1. Introduction
Machine searchable databases that contain structural properties of related materials, calculated electronic structure, and measured electromagnetic properties, are providing a new way to design advanced functional materials. In addition, consolidating structural and functional information may lead to a better understanding of the microscopic mechanisms of correlated electron materials. Herein, we detail the launch of a new database of crystalline organic materials, many of which are conducting or superconducting, with the goal of motivating data-centered research to enhance the understanding of lower dimensional correlated electron materials. The database can be accessed through a website at
osd.clarku.edu.
The crystalline organic materials (COM) are well suited to create this type of database first of all because they are interesting experimentally. Partially driven by their low dimensionality, this class of materials exhibits a variety of competing electronic behaviors [
1,
2,
3,
4,
5] including metallic conductivity, Mott insulators [
6,
7,
8], antiferromagnetic states [
9,
10,
11], and superconductivity [
12,
13]. Other forms of long range charge order have also been observed, such as charge density waves (CDWs) [
14,
15,
16] and spin density waves (SDWs) [
17,
18]. More exotic long range order, such as the quantum hall effect [
18] and some of the first believable evidence for field induced inhomogeneous superconductivity (the FFLO state) [
19,
20,
21,
22,
23] were also found in COM. There has also been discussion about the existence of tilted Dirac points [
24] and spin liquids [
25,
26,
27] in these organic salts.
In addition to their rich correlated electron behavior, COMs are easy to access theoretically because they form regular stoichiometric crystals based on a few common cation molecules held together with various anion complexes. It is the electron deficient cation layer that contains holes, which enables itinerant electron behavior within the layer. It is evident that the geometric arrangement of the cation molecules is a principal factor that determines the electronic ground state of the system. This so-called packing of the cation molecules can be altered either by placing different molecules into the anion layer, or by applying external pressure. By adjusting the physical parameters of the cation layer, competing correlated electron states are selectively enabled and a complex electronic phase diagram can be traversed [
28,
29,
30].
There has been much theoretical progress towards understanding how the various degrees of freedom of these materials lead to the bulk electronic states that are observed [
31,
32,
33]. From a theoretical perspective, the materials offer a unique window into the physics of correlated electrons, because (i) COMs span the full range of states from interaction-induced insulators to superconductors and metals, (ii) the electronic structures are relatively simple, with only one orbital per molecule typically being relevant, (iii) nearly all the materials are stoichiometric and exhibit a high degree of crystalline order, so that simple models may closely approximate experiments, and (iv) COMs are built from common molecular entities, so that variations in properties may be directly related to structural variations across vast numbers of compounds.
The theoretical study of these materials via the tight binding model, or density functional theory (DFT), is leading to a better understanding of the fundamental physics behind correlated electron systems and of quantum materials in general. Various packing sub-families are known, in which particular structural degrees of freedom, and their relationship with the underlying hopping integrals, are key inputs to predict the behavior of the system [
34,
35,
36,
37]. Highlights of the strong exchange between experiment and theory include, e.g., quantitative agreement between results of high level calculations (dynamical mean field theory) and measurements of Mott critical scaling [
7] and correlation-driven crossovers in the optical response [
38].
This new database was built to be used both as a way to find representative materials for targeted experiments investigating particular correlated electron states, or the proximity of competing states, and as a research tool for discovering structure–function relationships [
39,
40]. An aspirational use of the database would be to predict and design new materials with targeted electronic properties. To serve these functions, the database contains experimentally measured properties, crystal structures, and calculated electronic structures of quasi one and two-dimensional crystalline organic conductors and superconductors. We hope that the enhanced accessibility of information that our database provides will serve the scientific community and lead to new discoveries.
2. Website and Database
When arriving at the home page of the website, the user is presented with a list of available materials and a window in which one may specify a desired type of packing, cation molecule, and anion molecule in order to search for any matches in the database. Leaving a field blank will act as a wildcard. The user can then click on a material to navigate to the home page for that material, which shows its crystal parameters followed by an interactive set of graphics starting with rotatable views of the crystal structure, the calculated electronic structure, and the cation morphology. These views are followed by available measurements of the material properties. Individual pages for each measurement enable the user to view associated information in greater detail. When observing crystal structures, they can be viewed and filtered to include all of the atoms, only the cations, only the anions, or rectangles to represent the cations. Electronic structure diagrams can also be customized by the user to show different k-paths or results from various types of calculations.
