
How Do Small Differences in Geometries Affect Electrostatic Potentials of High-Energy Molecules? Critical News from Critical Points


Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Molecular Electrostatic Potential Calcuations
3.2. Interaction Energies Calculations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, G.; Wei, S.-H.; Zhang, C. Review of the Intermolecular Interactions in Energetic Molecular Cocrystals. Cryst. Growth Des. 2020, 20, 7065–7079. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Perspectives on the crystal densities and packing coefficients of explosive compounds. Struct. Chem. 2016, 27, 401–408. [Google Scholar] [CrossRef]
- Born, M.; Plank, J.; Klapötke, T.M. Energetic Polymers: A Chance for Lightweight Reactive Structure Materials? Prop. Explos. Pyrotech. 2022, 47, e202100368. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Some molecular/crystalline factors that affect the sensitivities of energetic materials: Molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume. J. Mol. Model. 2015, 21, 25. [Google Scholar] [CrossRef]
- Born, M.; Karaghiosoff, K.; Klapötke, T.M.; Voggenreiter, M. Oxetane Monomers Based On the Powerful Explosive LLM-116: Improved Performance, Insensitivity, and Thermostability. ChemPlusChem 2022, 87, e202200049. [Google Scholar] [CrossRef] [PubMed]
- Nešić, J.; Cvijetić, I.N.; Bogdanov, J.; Marinković, A. Synthesis and Characterization of Azido Esters as Green Energetic Plasticizers. Prop. Explos. Pyrotech. 2021, 46, 1537–1546. [Google Scholar] [CrossRef]
- Murray, J.S.; Concha, M.C.; Politzer, P. Links between surface electrostatic potentials of energetic molecules, impact sensitivities and C–NO2/N–NO2 bond dissociation energies. Mol. Phys. 2009, 107, 89–97. [Google Scholar]
- Politzer, P.; Murray, J.S. High Performance, Low Sensitivity: Conflicting or Compatible? Propellants Explos. Pyrotech. 2016, 41, 414–425. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Murray, J.S. Electrostatic Potentials, Intralattice Attractive Forces and Crystal Densities of Nitrogen-Rich C,H,N,O Salts. Crystals 2016, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Hammerl, A.; Klapötke, T.M.; Mayer, P.; Weigand, J.J. Synthesis, Structure, Molecular