
| Cubic | Fm3m | a = b = c = 5.062 | a = b = c = 5.277 | a = b = c = 5.08 [30] |
| Tetragonal | P42/nmc | a = b = 5.062 c = 5.22 | a = b = 5.283 c = 5.43 | a = b = 5.06 [31] c = 5.2 |
| Phase | HfO2 Formation Energy | HfO2 − x Formation Energy | Defect Formation Energy |
|---|---|---|---|
| Monoclinic P21/c | −3.915 | −3.630 | 0.285 |
| Orthorhombic Pca21 | −3.887 | −3.572 | 0.315 |
| Cubic Fm3m | −3.792 | −3.643 | 0.149 |
| Tetragonal P42/nmc | −3.813 | −3.592 | 0.221 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laudadio, E.; Stipa, P.; Pierantoni, L.; Mencarelli, D. Phase Properties of Different HfO2 Polymorphs: A DFT-Based Study. Crystals 2022, 12, 90. https://doi.org/10.3390/cryst12010090
Laudadio E, Stipa P, Pierantoni L, Mencarelli D. Phase Properties of Different HfO2 Polymorphs: A DFT-Based Study. Crystals. 2022; 12(1):90. https://doi.org/10.3390/cryst12010090
Chicago/Turabian StyleLaudadio, Emiliano, Pierluigi Stipa, Luca Pierantoni, and Davide Mencarelli. 2022. "Phase Properties of Different HfO2 Polymorphs: A DFT-Based Study" Crystals 12, no. 1: 90. https://doi.org/10.3390/cryst12010090
APA StyleLaudadio, E., Stipa, P., Pierantoni, L., & Mencarelli, D. (2022). Phase Properties of Different HfO2 Polymorphs: A DFT-Based Study. Crystals, 12(1), 90. https://doi.org/10.3390/cryst12010090