In order to identify relevant information to include in the database, the websites of journals were automatically searched for key phrases such as “BEDT-TTF”, “organic conductor”, and “organic superconductor”. The papers resulting from those searches were then recorded as potentially containing measurement information relevant to the database. Each paper of interest was parsed to determine its relevance, and encode any reported measurements into the database. Candidates for crystal structures were obtained from the Cambridge Structural Database (CSD) by performing a substructure search on the set of known cations. An algorithm was then used to analyze each crystal structure and determine if it was a lower dimensional charge transfer salt of interest. Relevant crystal structures were then added to the database, and their electronic structures were computed automatically. For a smaller selection of materials DFT (WIEN2k) was also used to calculate the electronic structure to compare to the tight binding results. The organization is depicted in
Figure 1. The database currently contains 110 materials, 184 crystal structures obtained from the CSD and 440 measured properties. A link is provided for each material to the corresponding CIF entry on the CSD website for the crystal structure information, and a link is provided to the paper where each measurement was found. Below we describe in more detail how the electronic calculations were made, and the methods for encoding the measurements into the database.
3. Crystal Structure Analysis Algorithm
To automatically identify relevant crystal structures from the Cambridge Structural Database, we created an algorithm to assess relevance, and to perform some preliminary diagnostics. The algorithm reads a CIF file, which contains a list of the atomic coordinates within the unit cell, and the lattice information. The distance between each pair of atoms is calculated, and if that distance is below the bonding threshold distance for the given atomic species, it is assumed that a bond exists between them. In this manner, the molecules within the unit cell are identified (see
Figure 2a,b). The structure of each molecule is then compared to a list of predefined structures of cation molecules such as BEDT-TTF. If the structures match, it is then known that the molecule is a cation molecule of interest. At this point it can be determined whether the material is quasi two-dimensional or not, and if so, what crystal axis is perpendicular to the layers. This is achieved by examining the overlap between cation and non-cation molecules for each Cartesian axis. If along a certain axis there are only overlapping cation molecules with no non-cation molecules, we make the assumption that this axis is in the plane of a conducting layer.
With the orientations and identities of the cations and anions automatically identified, this algorithm can automatically suggest the chemical formula and packing type of a given crystal structure. This is useful in grouping multiple structures for the same compound; however, we did not use this detected chemical formula as the compound label in the database. The name of a crystal structure is entered as denoted in the original paper to ensure that any special chemical naming conventions used by the authors are preserved in the database.
In order to display the packing geometry of the cation molecules to the user, the best fit plane of each cation is computed, and a minimum area rectangle algorithm is applied to generalize a cation molecule as a rectangle in three dimensional space. This type of generalization is common in cartoon diagrams of these materials. The resulting unit cell of cation-rectangles, shown in
Figure 2c–f, is useful to inspect the packing geometry of the material. Given that the overlap of the cations is the major determinant of the electronic structure of the crystalline organics, seeing the cations as blocks to visually show the morphology of the crystal symmetry of the cations is instructive. The relative distances and angles of the rectangles can then be calculated to quantitatively analyze the packing. We also generated two-dimensional diagrams, which more simply showed the geometric orientation of the cation molecules within the conducting layers based on the angles calculated before.
4. High-Throughput Electronic Structure Calculations
High-throughput electronic structure calculations using density functional theory (DFT) have found applications in several materials databases; however, the large unit cells of these materials and low crystal symmetries make full-scale DFT calculations with plane-wave basis sets computationally expensive. This is especially problematic for cases with open-shell anions, which feature localized unpaired electrons. Unless the local correlations in the anion layer are treated explicitly (via DFT + U), anion bands may appear erroneously near the Fermi energy, yielding incorrect Fermi surfaces. More importantly however, many of our crystal structures have missing or disordered atoms, especially in the anion layer. This is particularly prevalent in anion layers that contain solvents, which are often disordered across unit cells. It is therefore necessary to reduce the computational expense and focus exclusively on the cation layers.
