1. Introduction
Due to the inherent stability of crystalline materials and the positive impact of the crystallization processes on purification and isolation of chemical substances, typically, crystalline drugs are preferred to amorphous solid forms. However, due to the poor solubility in water of many Active Pharmaceutical Ingredients (APIs), enhanced interest in multi-component crystalline materials has been seen because they enable better drug bioavailability; these materials enable tuning the physicochemical properties of a drug, such as solubility and hence bioavailability, permeability, hydration, color, compaction, and tableting [
1]. Especially when the widely used salt formations are unable to provide adequate enhancement of drug bioavailability (or cannot be formed due to the absence of ionization sites), an attractive alternative for improving the said characteristics are cocrystals of APIs (for a rigorous description of APIs see [
2,
3]) and a pharmaceutically acceptable cocrystal former (which do not alternate the pharmacological behavior of the APIs); the cocrystals of APIs are very promising for increasing drugs’ poor aqueous solubility, slow dissolution rate in biological fluids (and hence insufficient bioavailability) and insufficient chemical stability of amorphous drugs, which results from their relatively high moisture uptake.
Cocrystals are homogeneous stoichiometric compositions of two or more neutral molecular constituents bound together in the same crystal lattice—but not a mixture of pure component crystalline phases. Cocrystals encompass many types of compounds, including clathrates (which are inclusion compounds in which the guest molecule is in a cage formed by the host molecule or by a lattice of host molecules). The cocrystals are neither solvates nor simple salts. The different components in the cocrystal are neutral in nature when compared to salts that have ionized components, the distinction being that the proton is shared between an acid and a base in cocrystals while it is transferred when forming salts. The components in the cocrystal exist in a definite stoichiometric molar ratio and assemble via non-covalent interactions, such as hydrogen bonds, ionic bonds, π–π or van der Waals interactions between the different molecular species. The molecules in the cocrystal superstructure are held together by supramolecular synthons [
4] that are basic structural units formed from the non-covalent interactions. Importantly, the cocrystals’ constituent components form a novel crystalline structure, which possesses unique properties [
5,
6].
Due to cocrystals’ applications in the pharmaceutical industry [
7,
8,
9], there is an active cocrystal research during these past few decades. There are many approaches to synthesize cocrystals [
10]. The most frequently used one is slow evaporation from solutions of cocrystal-components contained in stoichiometric relations. This method ensures formation of a small number of larger crystals (as opposed to a high number of smaller crystals produced by other methods). To produce co-crystals via solvent evaporation, 1:1 stoichiometric amounts of co-crystal components are dissolved usually, e.g., [
11]. Similar is the process of cooling co-crystallization. Another effective approach for cocrystal formation is the reaction co-crystallization method. In contrast to solvent evaporation, in the reaction crystallization method supersaturation with respect to the cocrystal is generated by reactant dissolution. Usually the drug, which is the least soluble component, is dissolved in solutions of the highly soluble component, which is usually the conformer [
12]. Used is also co-grinding the reactants in the presence of small quantities of organic solvents or water added to the reactants during grinding. Melt processes can also produce co-crystals.
To address physicochemical, biopharmaceutical, and mechanical properties and to expand solid form diversity of the API, drug development scientists are exploring diverse cocrystals. For instance, first-principles modeling was employed to explore the design space for co-crystallization of caffeine−glutaric acid [
13]. To account for uncertainty and variability in starting concentration, seed loading and seeding temperature, the operating ranges of process parameters on larger scales were determined via Monte Carlo simulation. (However, to the best of authors’ knowledge, the cocrystal formation per se is not mimicked by Monte Carlo simulations.) Finally, it is worth noting that cocrystals are also promising for the design and preparation of new explosives [
14].
The literature on cocrystals is vast and constantly increasing. For recent reviews on co-crystals see [
10,
15,
16]. A comprehensive book on the rational control of pharmaceutical cocrystals has also been published [
17]. However, despite the long history of cocrystals study, spanning more than 160 years, no theory for crystallization of pharmaceutical cocrystals has been devised until now. Importantly, the process of co-crystallization imposes additional levels of complexity when considering APIs behavior in solution and during crystallization. For instance, it is noted in Ref. [
16] that “the complexity of the thermodynamic landscape and the kinetics of co-crystallization offers fresh challenges which are not encountered in single component crystallization.” Therefore, both thermodynamics and kinetics of co-crystallization are considered in this paper.
Especially from an industrial perspective, there is an increased awareness of the need to understand the co-crystallization process in more details. However, due to the molecular scale of the processes involved, there is little fundamental understanding of what is happening in a solution as molecules interact and begin to form entities that eventually become crystals. Presently, chemical engineers in the pharmaceutical and chemical industries handle the transformation from the liquid to the solid state in an empirical fashion that is not based on rigorous thermodynamic or kinetic principles. This approach is limited as molecular details are omitted. For instance, although critical for obtaining a cocrystal from solution, the role of a solvent in nucleation of cocrystals remains poorly understood [
18]. (However, it is logical to suggest that changing the solvent will change the intermolecular interactions and enable cocrystal formation.)
In this paper, the theoretical consideration of co-crystallization is started by pointing out rules known from the kinetics of single component crystallization that also govern the co-crystallization process. Our aim is to contribute to elucidating the principles and the molecular-scale mechanisms of the co-crystallization process, imposing additional levels of understanding when considering APIs behavior in solution. With this end in view, different intermolecular interactions are assumed when modeling co-crystallization by 2D Monte Carlo simulation.
2. Theoretical Consideration of the Co-Crystallization Process
Importantly, co-crystallization is almost always a thermodynamically favorable process [
19]. Evidently, it is favored by the high conformational entropy (S) of the cocrystal. The famous Boltzmann equation renders the statistical definition of the entropy:
where
W is the number of different ways in which the energy of the system can be achieved by rearranging the molecules among their available states, i.e., their different configurations. Especially for the cocrystals,
W, and thus
S, are much higher as compared with the corresponding values for one-component crystals. Recalling that the second law of thermodynamics states that the entropy of an isolated system always increases for a spontaneous (i.e., a naturally occurring) process, i.e., Δ
S > 0, under properly selected conditions, the co-crystallization is a spontaneous process occurring due to a decrease in the system’s free energy. Therefore, it does not need to be driven by an outside source of energy. This explains the relatively ready co-crystallization and cocrystal formation in many systems.
