


Crystal Structure of a New 1:1 Acridine-Diclofenac Salt, Obtained with High Yield by a Mechanochemical Approach

 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Solid State Synthesis
2.2. X-ray Powder Diffraction and Structure Solution
2.3. Thermal Analysis
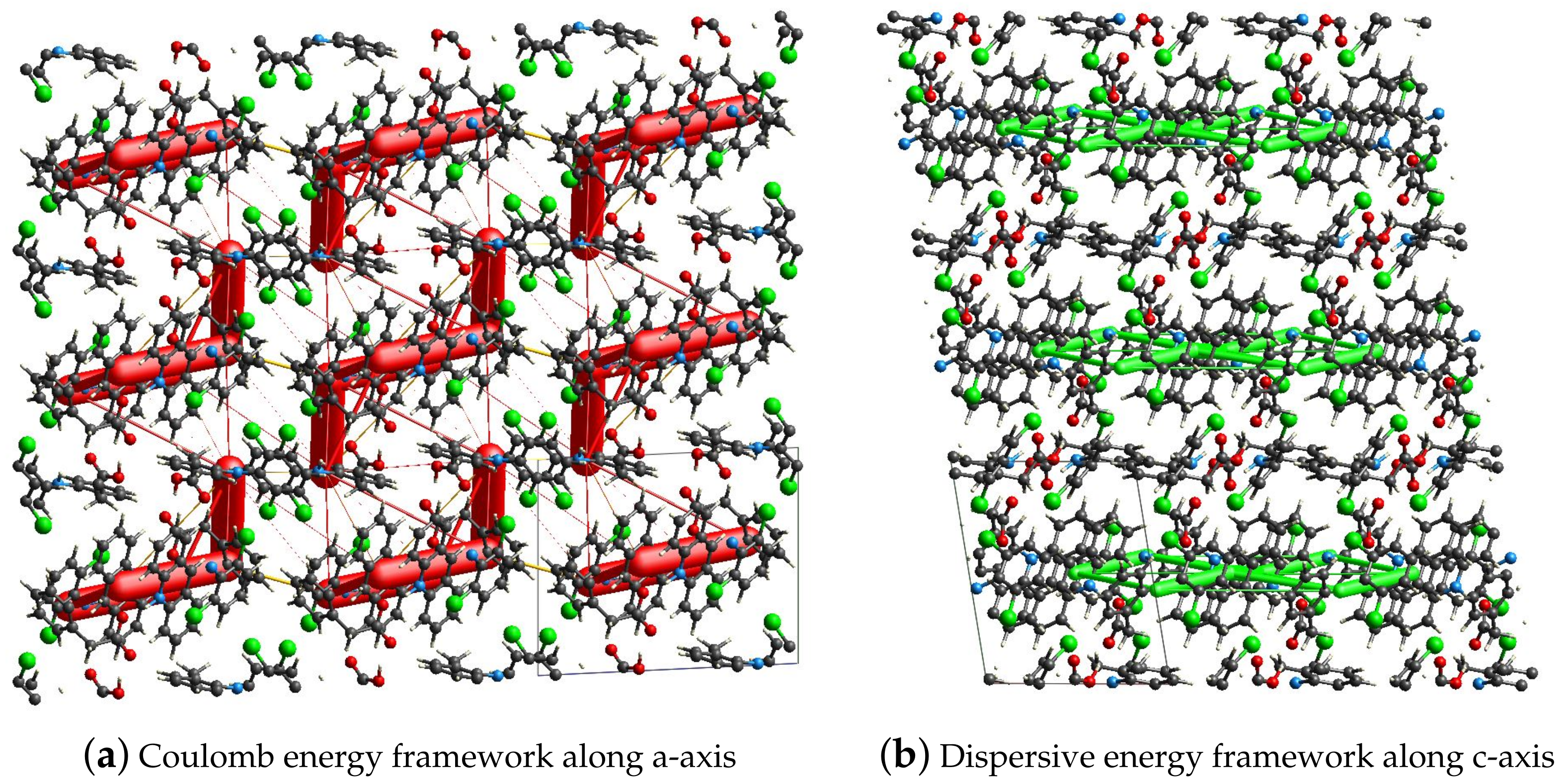
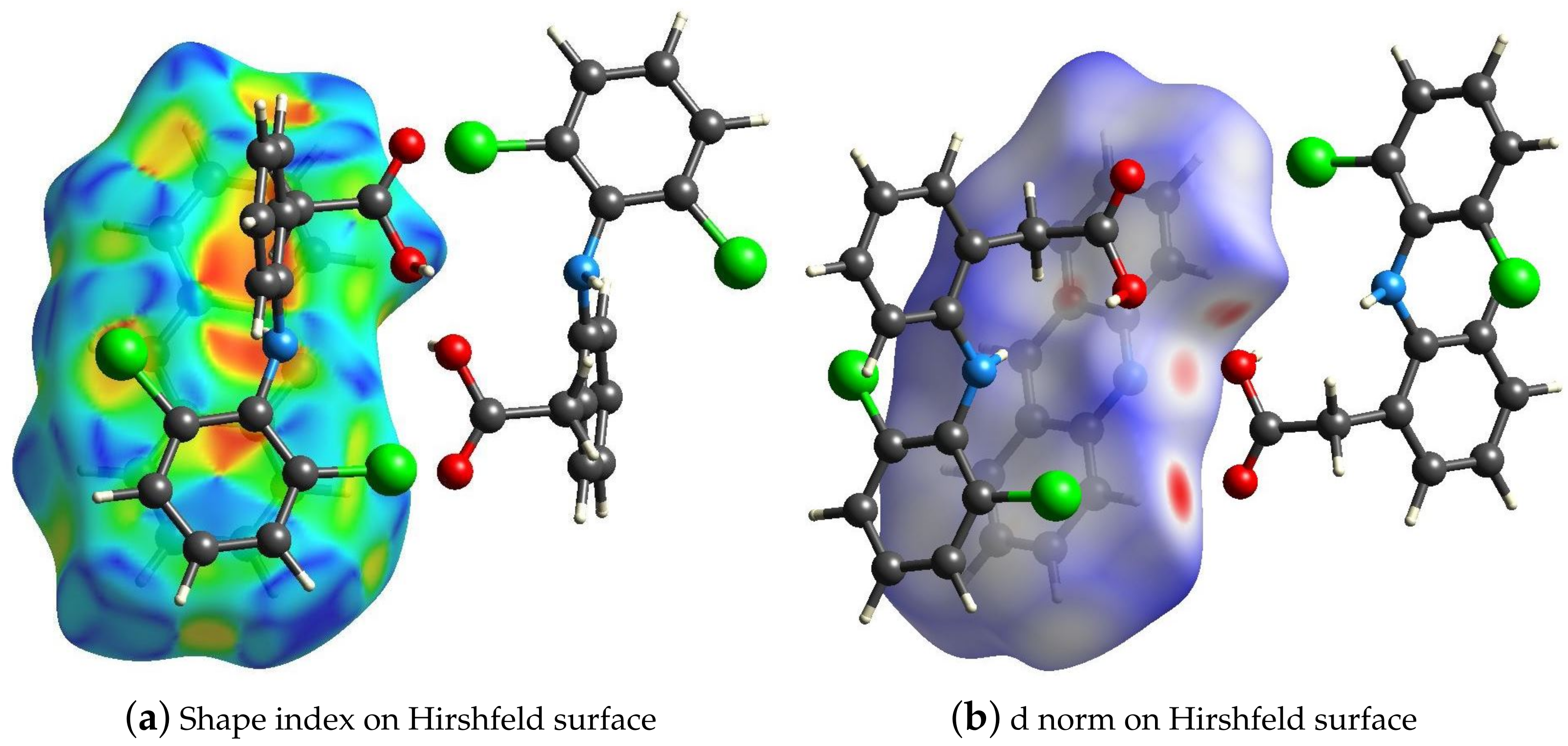
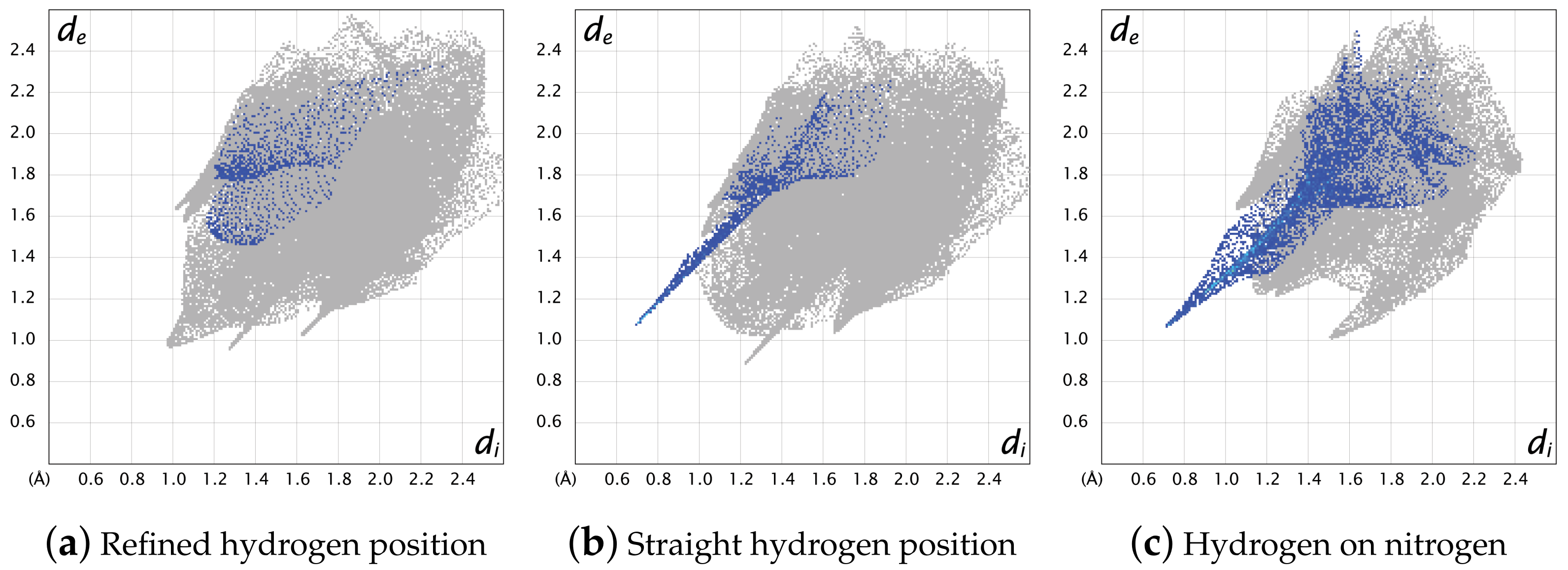
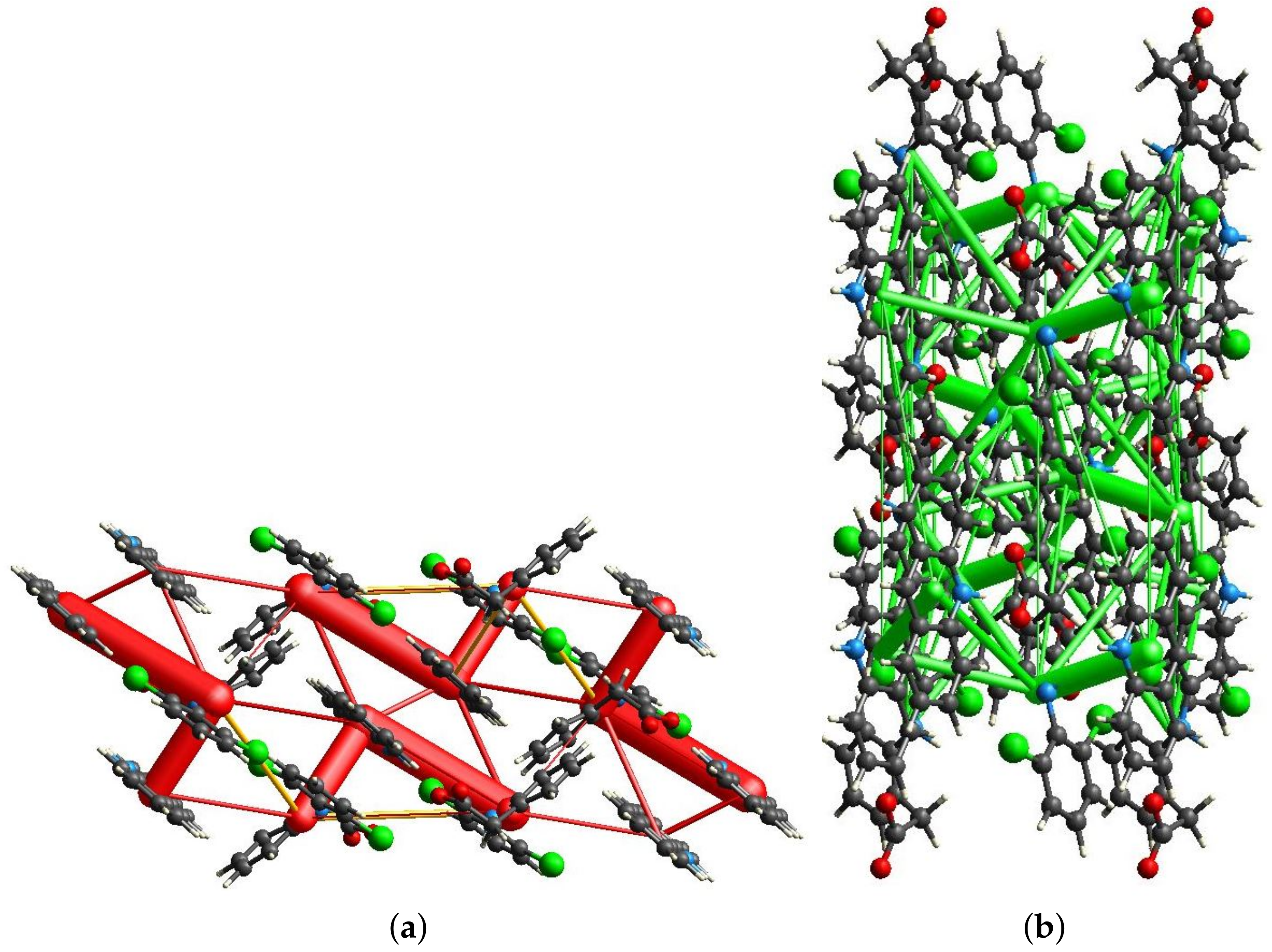
2.4. Hirshfeld Surface and the Energy Frameworks Ab Initio Calculations
3. Results and Discussion
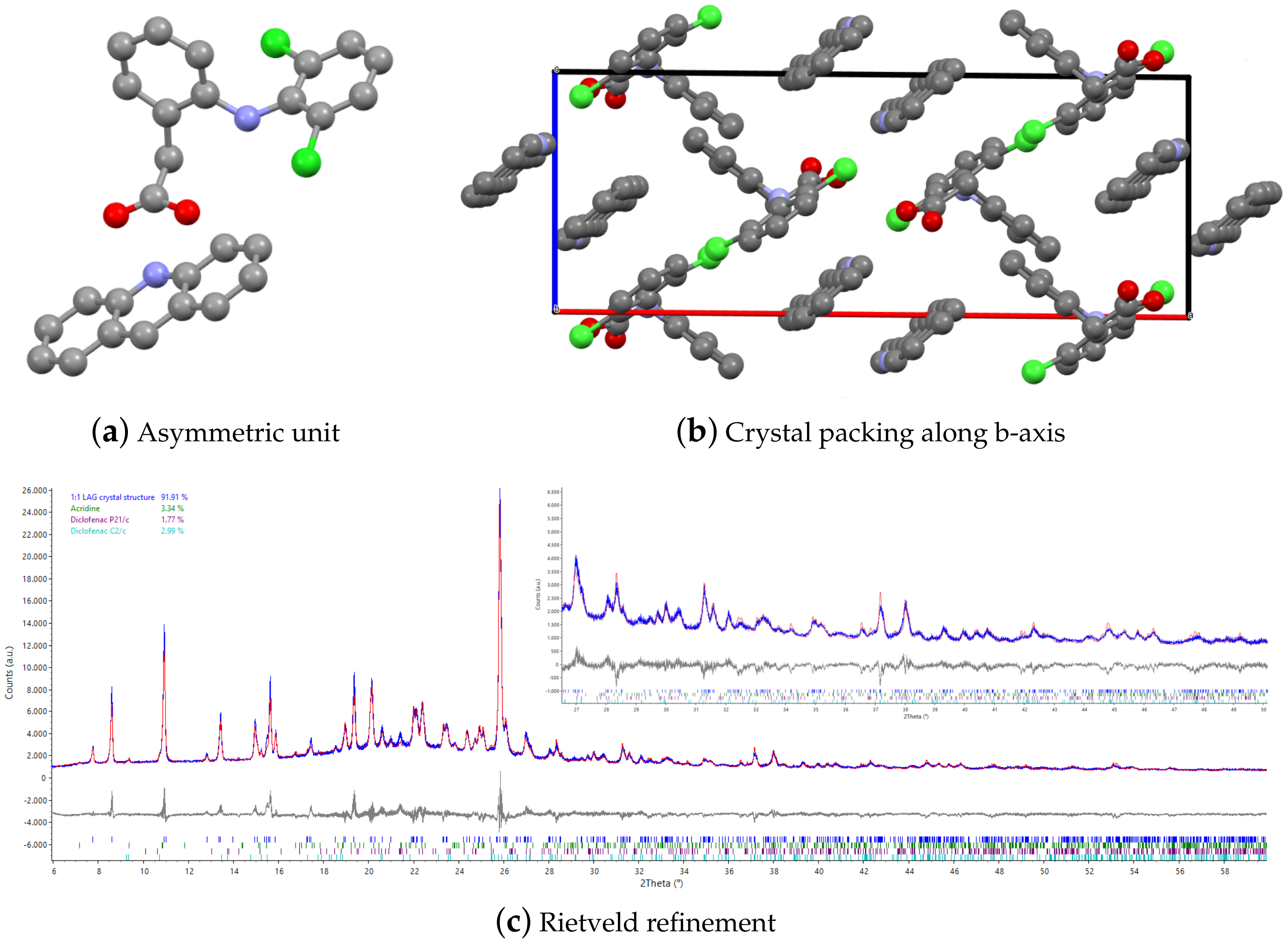
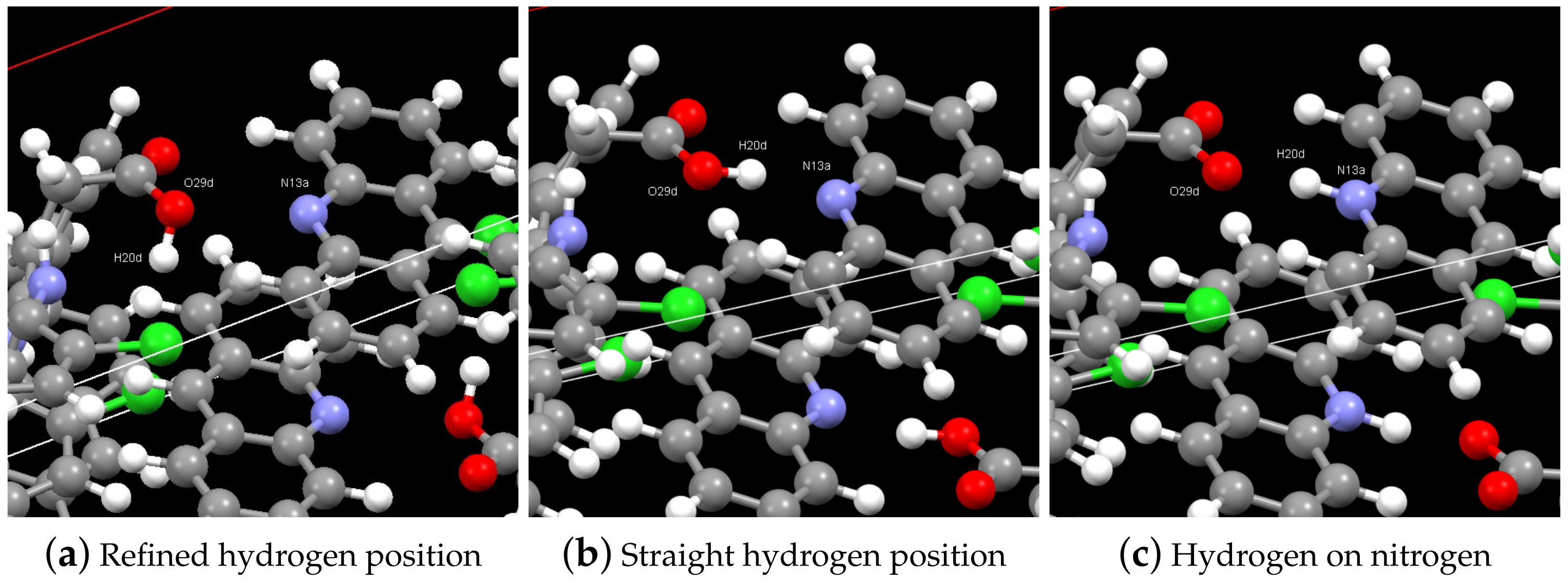
3.1. Structure Solution by X-ray Powder Diffraction and DSC Analysis
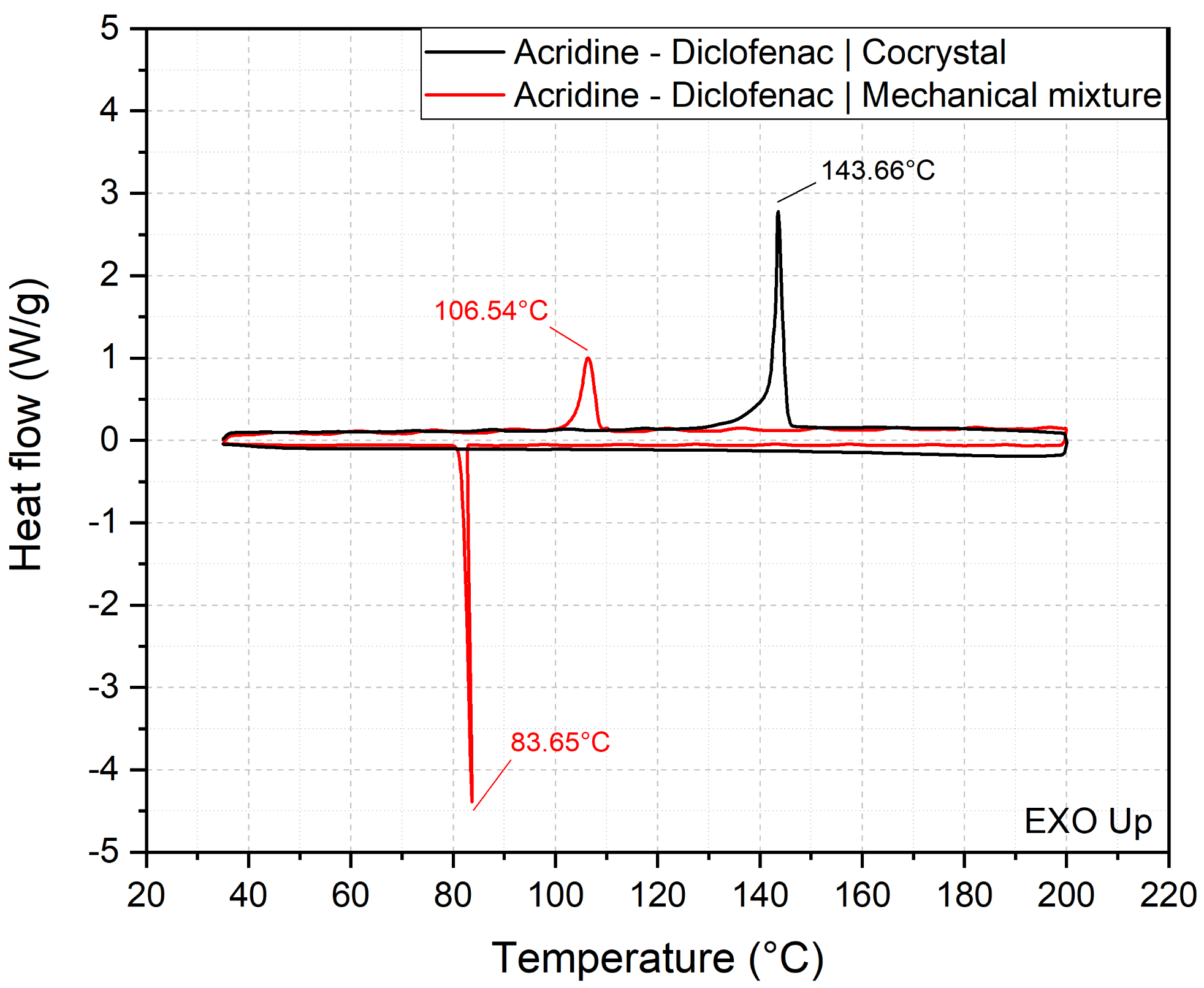
3.2. Thermal Analysis
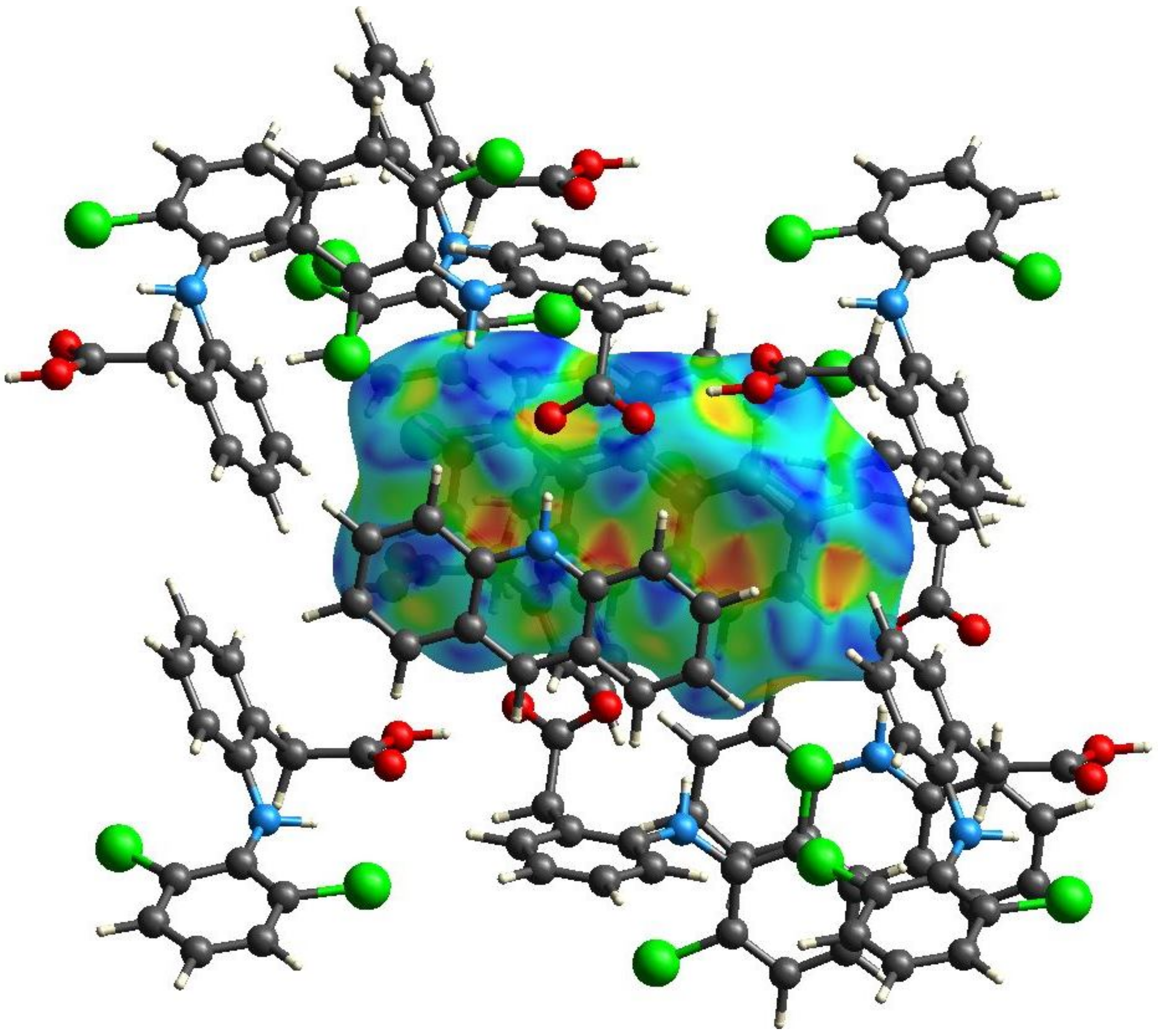
3.3. Hirshfeld Surface Calculations
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mirocki, A.; Lopresti, M.; Palin, L.; Conterosito, E.; Sikorski, A.; Milanesio, M. Exploring the molecular landscape of multicomponent crystals formed by naproxen drug and acridines. CrystEngComm 2022, 24, 6839–6853. [Google Scholar] [CrossRef]