Orbital Calculations and Decomposition Mechanism for Tetrazolylazide CHN7, its Phenyl Derivative PhCN7 and Tetrazolylpentazole CHN9. Prop. Explos. Pyrotech. 2005, 30, 17–26. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Nordheider, A.; Stierstorfer, J. Synthesis and reactivity of an unexpected highly sensitive 1-carboxymethyl-3-diazonio-5-nitrimino-1,2,4-triazole. New J. Chem. 2012, 36, 1463–1468. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Murray, J.S. Tricyclic polyazine n-oxides as proposed energetic compounds. Cent. Eur. J. Energetic Mater. 2013, 10, 305. [Google Scholar]
- Krishnapriya, V.U.; Suresh, C.H. The use of electrostatic potential at nuclei in the analysis of halogen bonding. New J. Chem. 2022, 46, 6158–6164. [Google Scholar] [CrossRef]
- Kim, C.K.; Cho, S.G.; Kim, C.K.; Park, H.-Y.; Zhang, H.; Lee, H.W. Prediction of densities for solid energetic molecules with molecular surface electrostatic potentials. J. Comput. Chem. 2008, 29, 1818–1824. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Lai, W.; Yu, T.; Ma, Y.; Ge, Z. A strategy for predicting the crystal structure of energetic N-oxides based on molecular similarity and electrostatic matching. CrystEngComm 2021, 23, 714–723. [Google Scholar] [CrossRef]
- Kretić, D.S.; Radovanović, J.I.; Veljković, D.Ž. Can the sensitivity of energetic materials be tuned by using hydrgen bonds? Another look at the role of hydrogen bonding in the design of high energetic compounds. Phys. Chem. Chem. Phys. 2021, 23, 7472–7479. [Google Scholar] [CrossRef] [PubMed]
- Đunović, A.B.; Veljković, D.Ž. Halogen bonds as a tool in the design of high energetic materials: Evidence from crystal structures and quantum chemical calculations. CrystEngComm 2021, 23, 6915–6922. [Google Scholar] [CrossRef]
- Eckhardt, C.J.; Gavezzotti, A. Computer Simulations and Analysis of Structural and Energetic Features of Some Crystalline Energetic Materials. J. Phys. Chem. B 2007, 111, 3430–3437. [Google Scholar] [CrossRef]
- Aina, A.A.; Misquitta, A.J.; Phipps, M.J.S.; Price, S.L. Charge Distributions of Nitro Groups Within Organic Explosive Crystals: Effects on Sensitivity and Modeling. ACS Omega 2019, 4, 8614−8625. [Google Scholar] [CrossRef] [Green Version]