As concerns the kinetics of co-crystallization, it is feasible to suggest that the general rules established for the growth of one-component crystals also rule the co-crystallization. For a diffusion-limited crystal growth, the growth rate is written as:
where
is crystal surface,
is the thickness of the Nernst diffusion layer;
denotes the solute concentration (which can differ for the two components) at any crystal growth moment of time
, and
being solubility, i.e., the equilibrium concentration with respect to an “infinitely” large crystal (activity coefficients equal to unity are assumed usually).
[cm
2/s] is diffusivity, which can also differ for the two components. Additionally, the rate of the face propagation normal to itself (
) is proportional to the supersaturation,
[
20]:
where
is the surface step density and
is the propagation rate of a step on the crystal face;
is the kinetic coefficient for step propagation, and
denotes the volume of the crystal building unit.
The rate
of step propagation during growth of the crystal is determined by the kink density (
/
) and the velocity
of kink propagation along the step:
where
is a typical molecular size,
is the average distance between the kinks, and
and
are the frequencies of molecular attachment to and detachment from a kink, respectively. Kaischew showed that the number density of kinks of the two components is proportional to its molar part [
21] (and on the temperature), but
and
,
and
must be determined separately for each cocrystal component.
Importantly, to design cocrystal screening experiments, it is vital to understand how the cocrystals’ bonding arise and how they form. The desirable physicochemical properties are based on an understanding of the intermolecular interactions, which dictate the molecular arrangement in the crystal lattice [
22]. In plain language, knowledge of the intermolecular interactions and their effects on crystal packing are indispensable for the engineering of cocrystals with desired physical and chemical properties.
To give a plausible explanation of the co-crystallization, the method of slow evaporation from solutions of cocrystal components will be considered first. It is evident that the two cocrystal ingredients must have a similar solubility. (Otherwise, solely one of them will crystallize.) Though, (exactly) the same solubility of two different substances is hardly possible—in general, slightly different solubilities are preferred. Therefore, the critical supersaturation, which is needed for crystal nucleation of one or the other of the cocrystal components is reached for one of the components somewhat earlier than for the other [
23]. Thus, the critical cocrystal nucleus should be formed, at least predominantly, from the molecules of the slightly less-soluble co-crystal component. Growing under diffusion limitations, the cocrystal nucleus exhausts the molecules of the same kind in its surrounding. Then, when the second cocrystal component overcomes the solubility threshold, the crystallization continues further with deposition of the second cocrystal component molecules on the existing crystal nucleus. Imaginably, the co-crystallization consists of repeated exhausting and enrichment of the two kinds of molecules. (In fact, periodic co-crystallization is observed by some of our Monte Carlo simulations—a work in progress which will be published elsewhere).
Secondly, for reaction co-crystallization, the mechanism of periodic precipitation resembles Liesegang patterning [
24]—in both cases the deposited material is formed due to chemical reactions; the difference being only the appearance of void of deposition bands which are typical for the Liesegang patterning, which is missing in cocrystals. However, the consideration of co-crystallization is complicated even in the case of solution having two cocrystal components. In principle, the concentration on the boundary of the Nernst diffusion layer can be calculated by solving the following diffusion-reaction differential equations:
where a and b denote the concentrations of the two components, while
Da and
Db are the corresponding diffusion coefficients;
R(
a) and
R(
b) are the cocrystal growth rates for the corresponding component. Unfortunately, such sets of nonlinear partial differential equations cannot be treated by standard analytical methods—the only viable way is the application of numerical methods. Yet, numerical solution of such systems of equations is computationally demanding, and even nowadays—computationally prohibitive. Therefore, to treat the before mentioned scenario of the cocrystal formation, we use Monte Carlo simulations, based on two types of “protein-like” patchy particles and computational model based on the widely used PPs model proposed by Doye and co-workers [
25].
3. Monte Carlo Simulations: Computational Model, Results, and Discussion
Up to date, Monte Carlo (MC) simulation has been used predominantly as an auxiliary tool for proper design of co-crystallization of practically interesting systems. For instance, to account for the limited solute diffusion velocity through the boundary layer, analytical solution of a simple model of the boundary layer was combined via an iterative process with the MC simulation of the interface [
26]. However, direct investigations of cocrystal formation by using MC simulation are relatively rear [
27]. Clathrate formation was simulated most recently, using standard Metropolis MC simulations in the canonical ensemble with equally likely single-particle translation and rotation moves [
28]. Self-assembly scenario in a 2D binary mixtures of patchy colloidal particles (PPs) with attractive patches where the two components are characterized by different numbers of patchy sites were studied by Doppelbauer et al. [
29].
In this paper, we perform standard Metropolis [
30] Monte Carlo (MC) simulations in the canonical (NVT) ensemble with (equally likely) single-particle translation and rotation moves (the so-called roto-translation moves). The latter permit the particles to bind together in a way that is consistent with the target structure, thus removing the possibility of competing forms and ensuring the kinetic accessibility of the target structure. While the attraction of our PPs depends on the relative orientation of the particles, simultaneously, they also interact with an isotropic core repulsion. The specificity encoded by patch positions, patch interaction selectivity, and restrictions on spatial orientations ensure PPs assembling into the desired structure. Because for directional interactions the computational effort is considerably enhanced with respect to particles with spherically symmetric interactions, we have restricted ourselves to a simple model, working in two dimensions and using patch interactions that are based on potentials proposed by Doye et al. [
25]. The PPs we use in our simulations are represented by a monomer, the circumference of which is “decorated” with two pairs of patches (four patches in total for a monomer). The first pair of patches are “strong and narrow” (SN), while the second patches are “weak and wide” (WW). Thus, the patches are arranged on the monomer in a sequence SN-SN-WW-WW with given values of the degrees between each two consecutive patches. Here strong or weak refer to the amplitude of the patch-patch interaction, while narrow or wide define the geometrically span of the patches, namely the extent of the corresponding patch in degrees (or the so-called patchy width) along the monomer circumference. A reasonable ground to use strong and weak patches in our simulations is the nature of the hydrogen bonds– because of their strength and directionality, hydrogen bonds are particularly amenable to formation of cocrystals. The “strong” (or “conventional”) hydrogen bond includes (N–H–O), (O–H–O), (–N–H–N), and (O–H–N), which have energies 20–40 kJ mol
−1, while the “weak” hydrogen bonds involves the –C–H–O and C–H–O=C, having energies 2–20 kJ mol
−1 [
4]. Thus, the strong and weak attractive patches in our simulations resemble the supramolecular synthons.
