
Understanding the Semiconducting-to-Metallic Transition in the CF2Si Monolayer under Shear Tensile Strain


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
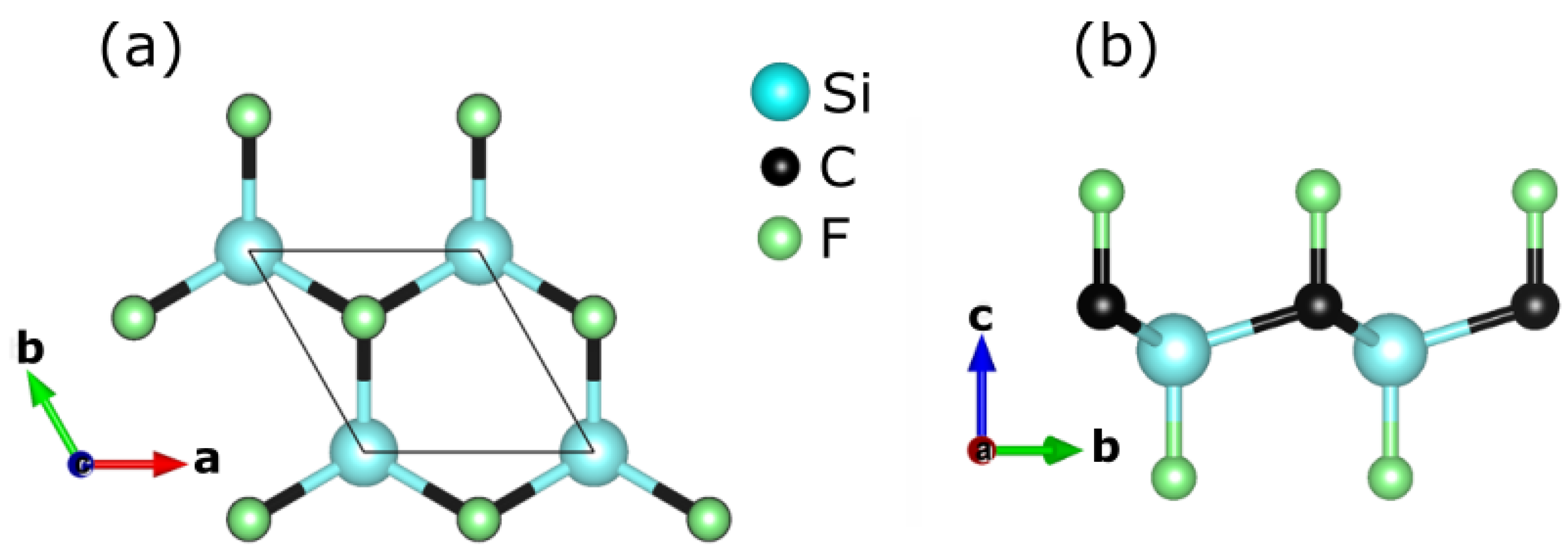
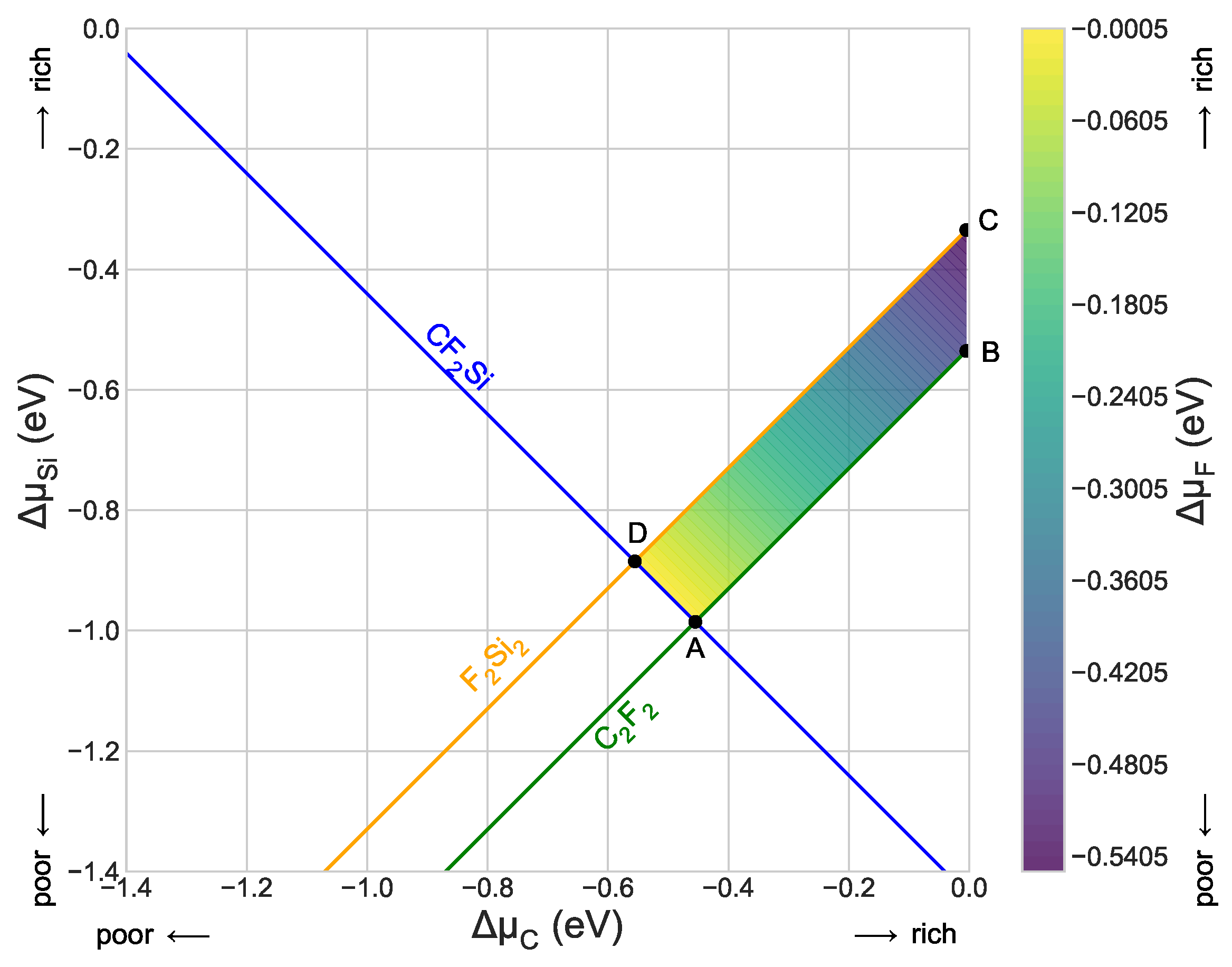
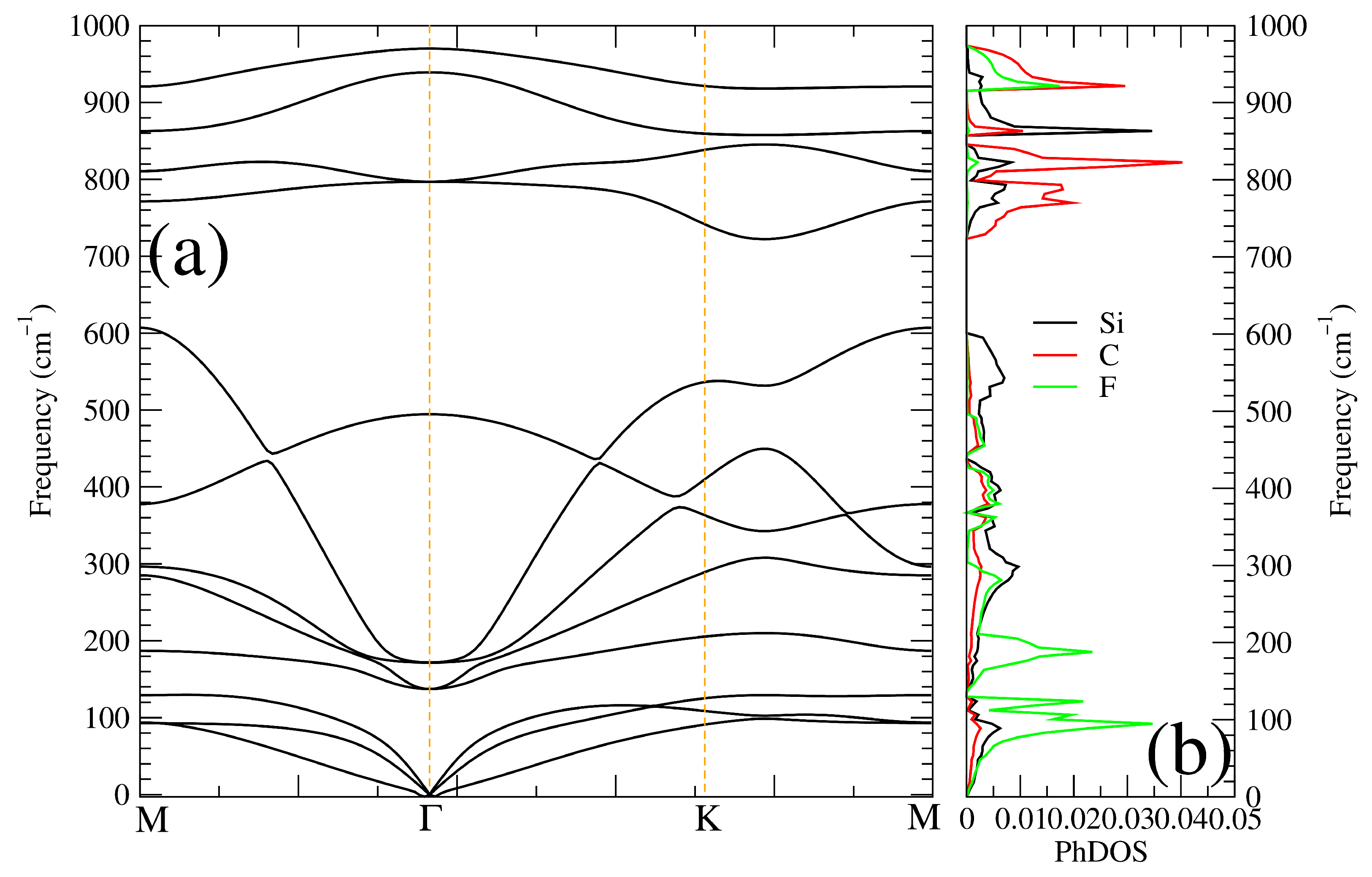
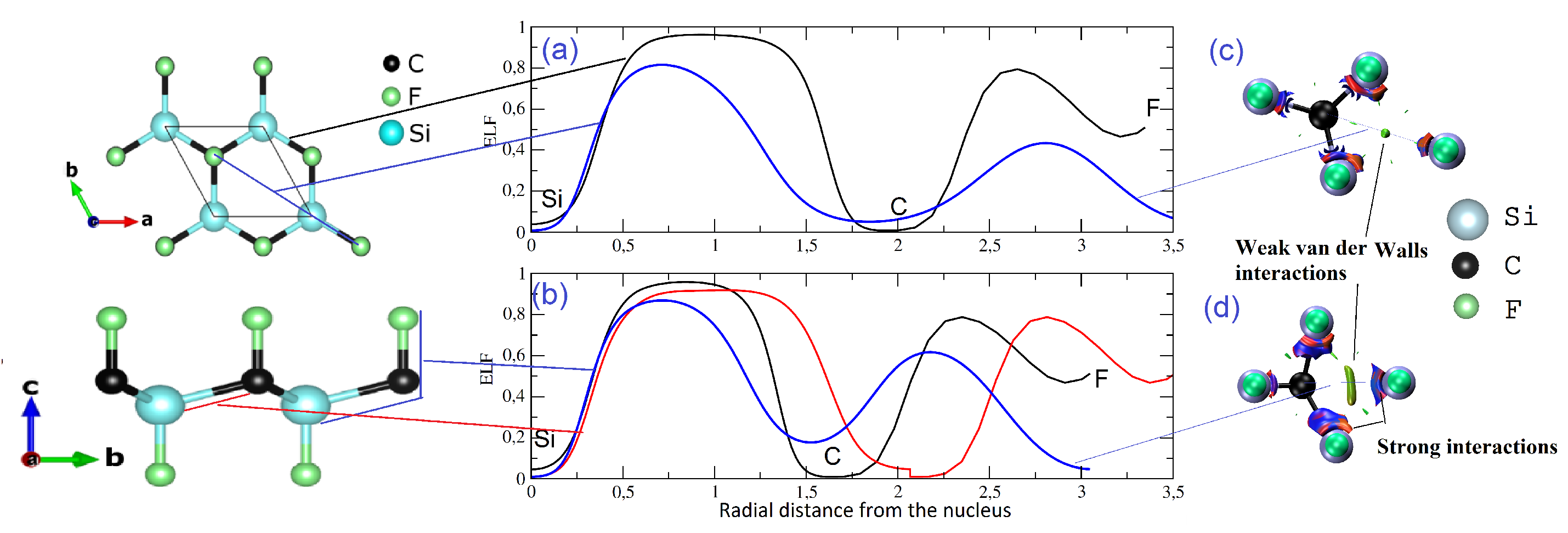
3.1. Optimized Structure and Its Stability
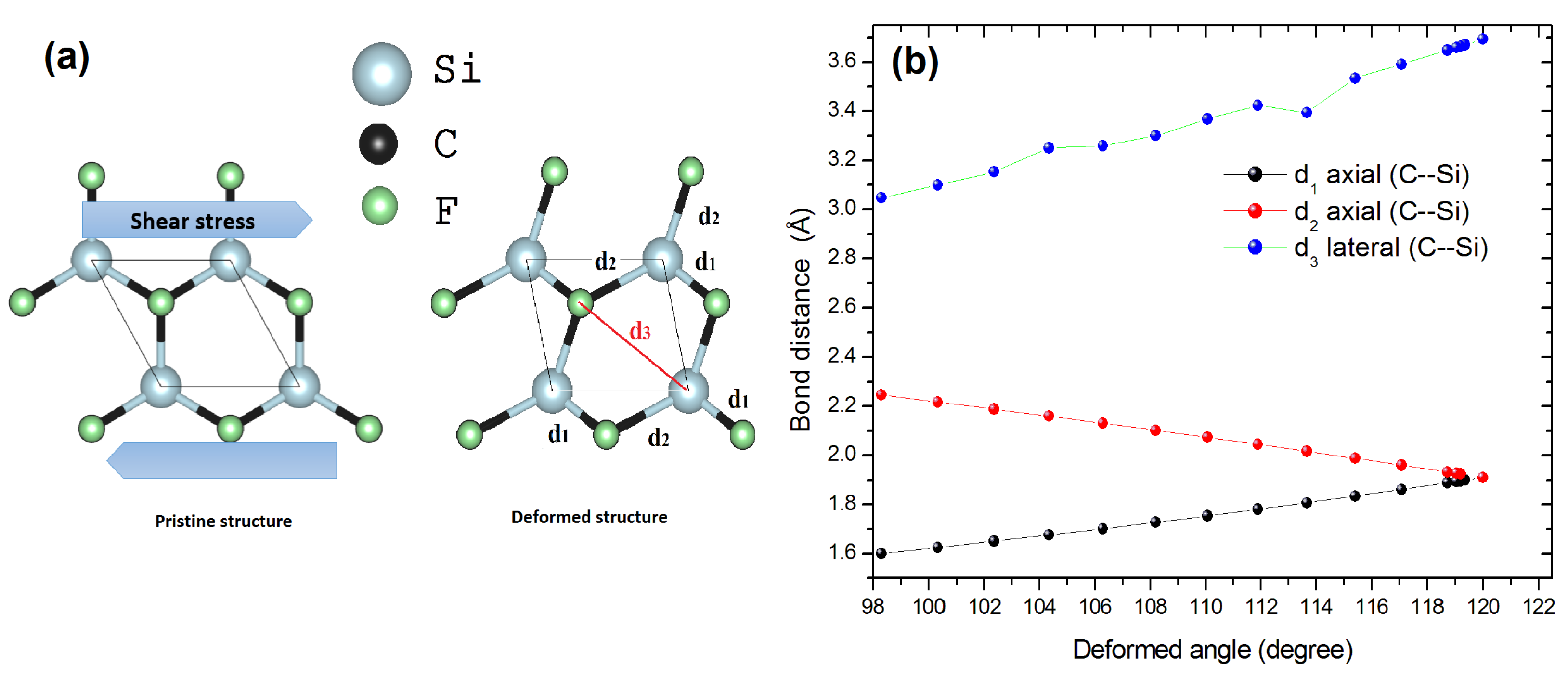
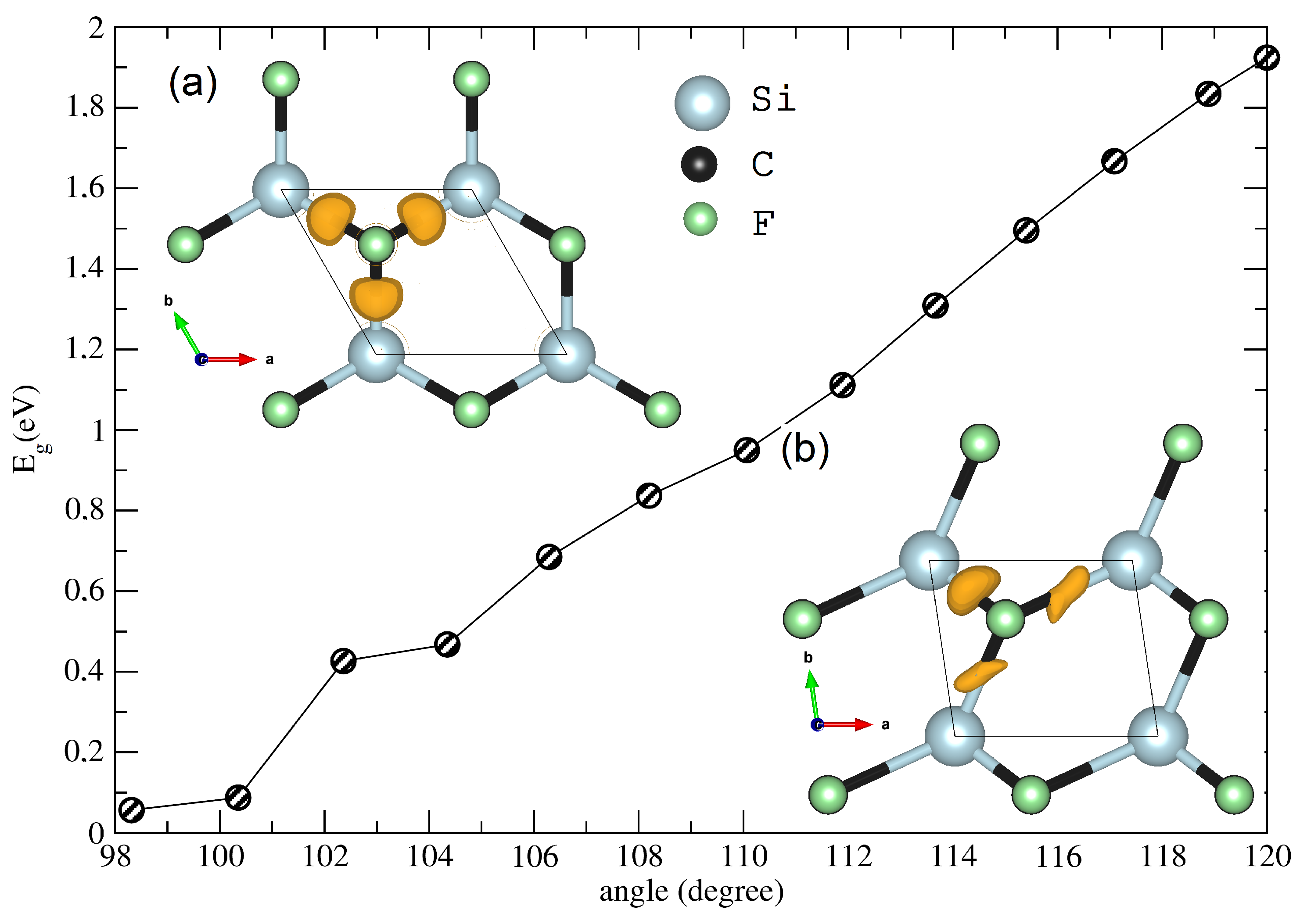
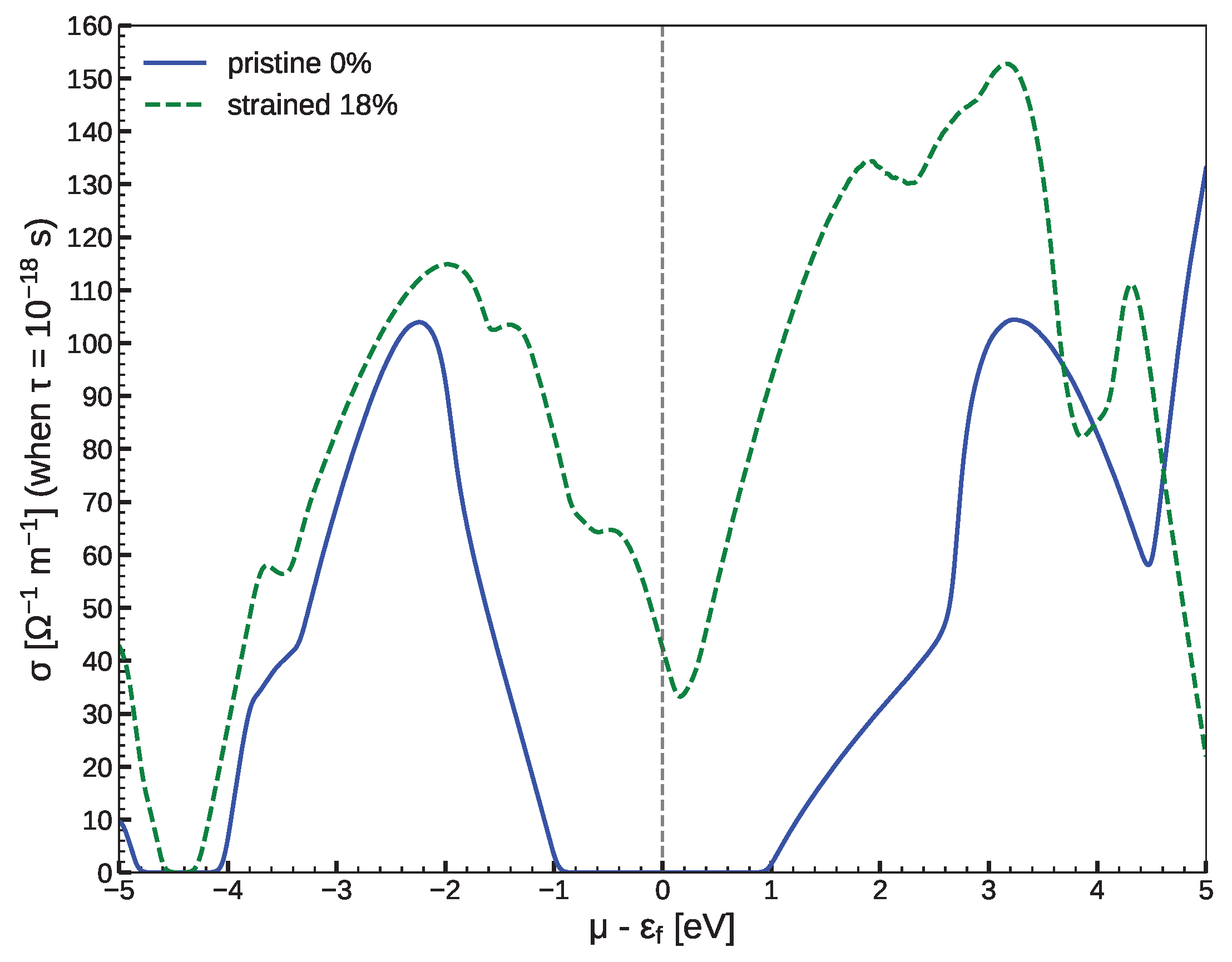
3.2. Low-Dimensional CFSi Structure under Shear Tensile Strain
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barreteau, C.; Michon, B.; Besnard, C.; Giannini, E. High-pressure melt growth and transport properties of SiP, SiAs, GeP, and GeAs 2D layered semiconductors. J. Cryst. Growth 2016, 443, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Schaack, S.; Depondt, P.; Huppert, S.; Finocc, H.F. Quantum driven proton diffusion in brucite-like minerals under high pressure. Sci. Rep. 2020, 10, 8123. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qian, X.; Li, J. Phase transitions in 2D materials. Nat. Rev. Mater. 2021, 6, 829–846. [Google Scholar] [CrossRef]