To calculate the electronic structure of every crystal structure in our database, we construct a two-dimensional tight binding model [
42] for highest occupied molecular orbitals in the layer of cation molecules using a series of local DFT calculations. Solving the tight-binding (TB) model produces the band structure. To carry out the calculation, we used the crystal structure analysis algorithm previously described to identify all symmetrically equivalent molecules and pairs of molecules with the cation layers. We then used the method employed in [
43] to estimate tight-binding hopping integrals using quantum chemistry packages (in this case ORCA [
44]). Results are shown in
Figure 3, for the example of
-(ET)
KHg(SCN)
. The method is based on calculations on pairs of molecules in which the local crystal environment is otherwise ignored, which significantly reduces computational expense. For this purpose, we used basis sets including 3-21G, 6-31G, 6-311G, and def2-SVP in conjunction with the B3LYP hybrid density functional. Localized Wannier molecular orbitals (MOs) are constructed for each molecule via maximizing the overlap with the corresponding orbital of the isolated molecules. The procedure is as follows:
Obtain Isolated MOs: For each molecular pair (labeled
), a calculation is first performed on the isolated molecules. From this, the MO coefficients (in the basis of Gaussian atomic orbitals) for each molecule are obtained as
and
. These are combined as:
Construct Wannier Functions: For each molecular pair, a calculation is then performed in the geometry corresponding to the crystal structure. This produces the diagonal MO energies
, the overlap matrix
, and the MO coefficients
. In ORCA,
is output in the atomic orbital basis. It is first rotated into the basis of the isolated MOs:
In this geometry, the basis of isolated MOs are no longer orthonormal. Thus, the local Wannier functions are constructed via symmetric orthornormalization, .
Rotate Fock Matrix: The diagonal orbital energies are then rotated into the above-defined localized MOs:
The resulting Fock matrix has the structure:
The on-site terms
and
now contain both the diagonal Wannier orbital energies, and small off-diagonal “crystal field” contributions. It is advantageous to remove the latter terms via unitary transformation:
where
and
are diagonal. The intersite hoppings can then be read from
.
We note, because this latter unitary transformation is different for every molecular pair, the hopping integrals obtained for different pairs represent slightly different definitions of the local Wannier functions. Nonetheless, this approximation is no more severe than the pairwise construction inherent to the method. Although this approach neglects the anion layer, the results agree well with full-scale calculations performed with Wien2k (at the GGA level) and experimental electronic structure as well (see
Figure 3c); however, the former approach is much faster. With Wien2k, for example, a full DFT calculation using GGA functionals can take several days to complete (with 100 processors), while construction of a TB model with ORCA calculations takes approximately five minutes per compound (with 10 processors), even when using more expensive hybrid functionals. Such a speed-up is desirable when making high-throughput calculations for each crystal structure entry in the database.
Because the pairwise calculations are made separately, we had to adjust the signs of the resulting charge transfer integrals such that the phase of the molecular orbital on each symmetrically equivalent molecule was the same. We used the centroids of the cation molecules for the positions of the sites, disregarding the out of layer component. The filling of each model was deduced from the stoichiometry and charge of the cation molecules. Solving the tight binding Hamiltonian at each point in k-space produced the energy eigenvalues that constitute the electronic structure. In this manner, the band structure, Fermi surface, and density of states are automatically computed and may be viewed on the website. Users can interact with these data directly by selecting the k-path to use and which basis sets to display. In addition, the computed hopping integrals, and their locations in the unit cell are presented to the user in order to serve as a basis for further theoretical modeling.
5. Measurements
Locations of phase transitions:
- –
Metal insulator.
- –
Superconductivity, , (where is the Pauli paramagnetic limit).
- –
Charge density wave.
- –
Spin density wave.
- –
Magnetic ordering.
As a function of:
- –
Temperature.
- –
Magnetic field.
- –
Pressure.
Lattice parameters.
Conductivity.
London penetration depth.
Coherence length.
Shubnikov—de Haas and de Haas—van Alphen frequencies.
Effective mass.
Dingle temperature.
Scattering time.