- Zaworotko, M. Crystal engineering of the composition of API’s: Understanding polymorphs and designing pharmaceutical co-crystals. Am. Pharm. Outsourcing 2004, 5, 16–23. [Google Scholar]
- Barbas, R.; Bofill, L.; de Sande, D.; Font-Bardia, M.; Prohens, R. Crystal engineering of nutraceutical phytosterols: New cocrystal solid solutions. CrystEngComm 2020, 22, 4210–4214. [Google Scholar] [CrossRef]
- Qiao, N.; Li, M.; Schlindwein, W.; Malek, N.; Davies, A.; Trappitt, G. Pharmaceutical cocrystals: An overview. Int. J. Pharm. 2011, 419, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G. Crystal Engineering: A Holistic View. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef]
- Bhowal, R.; Biswas, S.; Thumbarathil, A.; Koner, A.L.; Chopra, D. Exploring the Relationship between Intermolecular Interactions and Solid-State Photophysical Properties of Organic Co-Crystals. J. Phys. Chem. C 2018, 123, 9311–9322. [Google Scholar] [CrossRef]
- Walsh, R.D.B.; Bradner, M.W.; Fleischman, S.; Morales, L.A.; Moulton, B.; Rodríguez-Hornedo, N.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Chem. Commun. 2002, 2, 186–187. [Google Scholar] [CrossRef]
- Singh, G.; Fort, J.G.; Goldstein, J.L.; Levy, R.A.; Hanrahan, P.S.; Bello, A.E.; Andrade-Ortega, L.; Wallemark, C.; Agrawal, N.M.; Eisen, G.M.; et al. Celecoxib Versus Naproxen and Diclofenac in Osteoarthritis Patients: SUCCESS-I Study. Am. J. Med. 2006, 119, 255–266. [Google Scholar] [CrossRef]
- Brogden, R.; Heel, R.; Pakes, G.; Speight, T.; Avery, G. Diclofenac Sodium. Drugs 1980, 20, 24–48. [Google Scholar] [CrossRef]
- Goh, C.F.; Lane, M.E. Formulation of diclofenac for dermal delivery. Int. J. Pharm. 2014, 473, 607–616. [Google Scholar] [CrossRef]
- Mazumdar, K.; Dastidar, S.G.; Park, J.H.; Dutta, N.K. The anti-inflammatory non-antibiotic helper compound diclofenac: An antibacterial drug target. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 881–891. [Google Scholar] [CrossRef]