- Voiry, D.; Yamaguchi, H.; Li, J.; Silva, R.; Alves, D.C.B.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 2013, 12, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Eda, G.; Fujita, T.; Yamaguchi, H.; Voiry, D.; Chen, M.; Chhowalla, M. Coherent atomic and electronic heterostructures of single-layer MoS2. ACS Nano 2012, 6, 7311–7317. [Google Scholar] [CrossRef]
- Cai, X.; Ren, Y.; Wu, M.; Xu, D.; Luo, X. Strain-induced phase transition and giant piezoelectricity in monolayer tellurene. Nanoscale 2020, 12, 167. [Google Scholar] [CrossRef]
- Nayak, A.P.; Bhattacharyya, S.; Zhu, J.; Liu, J.; Wu, X.; Pandey, T.N.; Jin, C.; Singh, A.K.; Akinwande, D.; Lin, J.-F. Pressure-induced semiconducting to metallic transition in multilayered molybdenum disulphide. Nat. Commun. 2014, 5, 3731. [Google Scholar] [CrossRef] [Green Version]
- Fleurence, A.; Friedlein, R.; Ozaki, T.; Kawai, H.; Wang, Y.; Yamada-Takamura, Y. Experimental evidence for epitaxial silicene on diboride thin films. Phys. Rev. Lett. 2012, 108, 245501. [Google Scholar] [CrossRef]
- Li, Y.; Li, F.; Zhou, Z.; Chen, Z. SiC2 silagraphene and its one-dimensional derivatives: Where planar tetracoordinate silicon happens. J. Am. Chem. Soc. 2011, 133, 900–908. [Google Scholar] [CrossRef]