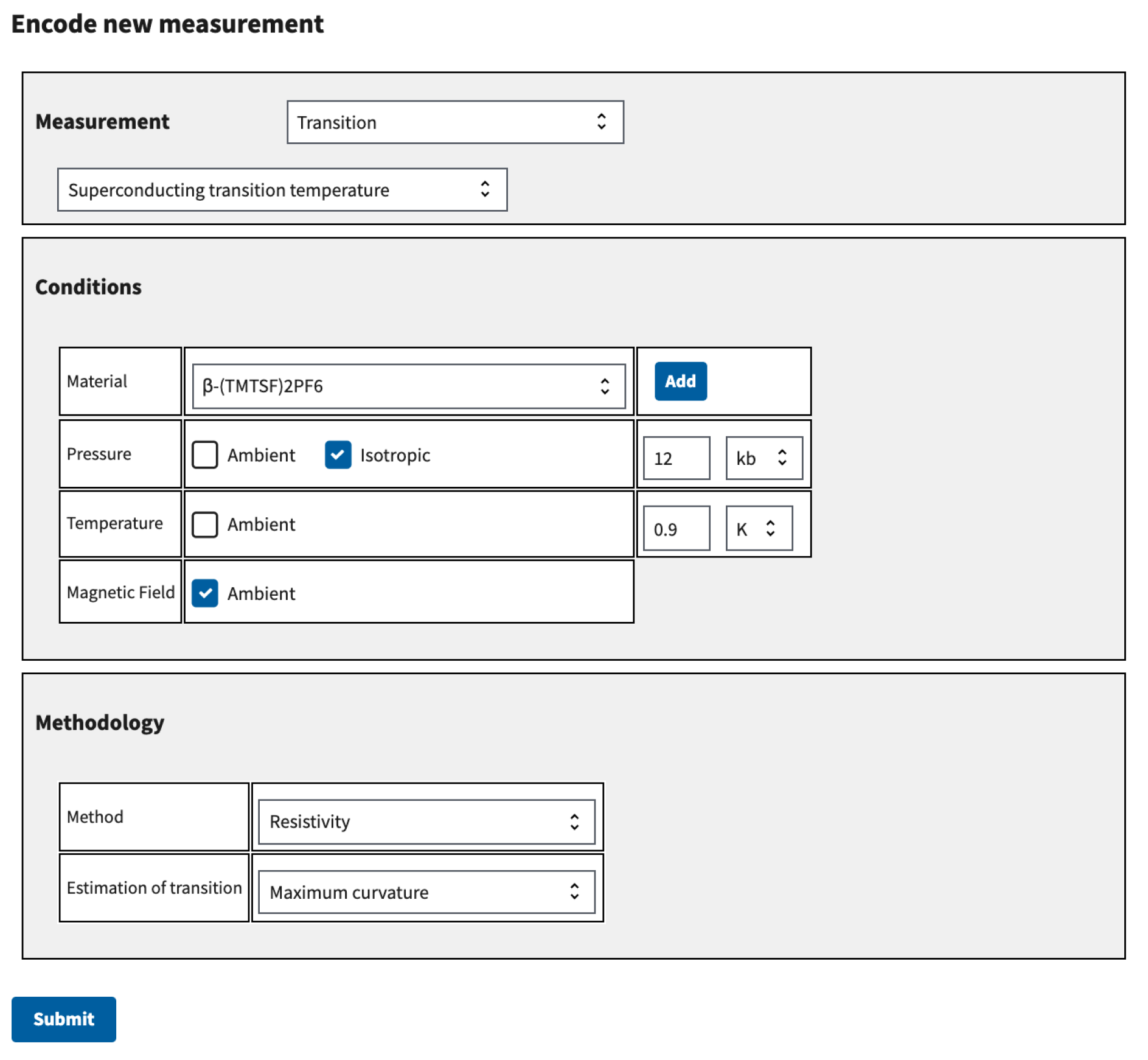
Each measurement entry in the database consists of three blocks of information: a block specifying the state of the system being measured, a block specifying the value of the measurement, including the error bars if available, and a block specifying the method by which the measurement was made. Our goal behind the data entry is to create a digital copy of measurement information in the precise manner in which it was specified by the authors who made the measurement. We implemented support for as many measurements as we could for this process. For example, the ability to specify a numeric value using an exact decimal, a range between two decimals, or an average decimal with a plus or minus value. Although laborious, we found that manual data entry was the most reliable way to extract measurement information from papers. We used a web application on the website for this purpose, shown in
Figure 4.
Once we had a sufficient number of measurements, there were many decisions that needed to be made about how they were presented. For a deep understanding of a single material, it is necessary to see a detailed view of the actual measurements labeled by the method of measurement; for example, the superconducting transition temperature, , found by resistance or specific heat, and the point on the transition curve, e.g., onset or midpoint, used to locate the transition, with citations for each measurement. For that reason, the details of the measurement method and the error bars, if given, are stored in the database. For that reason, it is also necessary to present an average value for a material when a number of materials are being compared to each other. We made the decision to discount some of the grossly outlying measurements in cases where we thought the data were not convincing. These rules for curating the data are constantly being reconsidered to present the most useful data to the community; however, the full collection of measurements will always be available so that a user of the database can analyze the published measurements with their own algorithms.
6. Discussion and Future Outlook
We present this database of crystalline organic conductors and superconductors as an evolving tool that will be continuously updated with new materials, features, and metrics. The goal of gathering all relevant calculation and measurement information into one central location is an arduous one, but the progress that we have made so far illustrates that it is possible. In further developing this database, we have two main goals.