Here we will use two types of patchy particles which will “generate” the building blocks of our patchy cocrystals. In both types of the PPs, the patches are arranged in a sequence SN-SN-WW-WW but with different values of the degrees between each two consecutive patches. PPs type I are constructed by angular distribution 60-120-60-120 (degrees), while in PPs type II the angular distribution is 120-60-120-60, see
Figure 1.
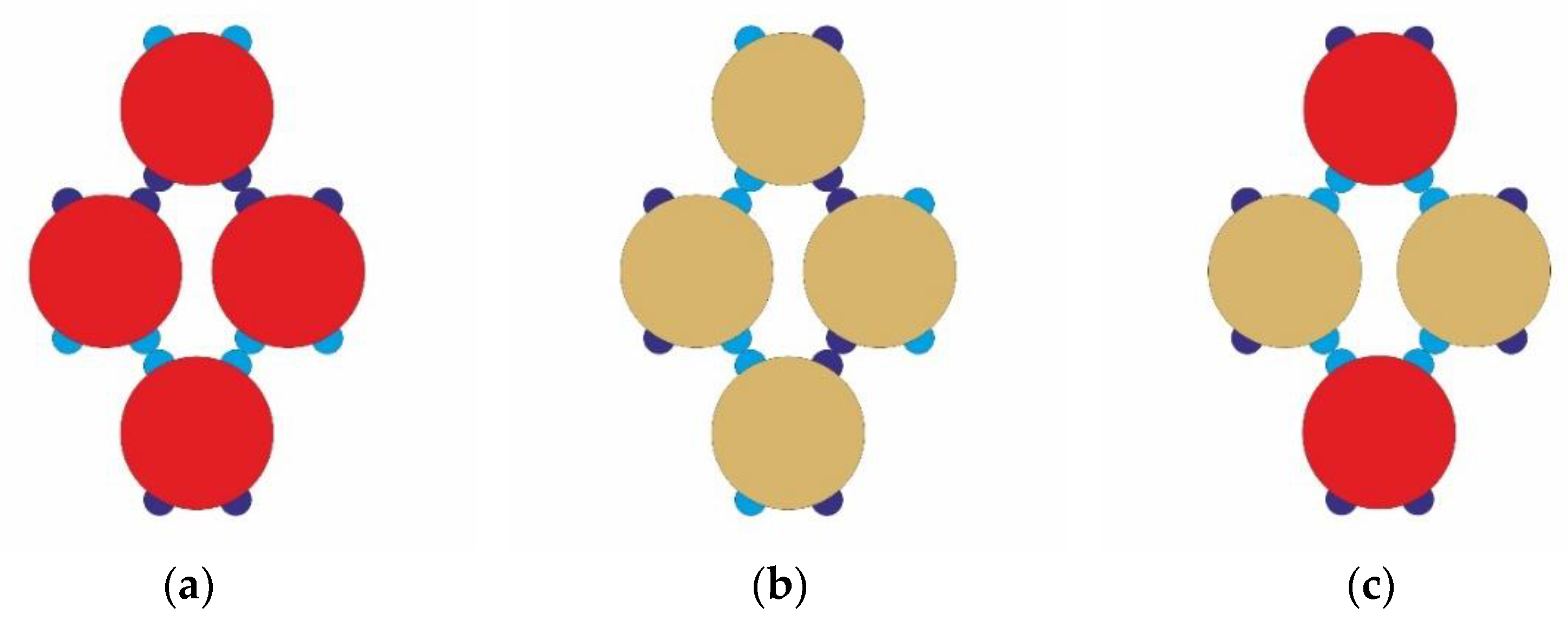
Having a lot in common, the two types of PPs presented in
Figure 1 have a very important difference, which is illustrated in
Figure 2 and
Figure 3. Both type I and type II can form their own metastable rhombohedral structures (see
Figure 2a,b), even they can form a mixed rhombohedral cocrystal structure in a ratio of 1:1 (see
Figure 2c). This mixed rhombohedral building block could be realized with four WW-WW bonds, as shown in
Figure 2c, or by four SN-SN bonds, too.
Stable trimers can be formed solely by PPs of type I (see
Figure 3a), while this is not possible for PPs of type II. However, it is possible for PPs of type II to be “included” in a mixed type of trimer in ratio 1:2 (see
Figure 3b) and hence to develop Kagome cocrystal structure, which is expected to be more stable in a given temperature interval.
Here we have to point out, that the mixed rhombohedral building block consists of two couples of PPs, connected by four identical bonds (this is the higher number of identical bonds in the so far presented building blocks from PPs type I and type II)–thus, this is rather a rare event, which explains why this mixed rhombohedral cocrystal phase is not well developed and monitored in our simulations.