- Zhou, L.J.; Zhang, Y.F.; Wu, L.M. SiC2 siligraphene and nanotubes: Novel donor materials in excitonic solar cells. Nano Lett. 2013, 13, 5431–5436. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, Y. Geometric and electronic structures of two-dimensional SiC3 compound. J. Phys. Chem. C. 2014, 118, 4509–4515. [Google Scholar] [CrossRef]
- Borlido, P.; Huran, A.W.; Marques, M.A.L.; Botti, S. Novel two-dimensional silicon-carbon binaries by crystal structure prediction. Phys. Chem. Chem. Phys. 2020, 22, 8442–8449. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Lu, S.; Guo, Y.; Hu, X. Novel bonding patterns and optoelectronic properties of the two-dimensional SixCy monolayers. J. Mater. Chem. C. 2017, 5, 3561–3567. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ning, Z.; Zhang, H.; Ni, G.; Shao, H.; Peng, B.; Zhang, X.; He, X. Anisotropic ultrahigh hole mobility in two-dimensional penta-SiC2 by strain-engineering: Electronic structure and chemical bonding analysis. RSC Adv. 2017, 7, 45705–45713. [Google Scholar] [CrossRef] [Green Version]
- Kilic, M.E.; Lee, K.-R. Tetrahex Carbides: Two-Dimensional Group-IV Materials for Nanoelectronics and Photocatalytic Water Splitting. Carbon 2021, 174, 15. [Google Scholar] [CrossRef]
- Feng, W.; Long, P.; Feng, Y.; Li, Y. Two-Dimensional Fluorinated Graphene: Synthesis, Structures, Properties and Applications. Adv. Sci. 2016, 3, 1500413. [Google Scholar] [CrossRef]
- Glass, C.W.; Oganov, A.R.; Hansen, N. USPEX—Evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175, 713. [Google Scholar] [CrossRef]
- Martonak, R.; Laio, A.; Bernasconi, M.; Ceriani, C.; Raiteri, P.; Parrinello, M. Simulation of structural phase transitions by metadynamics. Z. Krist. 2005, 220, 489. [Google Scholar]
- Ouahrani, T.; Merad-Boudia, I.; Baltache, H.; Khenata, R.; Bentalha, Z. Effect of pressure on the global and local properties of cubic perovskite crystals. Phys. Scr. 2011, 84, 025704. [Google Scholar] [CrossRef] [Green Version]
- Guedda, H.Z.; Ouahrani, T.; Morales-García, A.; Franco, R.; Salvado, M.A.; Pertierra, P.; Recio, J.M. Computer simulations of 3C-SiC under hydrostatic and non-hydrostatic stresses. Chem. Phys. Chem. Phys. 2016, 18, 8132–8139. [Google Scholar] [CrossRef]