Our first main goal is to populate the database with as many entries as possible. The limiting step in this process for inclusion of experimental measurements is data entry. Manual data entry of measurements from scientific articles is the only method with a high enough degree of reliability to be useful in our database. We have used members of our laboratory to perform data entry, and have trained undergraduate students as well. Currently, our database includes only a small fraction of all the relevant data that exist. In order to increase the number of measurements in our database, we will need to increase the size of our data entry team. We are considering crowdsourcing the process so that verified database users from across the world can also contribute. In addition, verified users will have the option of submitting CIF files for automatic calculation of electronic structure and tight-binding parameters. We would also like to populate the database with results from explicitly correlated theoretical methods suitable for high-throughput applications (such as density matrix embedding theory [
45]). Presently, a tight-binding electronic structure is provided for each crystal structure in the database. For some of these crystal structures, missing or corrupt atoms in the anion layer prevent the use of a full LAPW DFT calculation; however, we eventually plan to include full LAPW DFT calculations for as many of the crystal structures in the database as possible. Any persons interested in becoming involved with the project can click on the orange button at the bottom of the home page to request an account. An option is also available to provide anonymous feedback.
Our second main goal regarding development of the database is to implement new features. We plan to add an interactive web interface by which users can analyze the contents of the database as a whole by correlating various calculated and measured properties. We also would like to improve the search feature of the website so that more detailed searches can be performed. There are many different avenues by which our existing work can be further developed. We are trying to create as many tools as possible to perform simple visualization and analysis of data online, such as the feature shown in
Figure 5. A number of parameters can be extracted from the band structure calculations, such as the density of states at the Fermi level, and the cross sectional area of the Fermi surface. We are working on finding robust universal algorithms to calculate these and other representative values. Given this collection of the unit cell parameters, extracted electronic values, and measurement parameters, any set of data can be graphed against any other set of data, and scatter plots can be created including markers labeled with the material names. It is also possible to combine parameters with common arithmetic operations to create additional metrics. We will continue to enhance the user interface to create a more versatile and expansive analysis interface.
Following the invention and widespread availability of computers, an increasing trend towards digitization in science has taken place. Scientific databases have emerged in practically every area of study because they enable the analysis of many pieces of information, and the distribution of that information to individuals around the world. The field of data science has also grown to develop new ways of analyzing the large amount of data available. Many databases for materials science currently exist, particularly those that focus on electronic structure, crystal structure, and other measured properties [
46]. Our inspiration to create this database of organic conductors and superconductors was drawn in part by the success of other databases containing density functional theory electronic structure calculations for many crystal structures [
47,
48,
49,
50]. Our goal is to gather as many different types and pieces of information related to quasi two-dimensional organic conductors and superconductors as possible. We foresee this database as having a number of different applications. Primarily, it will serve as a useful reference tool for the scientific community of organic conductors and superconductors. Database users can easily find and view crystal structures, electronic structures, and other measured properties for the materials that they are interested in. We hope to cultivate a community of scientists from across the world who are interested in using the database, contributing data to the database, and requesting new features for the database. We also look forward to analyzing the data that are stored in the database. Many techniques in the field of data science are appropriate for this purpose. In particular, certain types of data mining and machine learning have proven useful in the analysis of other materials databases [
51,
52,
53,
54]. We hope that the identification of trends between various parameters will ultimately lead to a better understanding of the fundamental mechanisms of correlated electron systems in quasi two-dimensional organic conductors and superconductors.
Author Contributions
Conceptualization, O.G. and C.C.A.; Methodology, O.G., K.F., M.B.-d. and S.M.W.; Software, O.G.; Validation, K.F. and M.B.-d.; Investigation, O.G.; Resources, C.C.A. and S.M.W.; Data Curation, K.F., M.B.-d. and O.G.; Writing—Original Draft Preparation, O.G., C.C.A. and S.M.W.; Supervision, C.C.A.; Project Administration, C.C.A.; Funding Acquisition, C.C.A. and S.M.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NSF grant number DMR-1905950. S.M.W. and O.G. also acknowledge support from a pilot grant from the Center for Functional Materials, Wake Forest University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in the described database, accessible via
osd.clarku.edu.