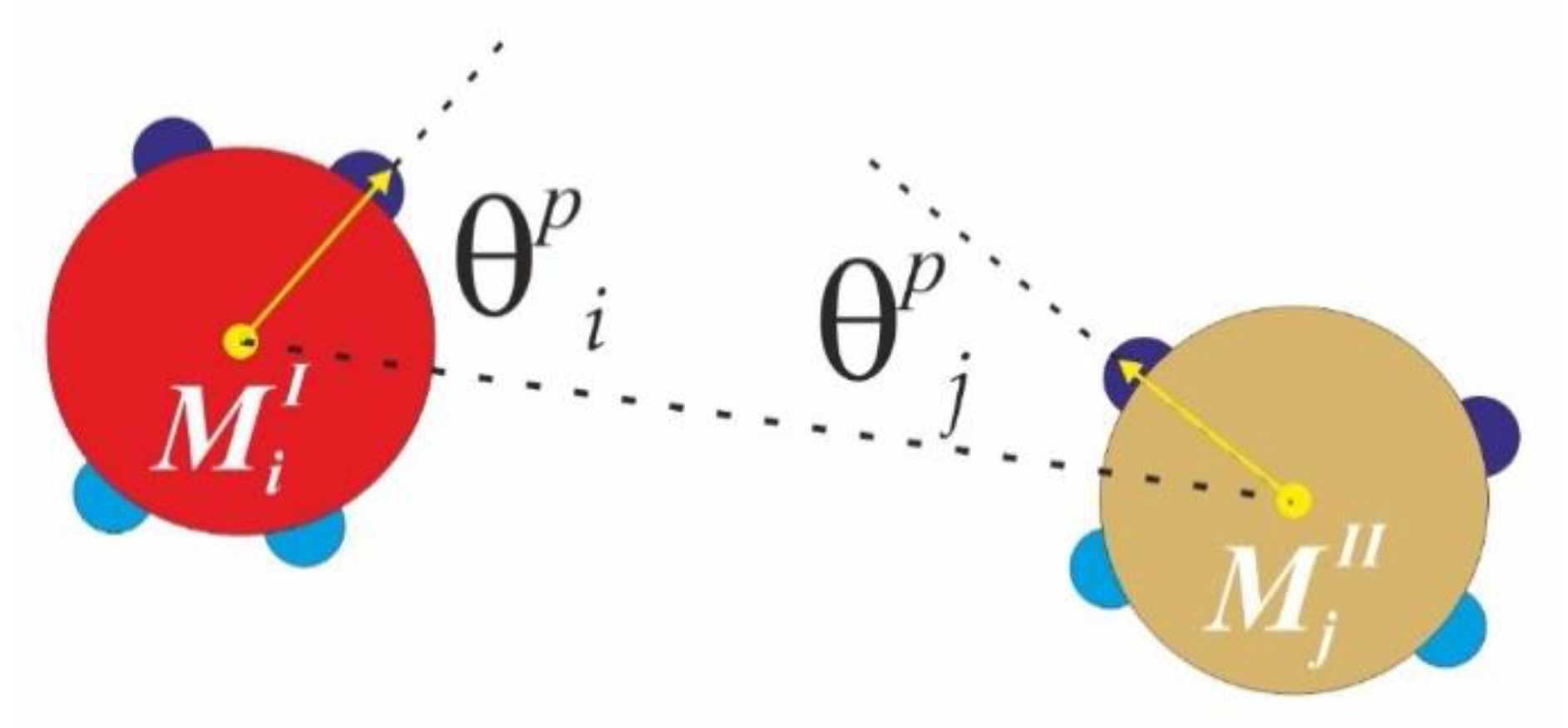
Our computational model is based on the model introduced by Doye [
25], slightly modified in order to include the two types of patches interactions–strong and narrow, and weak and wide. Patches of the same type interact via modulated 12-6 Lennard-Jones (LJ) pair potential with repulsive part for
, where no patch-patch interactions are available (excluded volume repulsion) and attractive part for
, which is multiplied by a number
between zero and one, depending on the mutual orientation of similar patches with respect to the inter-core vector connecting monomer
Mi and monomer
Mj–see
Figure 4 (irrespective to type I or type II). Thus, the 12-6 LJ pair potential is modulated according to the mutual orientation of similar patches on monomers type I and type II. The value of
is calculated for every step for every two monomers according to
, where index
accounts for the like-type patches (
SN-SN or
WW-WW only),
and
are the angles enclosed by the core particle vector and the corresponding patch vector, and parameter
gives the patchy extent in degrees (or, so called patchy width) along the particle circumference. In all simulations, the temperature is expressed in terms of the reduced temperature
, where
is the amplitude of the SN-SN patches attractive interaction. We use the following values of the model parameters:
.
Our task was to design a set of patchy particles, such that they can assemble into the targeted crystal structure. To do this, we used the following semi-empirical approach: Firstly, we drew the desired ordered structure and checked all possibilities that enable creating it by using patches with properties (geometry, interaction strength), needed to its assembly. In doing so, for each particle in our target structure we define patch vectors pointing at its nearest-neighboring particle; all particles of the same type have the same properties (number of patches, patch vectors, and all patch properties). Additionally, the MC simulation model selected by us [
31] demonstrated structural flexibility (it allows two polymorphic forms but is not locked into a single type of crystalline lattice or packing mode), it was also used for simulation of the co-crystallization process.
As we will show, it is remarkable that a relatively simple “design” of PPs–with only two pairs of attractive patches, but having different strengths–enable 2D Monte Carlo simulation of co-crystallization. Altering patch positions only–from 60–120–60–120 (type I) to 120–60–120-60 (type II) degrees–the bonding interpatch energies remain the same (including in the cocrystals). This means that, because of the same number of uniform connections, the enthalpy is the same for all resulting crystal structures. Thus, the processes are dictated by entropy only, i.e., by the high cocrystal conformational entropy.
Separately, each of the two types of PPs shown in
Figure 1 makes its “own” specific Rh-phase unit cell, characterized by different sorts of rhombuses (
Figure 2a,b and
Figure 5a,b). So, separately, each PPs type is expected to make its own Rh-crystal phase, as shown on
Figure 5. In fact, the PPs type I (60–120–60–120) generate a Rh-phase, which passing by through a polymorphic transition transforms into a Tri-hex (Kagome) lattice at specific temperature interval; and already beginning to make the Rh-structure, some PPs from type I start to create a relatively small number of stable trimers, formed solely by SN interactions (see
Figure 3a). Details on this polymorph transition are given in [
31]. In contrast, the PPs type II (120–60–120-60) cannot make its “own” stable trimers (and hence, no Kagome lattice from PPs type II could be built), but they generate their “own” Rh-phase only (see
Figure 2b for the single structure unit and
Figure 5b for the corresponding large crystal phase). Let’s point out that, strictly speaking, besides the Rh-phase, the PPs of type II can arrange in rings of six PPs. Formation of such rings is a rare event (because it requires very specific mutual in space and time orientation of type II monomers), and they are not stable either–when they reach the already grown Rh-phase they disintegrate into monomers, which are then incorporated sooner or later in the ridge of the growing Rh-phase.