- Belarouci, S.; Ouahrani, T.; Benabdallah, N.; Morales-García, A.; Belabbas, I. Two-dimensional silicon carbide structure under uniaxial strains, electronic and bonding analysis. Comp. Mater. Sci. 2018, 151, 288–295. [Google Scholar] [CrossRef]
- Bohorquez, H.; Boyd, R.J. A localized electrons detector for atomic and molecular systems. Theor. Chem. Acc. 2010, 127, 393. [Google Scholar] [CrossRef]
- Bohorquez, H.J.; Matta, C.F.; Boyd, R.J. The localized electrons detector as an ab initio representation of molecular structures. Int. J. Quantum Chem. 2010, 110, 2418. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5404. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, D.; Pilmé, J. New Insights in Quantum Chemical Topology Studies Using Numerical Grid-based Analyses. J. Comput. Chem. 2011, 32, 3207. [Google Scholar] [CrossRef] [PubMed]
- Ougherb, C.; Ouahrani, T.; Badawi, M.; Morales-García, A. Effect of the sulfur termination on the properties of Hf2CO2 MXene. Phys. Chem. Chem. Phys. 2022, 24, 7243–7252. [Google Scholar] [CrossRef] [PubMed]
- Born, M.; Huang, H. Dynamical Theory of Crystal Lattices; Oxford University Press: Clarendon, UK, 1954; 432p, ISBN 9780198503699. [Google Scholar]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B. 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Sekkal, A.; Benzair, A.; Ouahrani, T.; Faraoun, H.I.; Merad, G.; Aourag, H. Mechanical properties and bonding feature of the YAg, CeAg, HoCu, LaAg, LaZn, and LaMg rare-earth intermetallic compounds: An ab initio study. Intermetallics 2014, 45, 65–70. [Google Scholar] [CrossRef]
- Wei, Q.; Yang, Y.; Yang, G.; Peng, V. New stable two dimensional silicon carbide nanosheets. J. Alloys Compd. 2021, 868, 159201. [Google Scholar] [CrossRef]
- Chen, G.; Haire, R.G.; Peterson, J.R. Compressibilities of TbVO4 and DyVO4 Calculated from Spectroscopic Data. Appl. Spectrosc. 1992, 46, 1495–1497. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Savin, A.; Jepsen, O.; Flad, J.; Anderson, L.K.; Preuss, H.; von Schnering, H.G. Electron localization in solid-state structures of the elements: The diamond structure. Angew. Chem. Int. Ed. Engl. 1992, 32, 187–188. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Nyholm, R.S. Inorganic stereochemistry. Quart. Rev. 1957, 11, 339–380. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Limiting Inequalities | Point | Competing Phases |