Acknowledgments
The authors acknowledge discussions with Ben Powell. The authors thank the following people for their work in manually adding measurement entries to the database: Alireza Alipour, Abdulai Gassama, Ahad Ali Khan, Brett Laramee, Gwynnevieve Ramsey, Jade Consalvi, Meherab Hossain, and Raju Ghimire. Computations were performed on the Clark University High Performance Computing Cluster and the Wake Forest University DEAC Cluster, a centrally managed resource with support provided in part by Wake Forest University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jérome, D. Organic conductors: From charge density wave TTF- TCNQ to superconducting (TMTSF)2PF6. Chem. Rev. 2004, 104, 5565–5592. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Cui, H.; Kobayashi, A. Organic metals and superconductors based on BETS (BETS = bis (ethylenedithio) tetraselenafulvalene). Chem. Rev. 2004, 104, 5265–5288. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, K.; Kanoda, K.; Kawamoto, A. NMR studies on two-dimensional molecular conductors and superconductors: Mott transition in κ-(BEDT-TTF) 2X. Chem. Rev. 2004, 104, 5635–5654. [Google Scholar] [CrossRef] [PubMed]
- Geiser, U.; Schlueter, J.A. Conducting organic radical cation salts with organic and organometallic anions. Chem. Rev. 2004, 104, 5203–5242. [Google Scholar] [CrossRef]
- Kanoda, K.; Kato, R. Mott physics in organic conductors with triangular lattices. Annu. Rev. Condens. Matter Phys. 2011, 2, 167–188. [Google Scholar] [CrossRef]
- Powell, B.; McKenzie, R.H. Strong electronic correlations in superconducting organic charge transfer salts. J. Phys. Condens. Matter 2006, 18, R827. [Google Scholar] [CrossRef]
- Furukawa, T.; Miyagawa, K.; Taniguchi, H.; Kato, R.; Kanoda, K. Quantum criticality of Mott transition in organic materials. Nat. Phys. 2015, 11, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Gati, E.; Garst, M.; Manna, R.S.; Tutsch, U.; Wolf, B.; Bartosch, L.; Schubert, H.; Sasaki, T.; Schlueter, J.A.; Lang, M. Breakdown of Hooke’s law of elasticity at the Mott critical endpoint in an organic conductor. Sci. Adv. 2016, 2, e1601646. [Google Scholar] [CrossRef] [Green Version]
- Enoki, T.; Miyazaki, A. Magnetic TTF-based charge-transfer complexes. Chem. Rev. 2004, 104, 5449–5478. [Google Scholar] [CrossRef]
- Naka, M.; Ishihara, S. Magnetoelectric effect in organic molecular solids. Sci. Rep. 2016, 6, 20781. [Google Scholar] [CrossRef]
- Itaya, M.; Eto, Y.; Kawamoto, A.; Taniguchi, H. Antiferromagnetic Fluctuations in the Organic Superconductor κ-(BEDT-TTF)2Cu(NCS)2 under Pressure. Phys. Rev. Lett. 2009, 102, 227003. [Google Scholar] [CrossRef] [Green Version]
- Jérome, D.; Mazaud, A.; Ribault, M.; Bechgaard, K. Superconductivity in a synthetic organic conductor (TMTSF)2PF6. J. Phys. Lett. 1980, 41, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Wosnitza, J. Superconductivity of Organic Charge-Transfer Salts. J. Low Temp. Phys. 2019, 197, 250–271. [Google Scholar] [CrossRef]
- Latt, K.Z.; Schlueter, J.A.; Darancet, P.; Hla, S.W. Two-Dimensional Molecular Charge Density Waves in Single-Layer-Thick Islands of a Dirac Fermion System. ACS Nano 2020, 14, 8887–8893. [Google Scholar] [CrossRef] [PubMed]
- Andres, D.; Kartsovnik, M.V.; Biberacher, W.; Neumaier, K.; Sheikin, I.; Müller, H.; Kushch, N.D. Field-induced charge-density-wave transitions in the organic metal α-(BEDT-TTF)2KHg(SCN)4 under pressure. Low Temp. Phys. 2011, 37, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Kondo, R.; Higa, M.; Kagoshima, S.; Hanasaki, N.; Nogami, Y.; Nishikawa, H. Interplay of charge-density waves and superconductivity in the organic conductor β-(BEDT-TTF)2AuBr2. Phys. Rev. B 2010, 81, 024519. [Google Scholar] [CrossRef]
- Sasaki, T.; Lebed, A.; Fukase, T.; Toyota, N. Magnetic field response of the spin density wave in α-(BEDT-TTF)2KHg(SCN)4. Synth. Met. 1997, 86, 2063–2064. [Google Scholar] [CrossRef]