Different is the situation when the two types of PPs are mixed-up in a starting composite “solution”. Under properly selected conditions of these composite “solutions”, the system of the two types of PPs evolve toward a Kagome like patchy cocrystal. Especially pronounced is the cocrystal Kagome lattice structure when the concentration ratio of the two PPs types differs from 1:1. Taking into account the structure of the mixed trimer (see
Figure 3b) it is to be expected that solely Kagome cocrystal emerges when the concentration of PPs is one part of type I to two parts of type II. The sequence of steps of the Kagome cocrystal formation is shown by four snapshots in
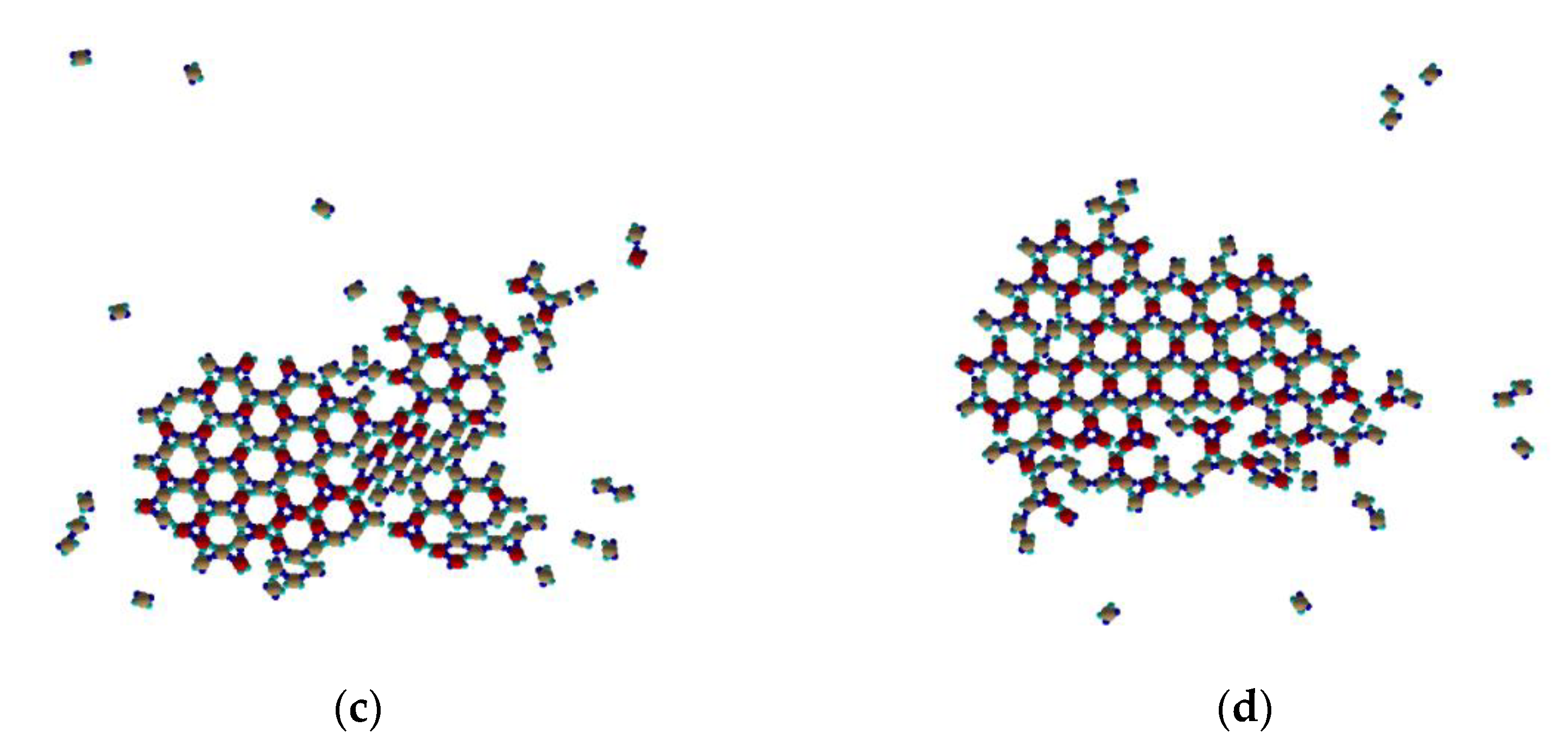
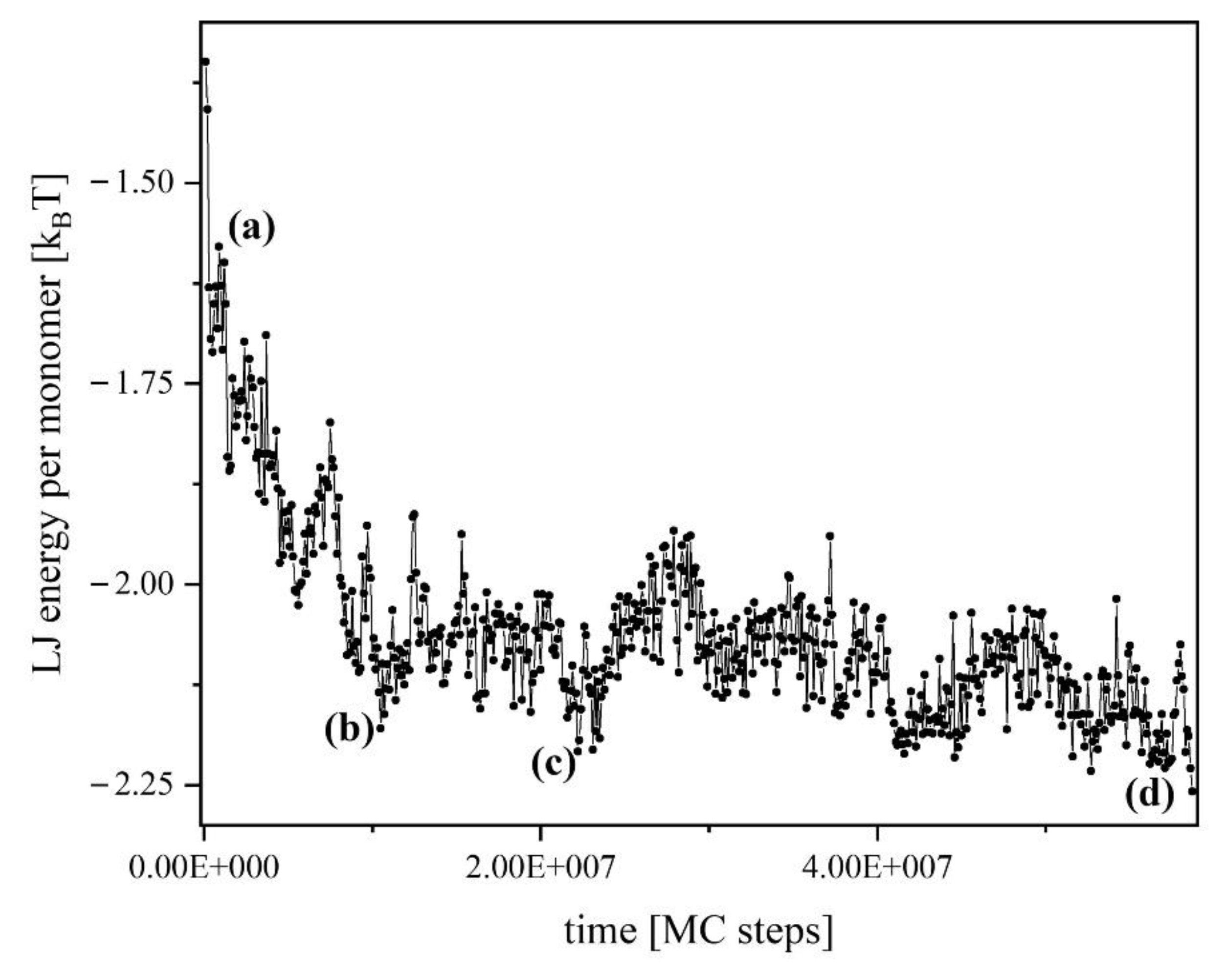
Figure 6, the corresponding energy of the system is shown in
Figure 7, and one can identify the following key steps:
Starting by randomly dispersed PPs type I and type II in ratio1:2, shortly after the start of the simulations both of them begin to form the four possible building blocks, which could be “constructed” from PPs type I and type II, and already schematically shown in
Figure 2 and
Figure 3–this is the Rh phase formed from PPs type I only, the Rh-phase formed from PPs type II only, the mixed Rh-phase building block, the Kagome phase trimer from PPs type I only and finally the most important Kagome mixed phase trimer from PPs type I and type II (see
Figure 6a). Let us point out, that the Kagome mixed trimers prevail in number over the “pure” type I trimers, and this is due to the chosen ratio1:2 of the monomers. As it was shown in [
31], the Rh-phase type I is not stable and it “releases” monomers type I, which are subsequently “catch” by monomers type II and thus continue to form the mixed phase trimers (see
Figure 6b), which begin to aggregate in Kagome cocrystal domains. This corresponds to a local minima of the system energy (see
Figure 7).