- Hauf, C.; Salvador, A.-A.H.; Holtz, M.; Woerner, M.; Elsaesser, T. Phonon driven charge dynamics in polycrystalline acetylsalicylic acid mapped by ultrafast x-ray diffraction. Struct. Dyn. 2019, 6, 014503. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Shu, Y.; Gao, S.; Chen, L.; Ma, Q.; Ju, X. Easy methods to study the smart energetic TNT/CL-20 co-crystal. J. Mol. Model. 2013, 19, 4909–4917. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bulat, F.A.; Toro-Labbe, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Kretić, D.S.; Veljković, I.S.; Đunović, A.B.; Veljković, D.Ž. Chelate Coordination Compounds as a New Class of High-Energy Materials: The Case of Nitro-Bis(Acetylacetonato) Complexes. Molecules 2021, 26, 5438. [Google Scholar] [CrossRef] [PubMed]
- Veljković, D.Ž.; Janjić, G.V.; Zarić, S.D. Are C–H•••O interactions linear? Case of aromatic CH donors. CrystEngComm 2011, 13, 5005–5010. [Google Scholar]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Babu, K.; Ganesh, V.; Gadre, S.; Ghermani, N.E. Tailoring approach for exploring electron densities and electrostatic potentials of molecular crystals. Theor. Chem. Acc. 2004, 111, 255–263. [Google Scholar] [CrossRef]