- Kang, W.; Hannahs, S.T.; Chaikin, P.M. Toward a unified phase diagram in (TMTSF)2X. Phys. Rev. Lett. 1993, 70, 3091–3094. [Google Scholar] [CrossRef]
- Tanatar, M.A.; Ishiguro, T.; Tanaka, H.; Kobayashi, H. Magnetic field–temperature phase diagram of the quasi-two-dimensional organic superconductor λ-(BETS)2GaCl4 studied via thermal conductivity. Phys. Rev. B 2002, 66, 134503. [Google Scholar] [CrossRef] [Green Version]
- Shimahara, H. Theory of the Fulde–Ferrell–Larkin–Ovchinnikov State and Application to Quasi-Low-dimensional Organic Superconductors. In The Physics of Organic Superconductors and Conductors; Lebed, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 687–704. [Google Scholar] [CrossRef]
- Cho, K.; Smith, B.E.; Coniglio, W.A.; Winter, L.E.; Agosta, C.C.; Schlueter, J.A. Upper critical field in the organic superconductor β”-(ET)2SF5CH2CF2SO3: Possibility of Fulde-Ferrell-Larkin-Ovchinnikov state. Phys. Rev. B 2009, 79, 220507. [Google Scholar] [CrossRef] [Green Version]
- Agosta, C. Inhomogeneous Superconductivity in Organic and Related Superconductors. Crystals 2018, 8, 285. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, S.; Isono, T.; Terashima, T.; Yasuzuka, S.; Schlueter, J.A.; Uji, S. Fulde-Ferrell-Larkin-Ovchinnikov and vortex phases in a layered organic superconductor. NPJ Quantum Mater. 2019, 4, 1–6. [Google Scholar] [CrossRef]
- Hirata, M.; Ishikawa, K.; Miyagawa, K.; Tamura, M.; Berthier, C.; Basko, D.; Kobayashi, A.; Matsuno, G.; Kanoda, K. Observation of an anisotropic Dirac cone reshaping and ferrimagnetic spin polarization in an organic conductor. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Miyagawa, K.; Kanoda, K.; Maesato, M.; Saito, G. Spin liquid state in an organic Mott insulator with a triangular lattice. Phys. Rev. Lett. 2003, 91, 107001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isono, T.; Kamo, H.; Ueda, A.; Takahashi, K.; Kimata, M.; Tajima, H.; Tsuchiya, S.; Terashima, T.; Uji, S.; Mori, H. Gapless Quantum Spin Liquid in an Organic Spin-1/2 Triangular-Lattice κ-H3(Cat-EDT-TTF)2. Phys. Rev. Lett. 2014, 112, 177201. [Google Scholar] [CrossRef] [PubMed]
- Miksch, B.; Pustogow, A.; Rahim, M.J.; Bardin, A.A.; Kanoda, K.; Schlueter, J.A.; Hübner, R.; Scheffler, M.; Dressel, M. Gapped magnetic ground state in quantum spin liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Science 2021, 372, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Rösslhuber, R.; Löhle, A.; Sanz Alonso, M.; Wenzel, M.; Kawamoto, A.; Pustogow, A.; Dressel, M. Chemical tuning of molecular quantum materials κ-[(BEDT-TTF)1-x(BEDT-STF)x]2Cu2(CN)3: From the Mott-insulating quantum spin liquid to metallic Fermi liquid. J. Mater. Chem. C 2021, 9, 10841–10850. [Google Scholar] [CrossRef]
- Faltermeier, D.; Barz, J.; Dumm, M.; Dressel, M.; Drichko, N.; Petrov, B.; Semkin, V.; Vlasova, R.; Meźière, C.; Batail, P. Bandwidth-controlled Mott transition in κ-(BEDT-TTF)2Cu[N(CN)2]BrxCl1-x: Optical studies of localized charge excitations. Phys. Rev. B 2007, 76, 165113. [Google Scholar] [CrossRef] [Green Version]
- Kanoda, K. Mott Transition and Superconductivity in Q2D Organic Conductors. In The Physics of Organic Superconductors and Conductors; Lebed, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 623–642. [Google Scholar] [CrossRef]
- Seo, H.; Hotta, C.; Fukuyama, H. Toward systematic understanding of diversity of electronic properties in low-dimensional molecular solids. Chem. Rev. 2004, 104, 5005–5036. [Google Scholar] [CrossRef]
- Lebed, A.G. The Physics of Organic Superconductors and Conductors; Springer: Berlin/Heidelberg, Germany, 2008; Volume 110. [Google Scholar] [CrossRef]
- Powell, B.; McKenzie, R.H. Quantum frustration in organic Mott insulators: From spin liquids to unconventional superconductors. Rep. Prog. Phys. 2011, 74, 056501. [Google Scholar] [CrossRef]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors I. Parallel Molecules: β and β" Phases. Bull. Chem. Soc. Jpn. 1998, 71, 2509–2526. [Google Scholar] [CrossRef]