|---|---|---|
| A(−0.4550, −0.9855, 0.0000) | CFSi, CF | |
| B(−0.0050, −0.5355, −0.4500) | CF | |
| C(−0.0050, −0.3345, −0.5505) | FSi | |
| D(−0.5555, −0.8850, 0.0000) | CFSi, FSi | |
| Symmetry | (cm) | Activity |
|---|---|---|
| A | 494.5 | IR/R |
| A | 939.2 | IR/R |
| A | 970.0 | IR/R |
| E | 137.2 | IR/R |
| E | 171.7 | IR/R |
| E | 797.0 | IR/R |
| a (Å) | C (N/m) | C (N/m) | E (N/m) | G (N/m) | K (N/m) | |
|---|---|---|---|---|---|---|
| 3.16086 | 118.543 | 22.410 | 114.306 | 48.067 | 70.477 | 0.189 |
| Basin | V() (Bohr) | q (Electrons) | ELF | Bond Metallicity () | (Atomic Units) |
|---|---|---|---|---|---|
| pristine | |||||
| V(C,Si) | 67.784 | −2.1654 | 0.9617 | −0.4906 | 0.12872651 |
| V(Si,C) | 71.167 | −2.2023 | 0.9617 | −0.4906 | 0.12872651 |
| V(F) | 57.249 | −2.4110 | 0.7945 | −1.2574 | 2.07097897 |
| V(F,C) | 3.137 | −0.7520 | 0.8466 | 0.2796 | 0.37723410 |
| strained | |||||
| V(C,Si) | 19.022 | −1.9622 | 0.9608 | −0.3428 | 0.23750961 |
| V(C,Si) | 35.711 | −0.6928 | 0.9252 | −0.5682 | 0.07178674 |
| V(Si,C) | 33.542 | −1.4756 | 0.9236 | −0.5340 | 0.09279469 |
| V(C,Si) | 33.642 | −1.4252 | 0.9175 | −2.4263 | 0.10376149 |