- Maitra, A.; Bates, S.; Shaik, M.; Evangelopoulos, D.; Abubakar, I.; McHugh, T.D.; Lipman, M.; Bhakta, S. Repurposing drugs for treatment of tuberculosis: A role for non-steroidal anti-inflammatory drugs. Br. Med. Bull. 2016, 118, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Kroesen, V.M.; Gröschel, M.I.; Martinson, N.; Zumla, A.; Maeurer, M.; van der Werf, T.S.; Vilaplana, C. Non-Steroidal Anti-inflammatory Drugs As Host-Directed Therapy for Tuberculosis: A Systematic Review. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Todd, P.A.; Sorkin, E.M. Diclofenac Sodium. Drugs 1988, 35, 244–285. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Grommet, A.B.; Desper, J. Co-Crystal Screening of Diclofenac. Pharmaceutics 2011, 3, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Bodart, L.; Prinzo, M.; Derlet, A.; Tumanov, N.; Wouters, J. Taking advantage of solvate formation to modulate drug–drug ratio in clofaziminium diclofenac salts. CrystEngComm 2021, 23, 185–201. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Mondal, A.; Soni, S.R.; Das, S.; Bhunia, S.; Raju, K.B.; Ghosh, A.; Reddy, C.M. Multidrug salt forms of norfloxacin with non-steroidal anti-inflammatory drugs: Solubility and membrane permeability studies. CrystEngComm 2018, 20, 6420–6429. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific features of supramolecular organisation and hydrogen bonding in proline cocrystals: A case study of fenamates and diclofenac. CrystEngComm 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Nugrahani, I.; Kumalasari, R.A.; Auli, W.N.; Horikawa, A.; Uekusa, H. Salt Cocrystal of Diclofenac Sodium-L-Proline: Structural, Pseudopolymorphism, and Pharmaceutics Performance Study. Pharmaceutics 2020, 12, 690. [Google Scholar] [CrossRef]
- Wang, R.; Yuan, P.; Yang, D.; Zhang, B.; Zhang, L.; Lu, Y.; Du, G. Structural features and interactions of new sulfamethazine salt and cocrystal. J. Mol. Struct. 2021, 1229, 129596. [Google Scholar] [CrossRef]
- Mirocki, A.; Sikorski, A. Structural Characterization of Multicomponent Crystals Formed from Diclofenac and Acridines. Materials 2022, 15, 1518. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef] [Green Version]