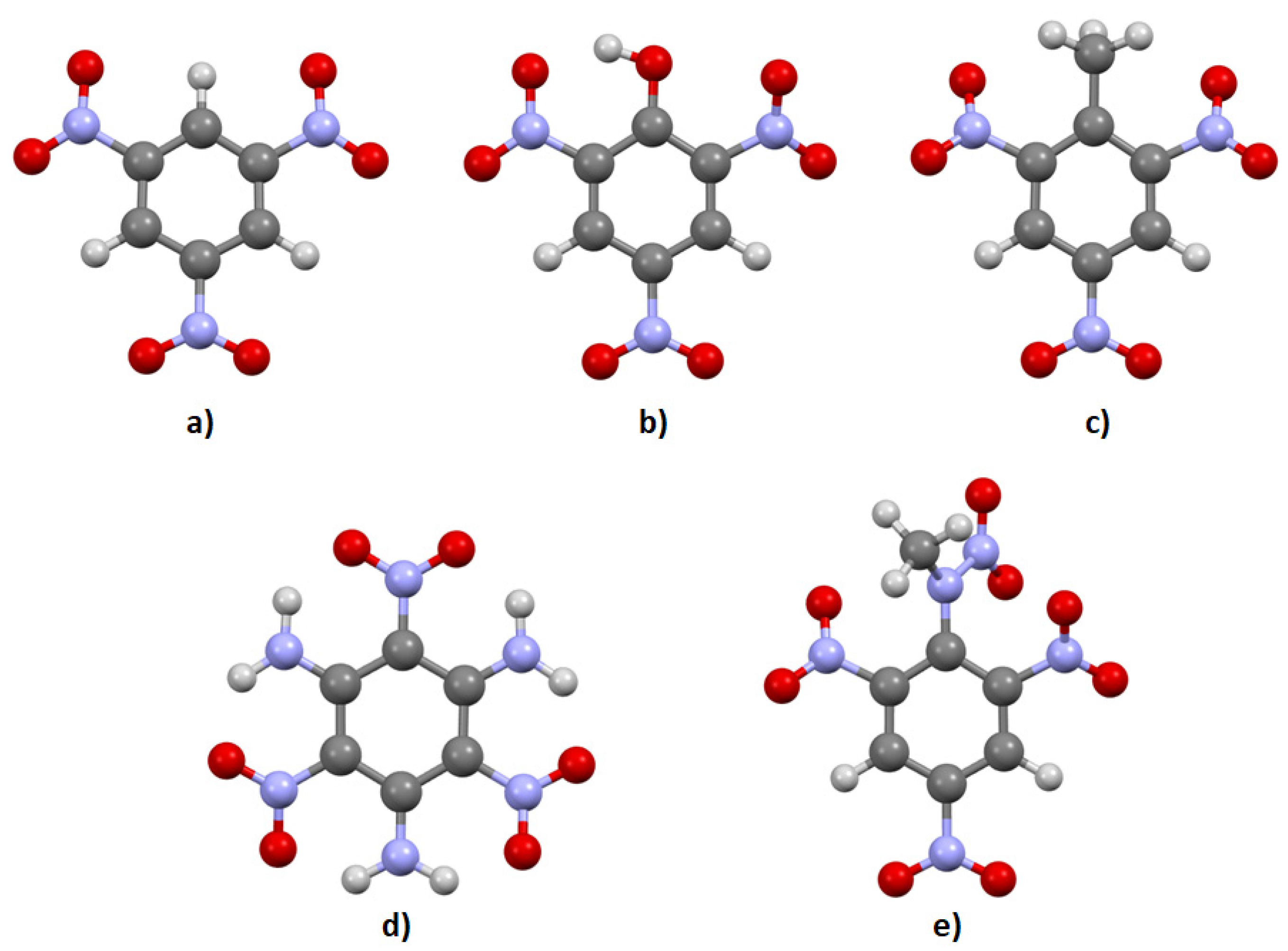{kind=link}
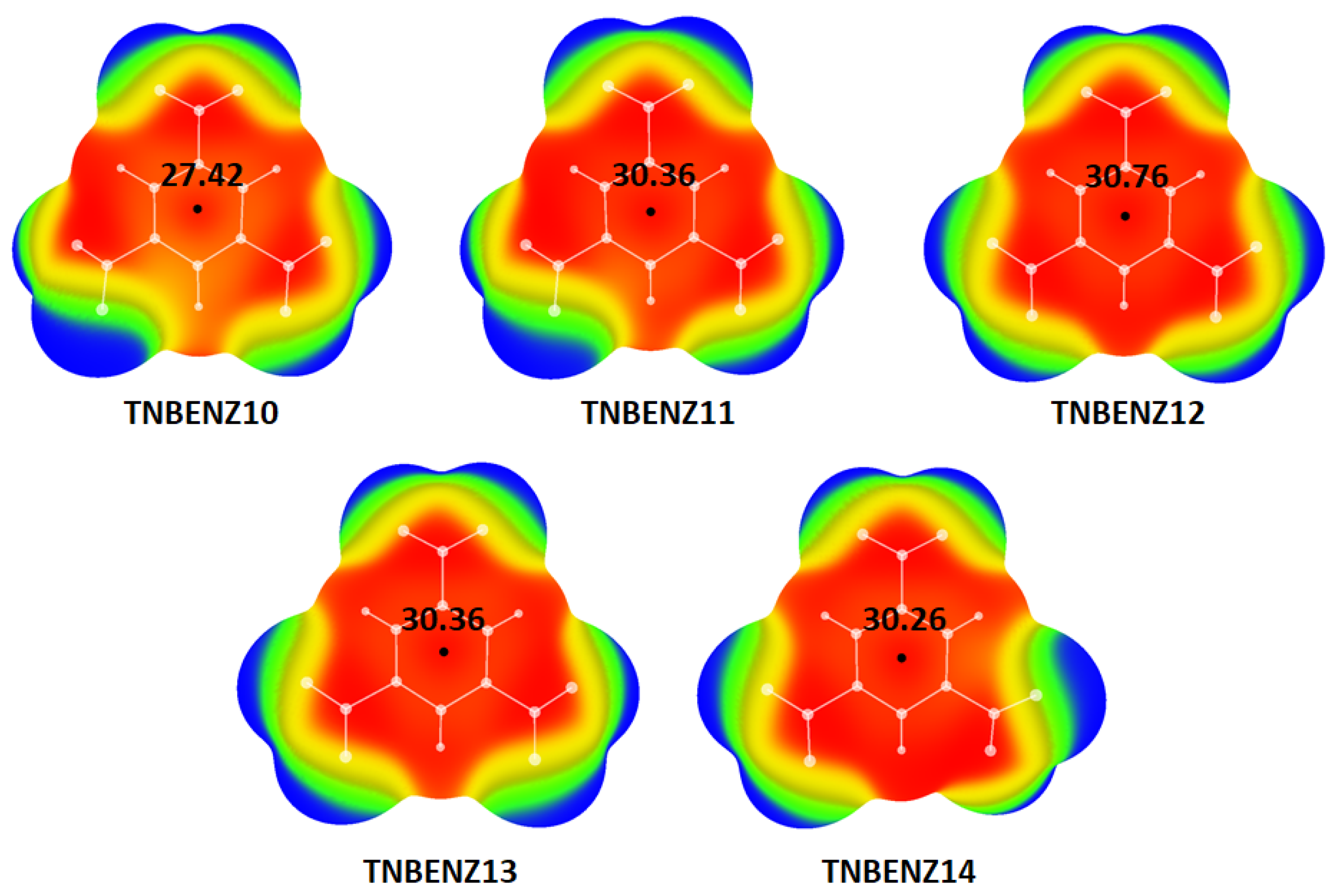{kind=link}
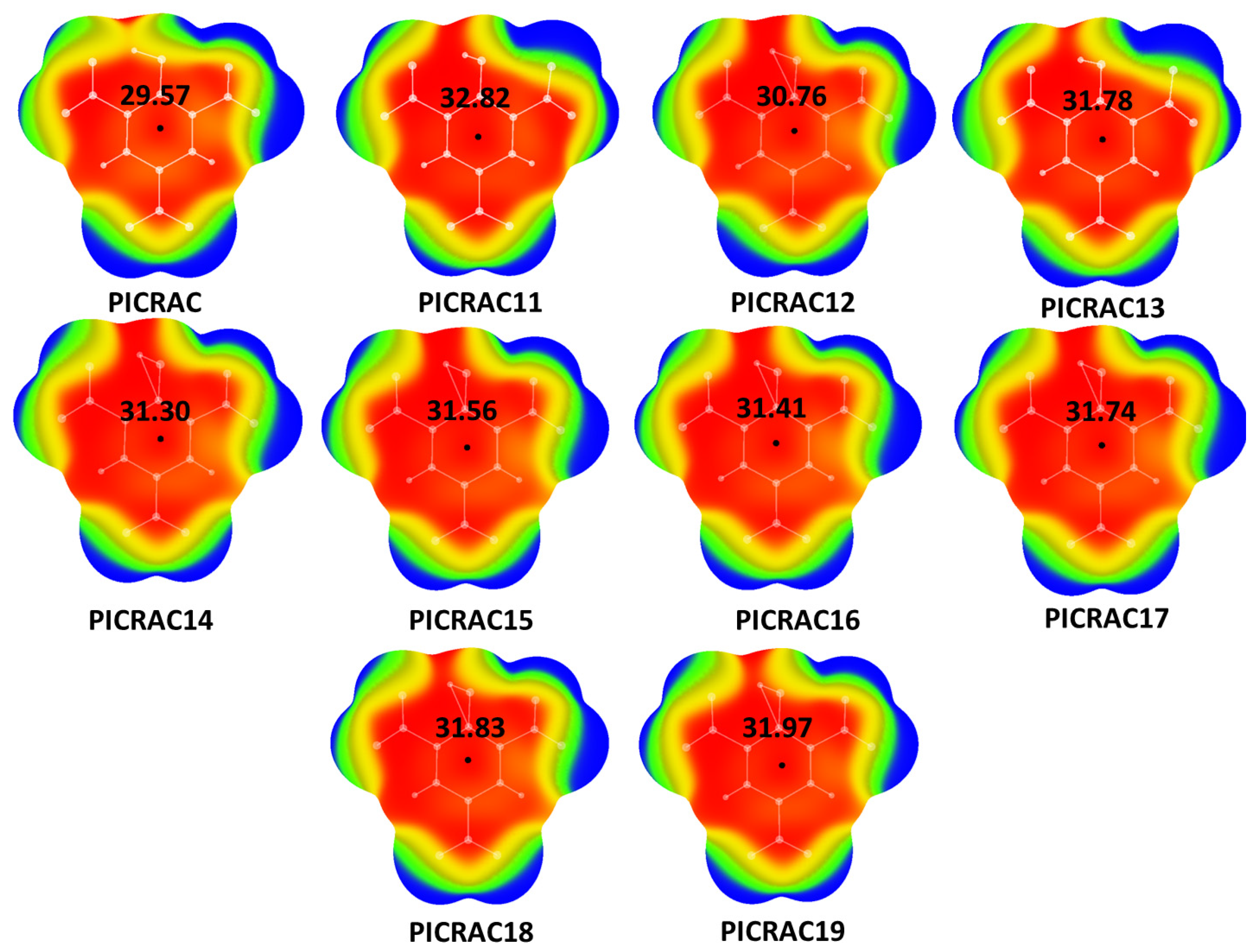{kind=link}
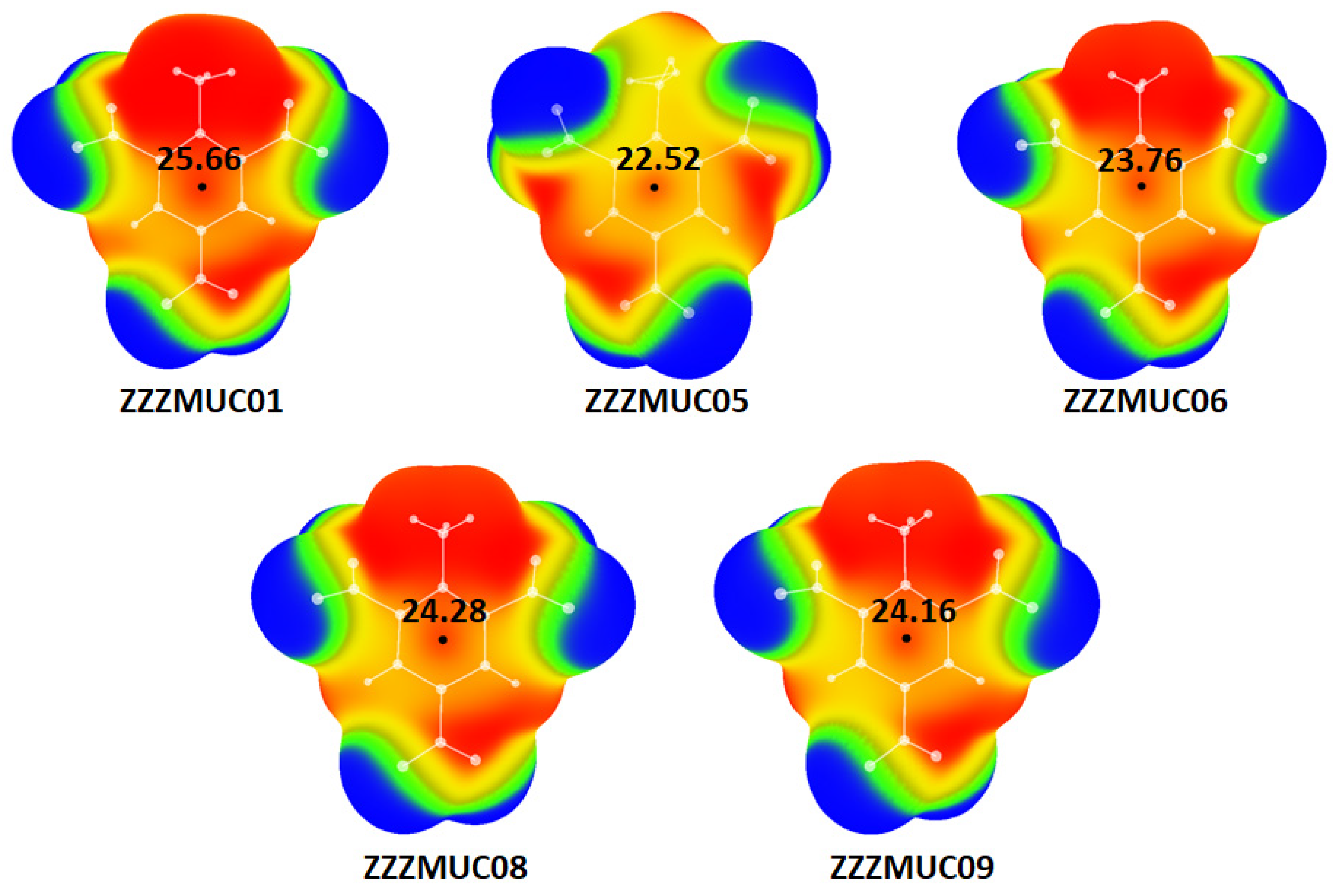{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Vmax 1 (kcal/mol) | E (Hartree) | Vmax Deviation 2 (%) |
|---|---|---|---|
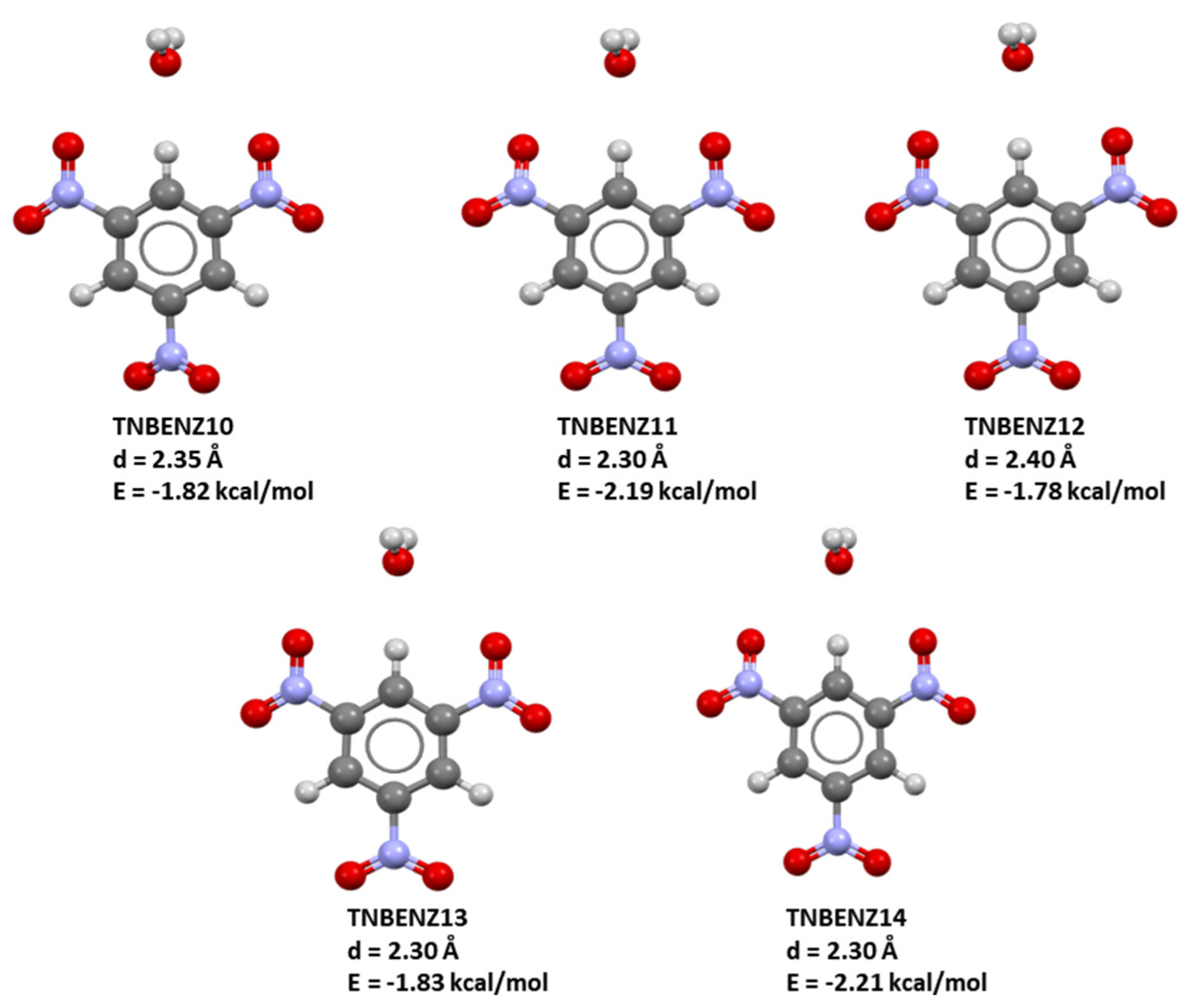
| TNBENZ10 | 27.42 | −845.3461488 | 0.35 |
| TNBENZ11 | 30.36 | −845.3002792 | 11.08 |
| TNBENZ12 | 30.76 | −845.2847208 | 12.56 |
| TNBENZ13 | 30.36 | −845.3030868 | 11.10 |
| TNBENZ14 | 30.26 | −845.3006406 | 10.72 |
| Optimized TNB 3 | 27.33 | −845.35482453 | / |
| Structure | Vmax 1 (kcal/mol) | E (kcal/mol) | Vmax Deviation 2 (%) |
|---|---|---|---|
| PICRAC | 29.57 | −920.4130444 | 7.56 |
| PICRAC11 | 32.82 | −920.2254342 | 19.40 |
| PICRAC12 | 31.51 | −920.5205850 | 14.64 |
| PICRAC13 | 31.78 | −920.4575628 | 15.62 |
| PICRAC14 | 31.30 | −920.4979973 | 13.86 |
| PICRAC15 | 31.56 | −920.4979960 | 14.82 |