Figure 6c shows further development of the Kagome cocrystal by almost merging together the small cocrystal domains and repetitively creating and breaking bonds toward an increasingly stable and a less defect structure. Because of the relative low number of “pure” type I trimers, they are not able to “built” their own “pure” Kagome lattice and they are incorporated into the growing cocrystal. Mixed Rh-phase cocrystal small domains are presented, too.
The final cocrystal Kagome structure is shown in
Figure 6d, which possesses minimal system energy. Of course, due to the inevitable presence of the stable type I trimers, the cocrystal presents some defects, especially at its edges. Just one building block of the mixed Rh-phase is presented in
Figure 6d, too. This is due to the fact that such a complex is a very rare event to be formed, as it requires four identical bonds to be properly oriented.
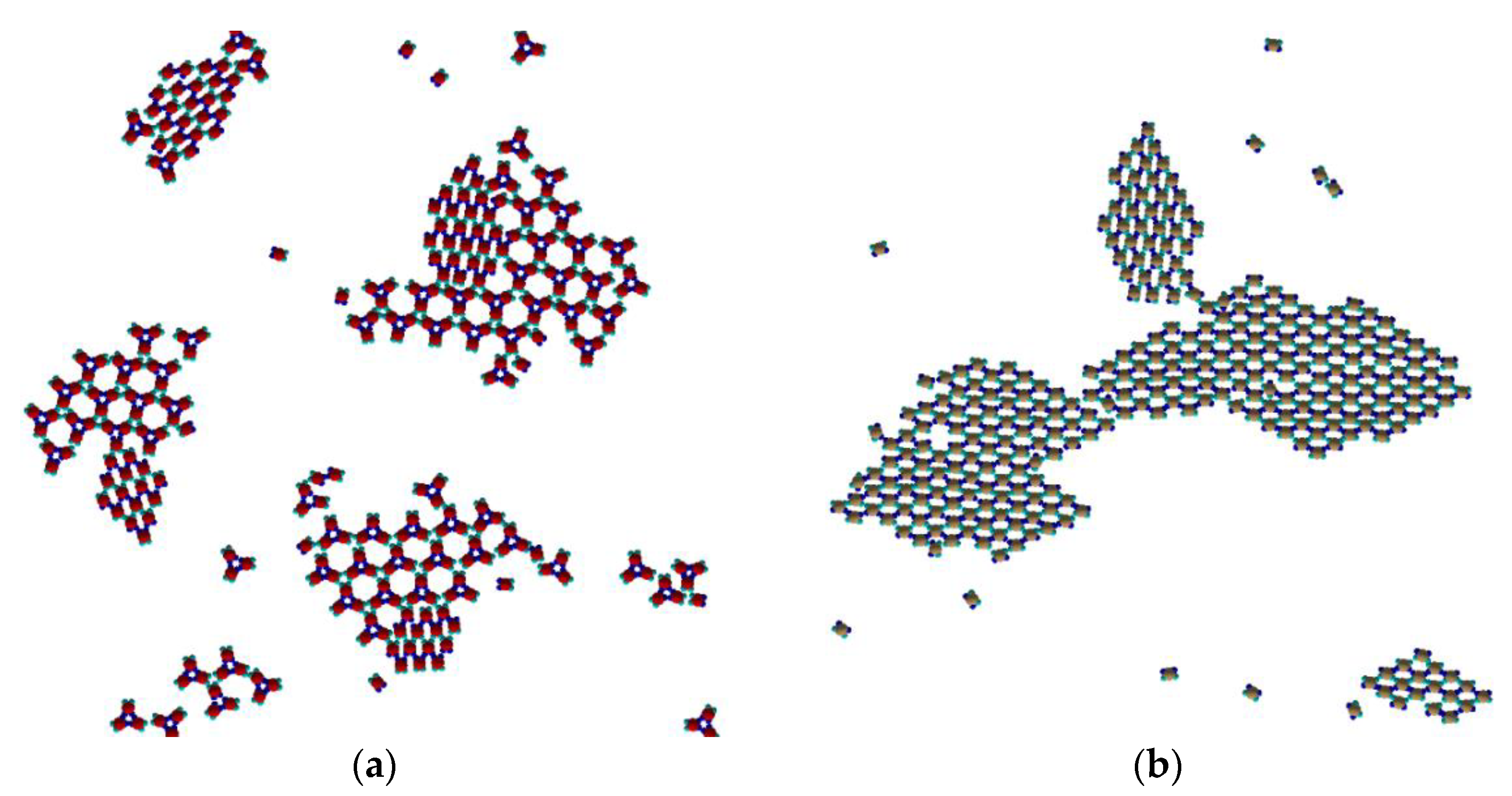
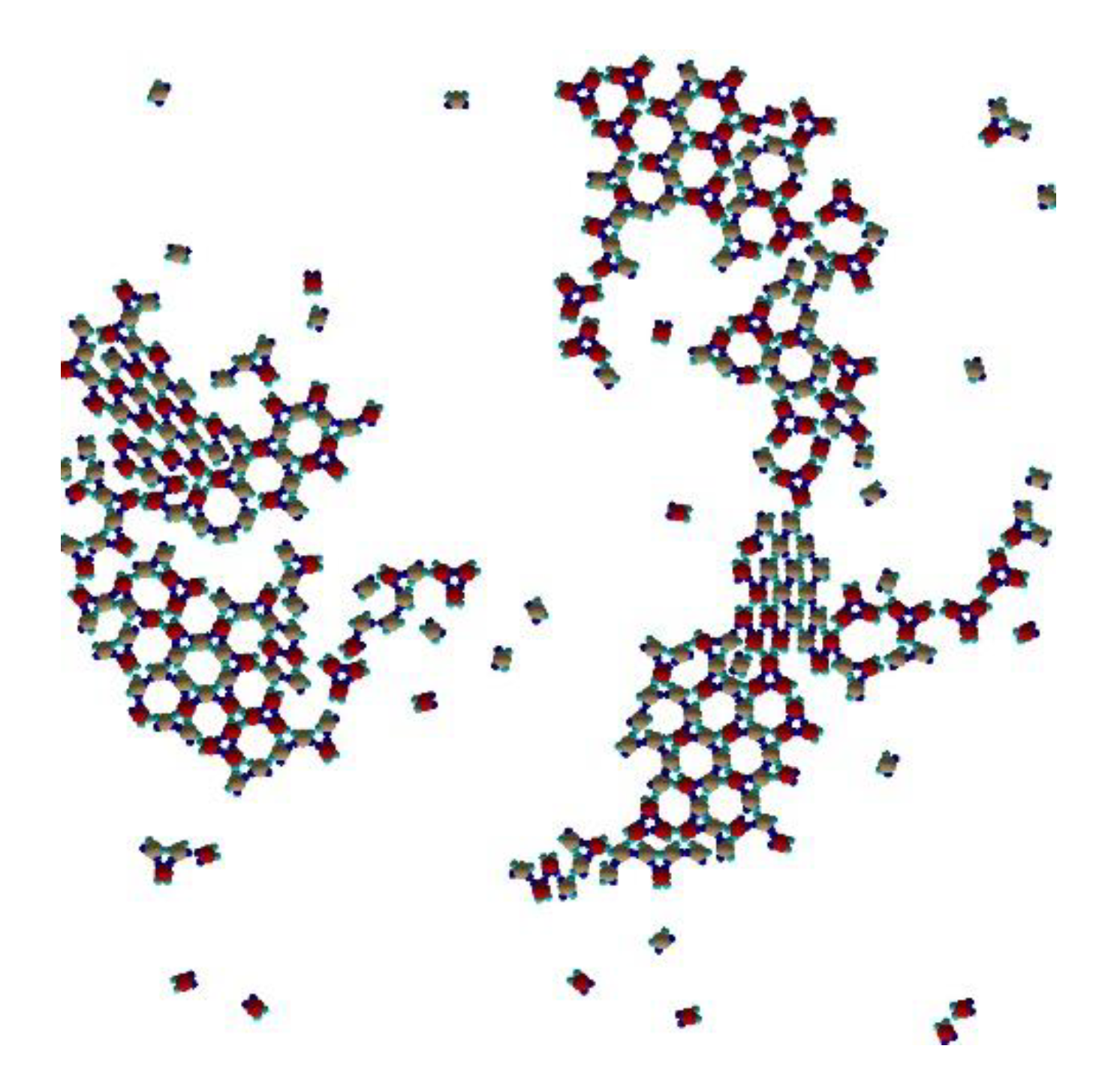
Keeping the temperature T* = 0.10, concentrations different from of 1:2 cannot produce “large” periodic cocrystal Kagome or even Rh-like structures (see
Figure 8). One can find several domains, consisting of rhombohedral-like placement of PPs type I and type II, without defined periodicity, and Kagome-like placement of both type trimers, also without defined periodicity.
In conclusion, controlling the concentration ratio of the two PPs types at fixed T* = 1.0, it is possible to simulate stable Kagome structures, which are of enhanced interest recently, e.g., [
32]. However, besides the “solution” composition, the cocrystal stability strongly depends on temperature, T*. Recall that T* is interrelated with the interparticle bonding energy: evidently, the higher T* corresponds to a relatively lower intermolecular bonding in the cocrystals. Practically important is that the weakened intermolecular bonding in cocrystals results in higher water-solubility, and hence in higher bioavailability of pharmaceutical cocrystals. Therefore, the dependence of the Kagome cocrystal stability on T* was checked carefully. It turns out that “optimal” are temperatures in a very narrow interval at approximately T* = 0.10.