- Mori, T.; Mori, H.; Tanaka, S. Structural Genealogy of BEDT-TTF-Based Organic Conductors II. Inclined Molecules: θ, α, and κ Phases. Bull. Chem. Soc. Jpn. 1999, 72, 179–197. [Google Scholar] [CrossRef]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors III. Twisted Molecules: δ and α′ Phases. Bull. Chem. Soc. Jpn. 1999, 72, 2011–2027. [Google Scholar] [CrossRef]
- Hotta, C. Classification of Quasi-Two Dimensional Organic Conductors Based on a New Minimal Model. J. Phys. Soc. Jpn. 2003, 72, 840–853. [Google Scholar] [CrossRef]
- Ferber, J.; Foyevtsova, K.; Jeschke, H.O.; Valentí, R. Unveiling the microscopic nature of correlated organic conductors: The case of κ-(ET)2Cu[N(CN)2]BrxCl1-x. Phys. Rev. B 2014, 89, 205106. [Google Scholar] [CrossRef]
- Mori, H.; Tanaka, S.; Oshima, M.; Saito, G.; Mori, T.; Maruyama, Y.; Inokuchi, H. Crystal and Electronic Structures of (BEDT–TTF)2[MHg(SCN)4](M=K and NH4). Bull. Chem. Soc. Jpn. 1990, 63, 2183–2190. [Google Scholar] [CrossRef] [Green Version]
- Caulfield, J.; Lubczynski, W.; Pratt, F.L.; Singleton, J.; Ko, D.Y.K.; Hayes, W.; Kurmoo, M.; Day, P. Magnetotransport studies of the organic superconductor κ-(BEDT-TTF)2Cu(NCS)2 under pressure: The relationship between carrier effective mass and critical temperature. J. Phys. Condens. Matter 1994, 6, 2911. [Google Scholar] [CrossRef]
- Rashid, S.; Turner, S.S.; Day, P.; Light, M.E.; Hursthouse, M.B.; Firth, S.; Clark, R.J.H. The first molecular charge transfer salt containing proton channels. Chem. Commun. 2001, 1462–1463. [Google Scholar] [CrossRef]
- Mori, T.; Kobayashi, A.; Sasaki, Y.; Kobayashi, H.; Saito, G.; Inokuchi, H. The Intermolecular Interaction of Tetrathiafulvalene and Bis(ethylenedithio)tetrathiafulvalene in Organic Metals. Calculation of Orbital Overlaps and Models of Energy-band Structures. Bull. Chem. Soc. Jpn. 1984, 57, 627–633. [Google Scholar] [CrossRef]
- Winter, S.M.; Riedl, K.; Valenti, R. Importance of spin-orbit coupling in layered organic salts. Phys. Rev. B 2017, 95, 060404. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Knizia, G.; Chan, G.K.L. Density matrix embedding: A simple alternative to dynamical mean-field theory. Phys. Rev. Lett. 2012, 109, 186404. [Google Scholar] [CrossRef] [Green Version]
- Lin, L. Materials Databases Infrastructure Constructed by First Principles Calculations: A Review. Mater. Perform. Charact. 2015, 4. [Google Scholar] [CrossRef]
- Borysov, S.S.; Geilhufe, R.M.; Balatsky, A.V. Organic materials database: An open-access online database for data mining. PLoS ONE 2017, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Topological Materials Database. Available online: https://www.topologicalquantumchemistry.org/ (accessed on 23 April 2022).
- Rutgers DFT & DMFT Materials Database. Available online: http://hauleweb.rutgers.edu/database_w2k/ (accessed on 23 April 2022).
- Stanev, V.; Oses, C.; Kusne, A.G.; Rodriguez, E.; Paglione, J.; Curtarolo, S.; Takeuchi, I. Machine learning modeling of superconducting critical temperature. NPJ Comput. Mater. 2018, 4, 29. [Google Scholar] [CrossRef]
- Xie, S.R.; Stewart, G.R.; Hamlin, J.J.; Hirschfeld, P.J.; Hennig, R.G. Functional form of the superconducting critical temperature from machine learning. Phys. Rev. B 2019, 100, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Geilhufe, R.M.; Borysov, S.S.; Kalpakchi, D.; Balatsky, A.V. Towards novel organic high-Tc superconductors: Data mining using density of states similarity search. Phys. Rev. Mater. 2018, 2, 024802. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ling, C. A strategy to apply machine learning to small datasets in materials science. NPJ Comput. Mater. 2018, 4, 25. [Google Scholar] [CrossRef] [Green Version]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