| PICRAC16 | 31.41 | −920.4980897 | 14.27 |
| PICRAC17 | 31.74 | −920.4872273 | 15.48 |
| PICRAC18 | 31.83 | −920.4745691 | 15.78 |
| PICRAC19 | 31.97 | −920.4732924 | 16.31 |
| Optimized TNP 3 | 27.49 | −920.55863946 | / |
| Structure | Vmax 1 (kcal/mol) | E (kcal/mol) | Vmax Deviation 2 (%) |
|---|---|---|---|
| ZZZMUC01 | 25.66 | −884.49156794 | 7.98 |
| ZZZMUC05 | 22.52 | −884.489125884 | 5.20 |
| ZZZMUC06 | 23.76 | −884.557859799 | 0.00 |
| ZZZMUC08 | 24.28 | −884.554014599 | 2.19 |
| ZZZMUC09 | 24.16 | −884.562118641 | 1.70 |
| Optimized TNT 3 | 23.76 | −884.581835322 | / |
| Scheme 1. | Vmax 1 (kcal/mol) | E (kcal/mol) | Vmax Deviation 2 (%) |
|---|---|---|---|
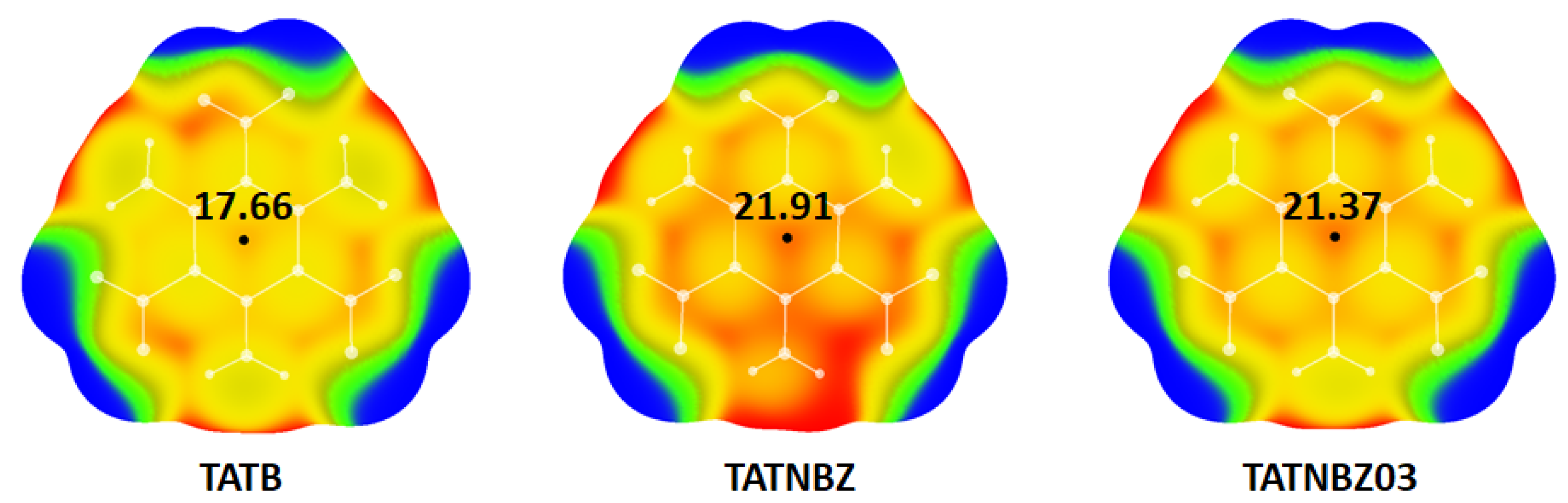
| TATNBZ | 25.66 | −1011.1872097 | 21.01 |
| TATNBZ03 | 22.52 | −1011.3807914 | 24.07 |
| Optimized TATB | 23.76 | −1011.3867634 |
| Structure | Vmax 1 (kcal/mol) | E (kcal/mol) | Vmax Deviation 2 (%) |
|---|---|---|---|
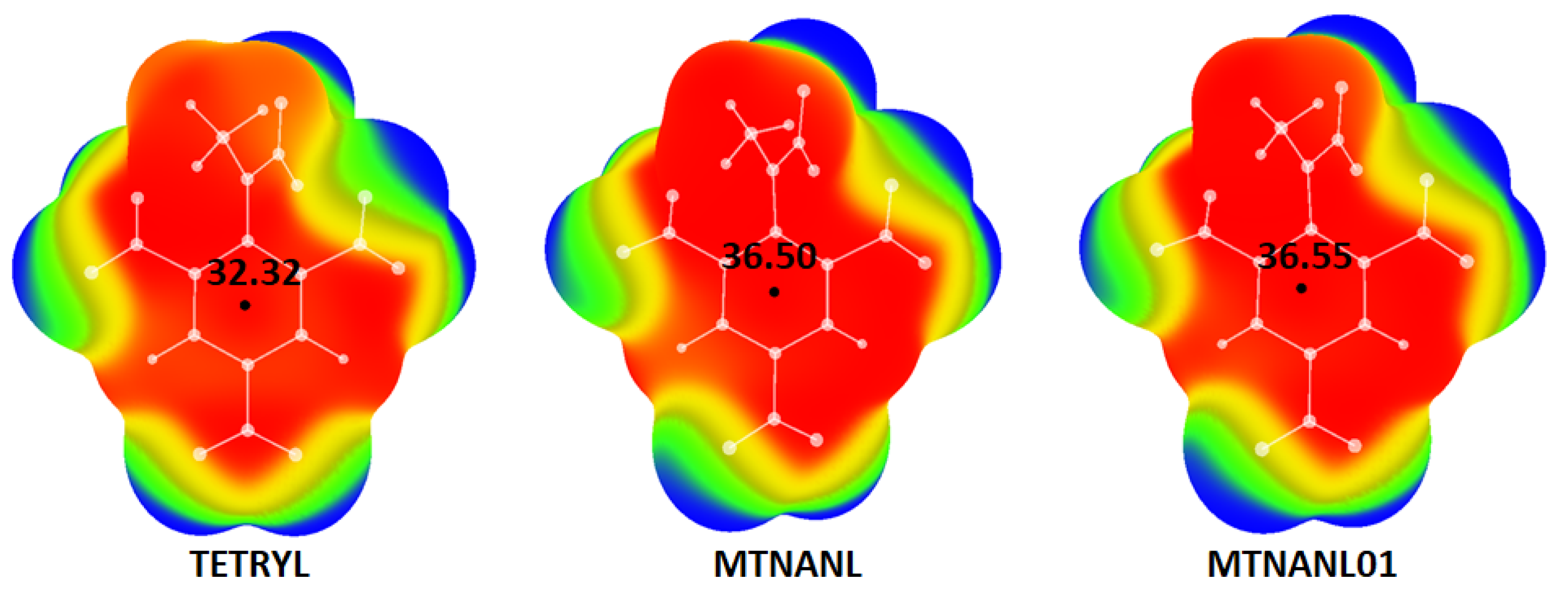
| MTNANL | 36.50 | −1144.2011083 | 12.93 |
| MTNANL01 | 36.55 | −1144.2184993 | 13.09 |
| Optimized TETRYL | 32.32 | −1144.3552437 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretić, D.S.; Medaković, V.B.; Veljković, D.Ž. How Do Small Differences in Geometries Affect Electrostatic Potentials of High-Energy Molecules? Critical News from Critical Points. Crystals 2022, 12, 1455. https://doi.org/10.3390/cryst12101455
Kretić DS, Medaković VB, Veljković DŽ. How Do Small Differences in Geometries Affect Electrostatic Potentials of High-Energy Molecules? Critical News from Critical Points. Crystals. 2022; 12(10):1455. https://doi.org/10.3390/cryst12101455
Chicago/Turabian StyleKretić, Danijela S., Vesna B. Medaković, and Dušan Ž. Veljković. 2022. "How Do Small Differences in Geometries Affect Electrostatic Potentials of High-Energy Molecules? Critical News from Critical Points" Crystals 12, no. 10: 1455. https://doi.org/10.3390/cryst12101455
APA StyleKretić, D. S., Medaković, V. B., & Veljković, D. Ž. (2022). How Do Small Differences in Geometries Affect Electrostatic Potentials of High-Energy Molecules? Critical News from Critical Points. Crystals, 12(10), 1455. https://doi.org/10.3390/cryst12101455