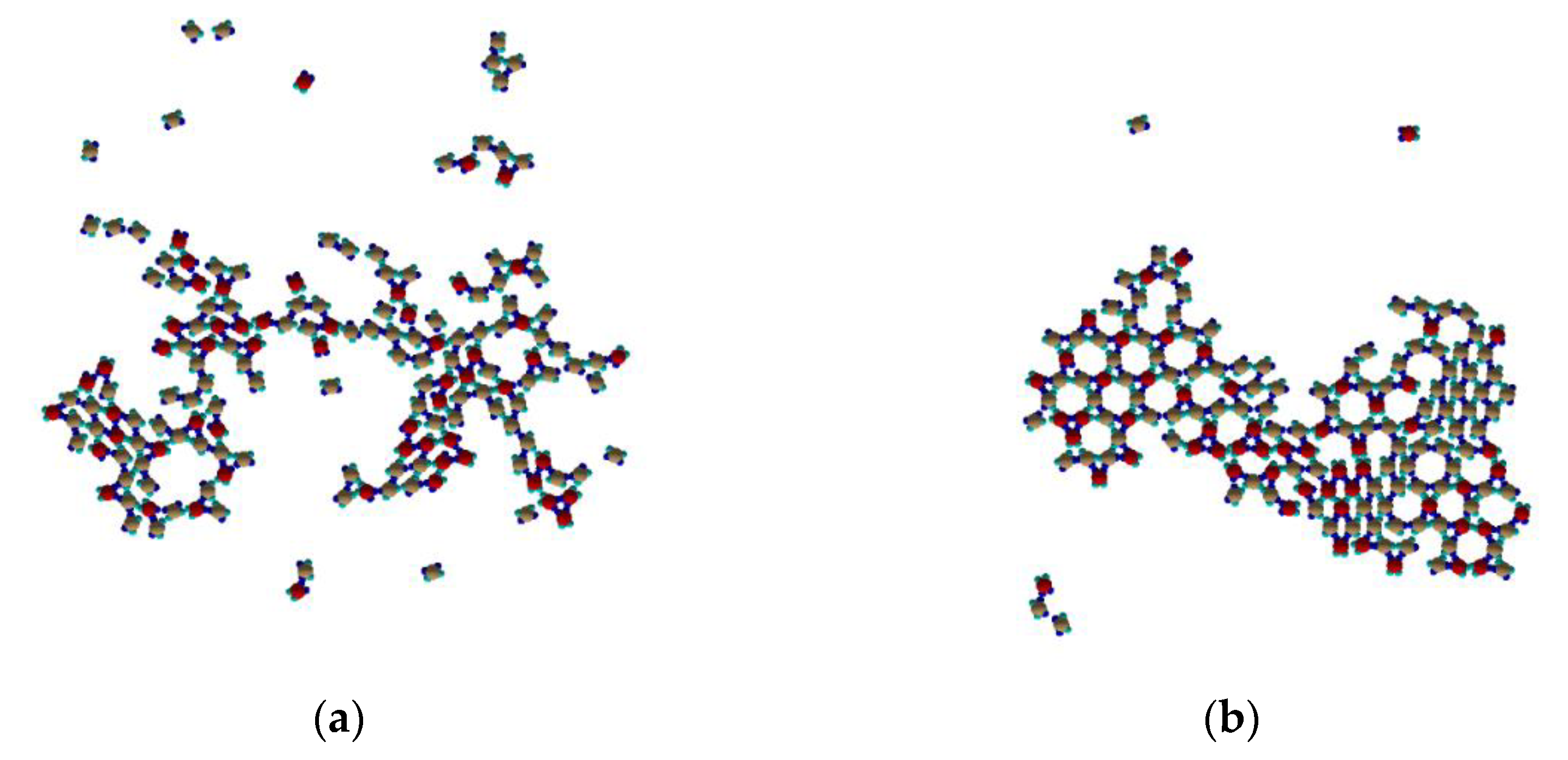
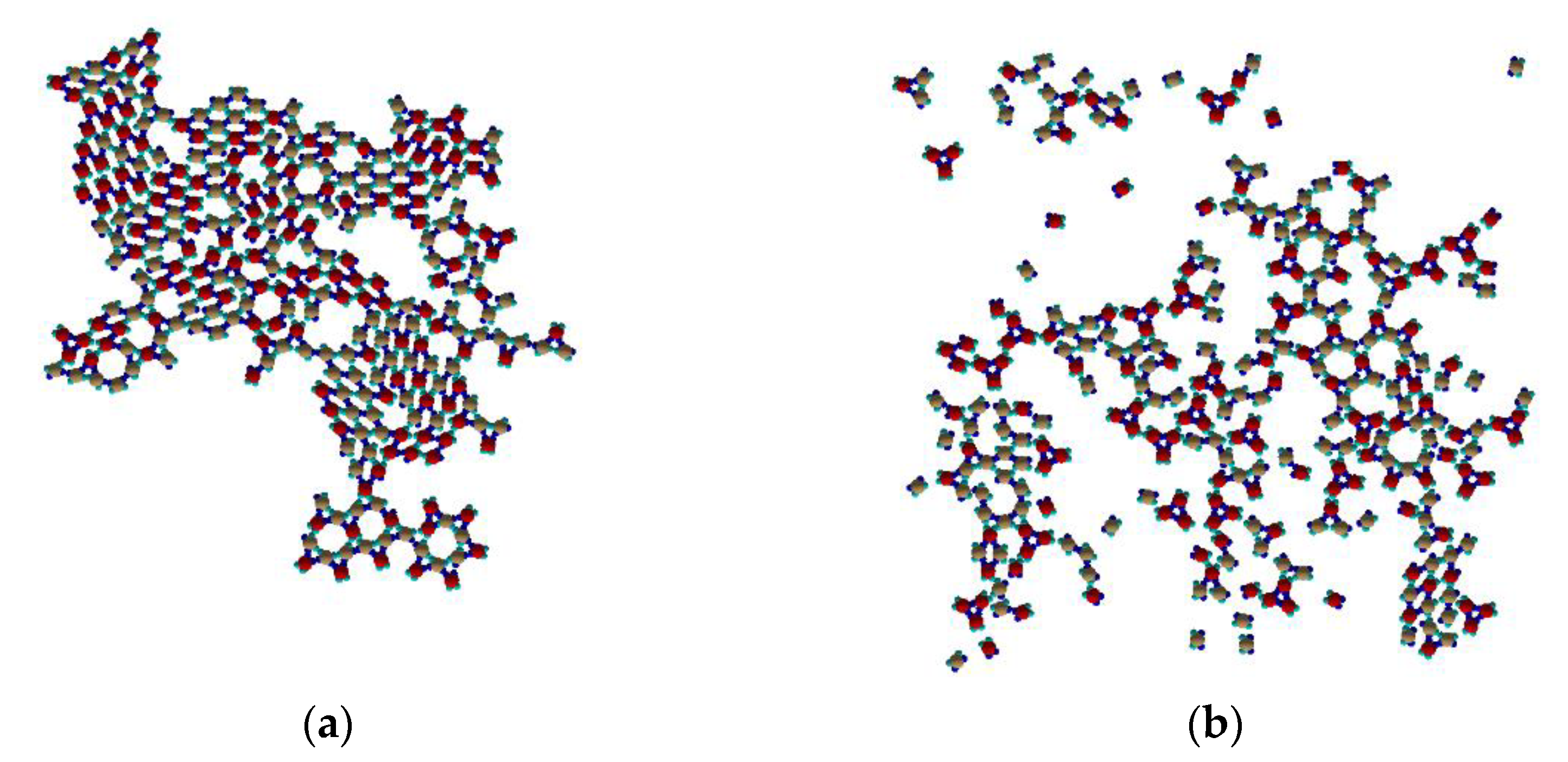
At temperatures below 0.10 (see
Figure 9a) Rh-phase like placement of PPs is dominant without defined periodicity. Beside it, some type I and mixed trimers, and some mixed ring structures as well can be identified. At temperatures slightly above 0.10 (see
Figure 9b) the dominant structures are type I and mixed trimers. At such “elevated” temperature the trimers are not able to form stable Kagome cocrystal.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