- Braga, D.; Casali, L.; Grepioni, F. The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools? Int. J. Mol. Sci. 2022, 23, 9013. [Google Scholar] [CrossRef] [PubMed]
- Acebedo-Martínez, F.J.; Alarcón-Payer, C.; Barrales-Ruiz, H.M.; Niclós-Gutiérrez, J.; Domínguez-Martín, A.; Choquesillo-Lazarte, D. Towards the Development of Novel Diclofenac Multicomponent Pharmaceutical Solids. Crystals 2022, 12, 1038. [Google Scholar] [CrossRef]
- Saikia, B.; Pathak, D.; Sarma, B. Variable stoichiometry cocrystals: Occurrence and significance. CrystEngComm 2021, 23, 4583–4606. [Google Scholar] [CrossRef]
- Losev, E.A.; Boldyreva, E.V. A salt or a co-crystal – when crystallization protocol matters. CrystEngComm 2018, 20, 2299–2305. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Coelho, A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 2018, 51. [Google Scholar] [CrossRef] [Green Version]
- Coelho, A. TOPAS-Academic V7; Coelho Software: Brisbane, Australia, 2020. [Google Scholar]
- Lopresti, M.; Mangolini, B.; Milanesio, M.; Caliandro, R.; Palin, L. Multivariate versus traditional quantitative phase analysis of X-ray powder diffraction and fluorescence data of mixtures showing preferred orientation and microabsorption. J. Appl. Crystallogr. 2022, 55, 837–850. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In Computational Science—ICCS 2003, Proceedings of the International Conference, Melbourne, Australia and St. Petersburg, Russia, 2–4 June 2003; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2003; pp. 142–151. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.P.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14, 1614–1623. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Majerz, I.; Gutmann, M.J. Intermolecular OHN hydrogen bond with a proton moving in 3-methylpyridinium 2, 6-dichloro-4-nitrophenolate. RSC Adv. 2015, 5, 95576–95584. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 90th ed.; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirocki, A.; Conterosito, E.; Palin, L.; Sikorski, A.; Milanesio, M.; Lopresti, M. Crystal Structure of a New 1:1 Acridine-Diclofenac Salt, Obtained with High Yield by a Mechanochemical Approach. Crystals 2022, 12, 1573. https://doi.org/10.3390/cryst12111573
Mirocki A, Conterosito E, Palin L, Sikorski A, Milanesio M, Lopresti M. Crystal Structure of a New 1:1 Acridine-Diclofenac Salt, Obtained with High Yield by a Mechanochemical Approach. Crystals. 2022; 12(11):1573. https://doi.org/10.3390/cryst12111573
Chicago/Turabian StyleMirocki, Artur, Eleonora Conterosito, Luca Palin, Artur Sikorski, Marco Milanesio, and Mattia Lopresti. 2022. "Crystal Structure of a New 1:1 Acridine-Diclofenac Salt, Obtained with High Yield by a Mechanochemical Approach" Crystals 12, no. 11: 1573. https://doi.org/10.3390/cryst12111573
APA StyleMirocki, A., Conterosito, E., Palin, L., Sikorski, A., Milanesio, M., & Lopresti, M. (2022). Crystal Structure of a New 1:1 Acridine-Diclofenac Salt, Obtained with High Yield by a Mechanochemical Approach. Crystals, 12(11), 1573. https://doi.org/10.3390/cryst12111573