| V(Si) | 52.592 | −2.2627 | 0.4977 | −0.3529 | 2.07237211 |
| V(F) | 73.435 | −3.3421 | 0.8837 | −0.5505 | 0.06132580 |
| V(F) | 42.653 | −3.3972 | 0.8746 | −0.5486 | 0.05989116 |
| V(F) | 48.171 | −1.9589 | 0.8696 | 0.2769 | 0.37582815 |
| V(F,C) | 3.124 | −0.7088 | 0.8478 | 0.2791 | 0.37814979 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouahrani, T.; Boufatah, R.M. Understanding the Semiconducting-to-Metallic Transition in the CF2Si Monolayer under Shear Tensile Strain. Crystals 2022, 12, 1476. https://doi.org/10.3390/cryst12101476
Ouahrani T, Boufatah RM. Understanding the Semiconducting-to-Metallic Transition in the CF2Si Monolayer under Shear Tensile Strain. Crystals. 2022; 12(10):1476. https://doi.org/10.3390/cryst12101476
Chicago/Turabian StyleOuahrani, Tarik, and Reda M. Boufatah. 2022. "Understanding the Semiconducting-to-Metallic Transition in the CF2Si Monolayer under Shear Tensile Strain" Crystals 12, no. 10: 1476. https://doi.org/10.3390/cryst12101476
APA StyleOuahrani, T., & Boufatah, R. M. (2022). Understanding the Semiconducting-to-Metallic Transition in the CF2Si Monolayer under Shear Tensile Strain. Crystals, 12(10), 1476. https://doi.org/10.3390/cryst12101476