

Charge Transport in Organic Semiconducting Crystals Exhibiting TADF: Insight from Quantum Chemical Calculations

Abstract
:1. Introduction
2. Methods
2.1. Crystal Structure Analysis
2.2. Charge Mobility
3. Results and Discussion
3.1. Molecular Properties
3.2. Crystal Properties
3.3. Charge Transport
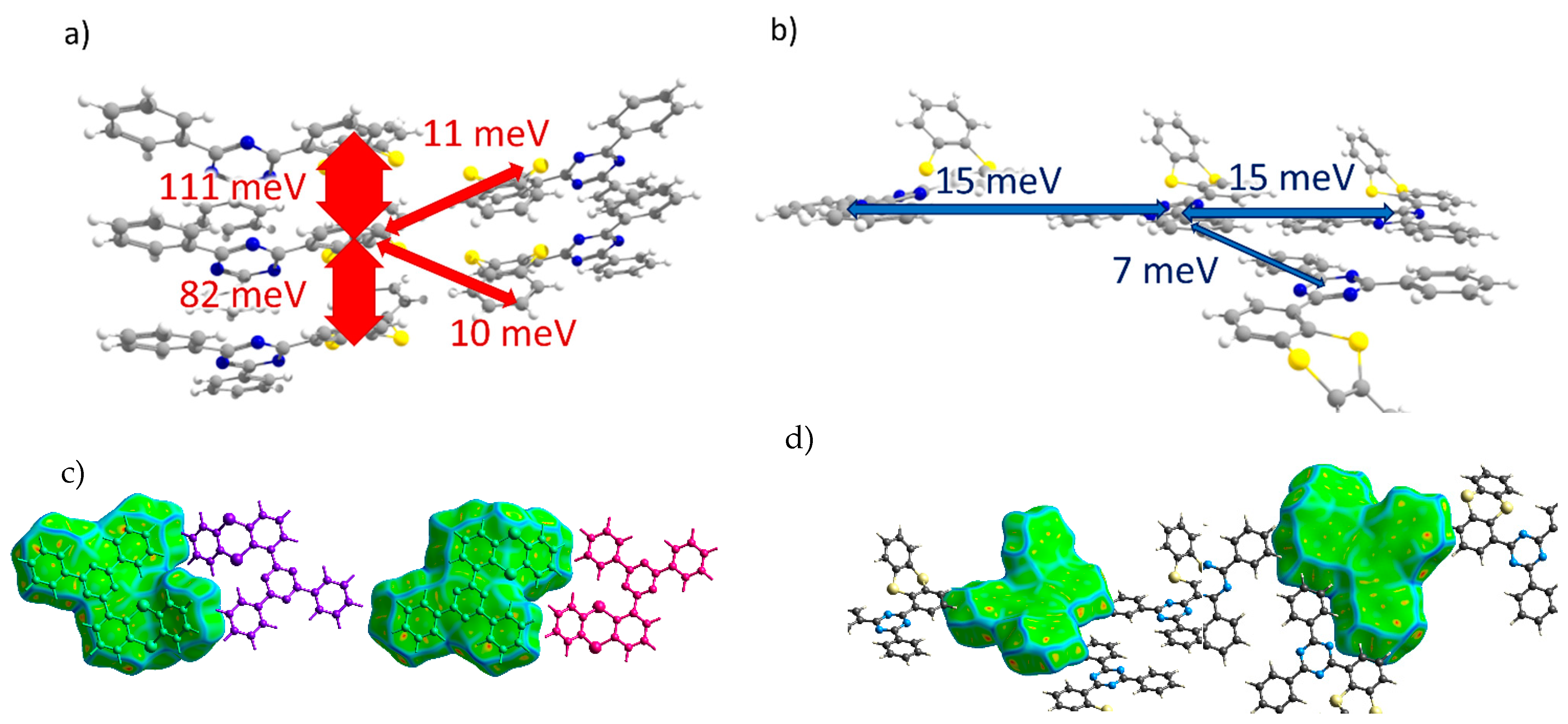
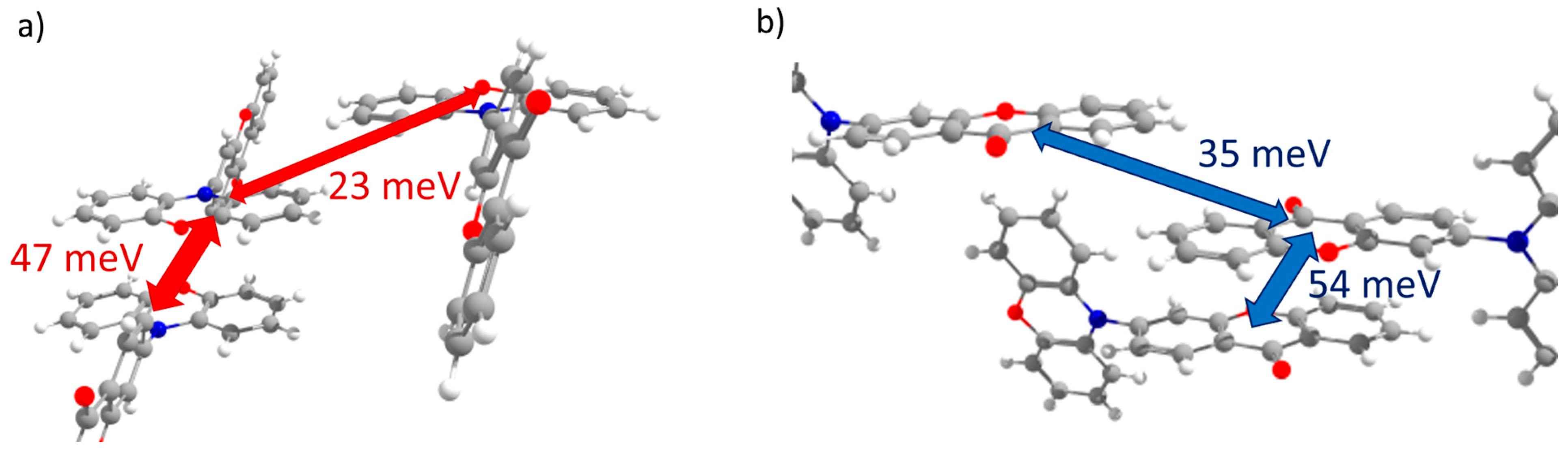
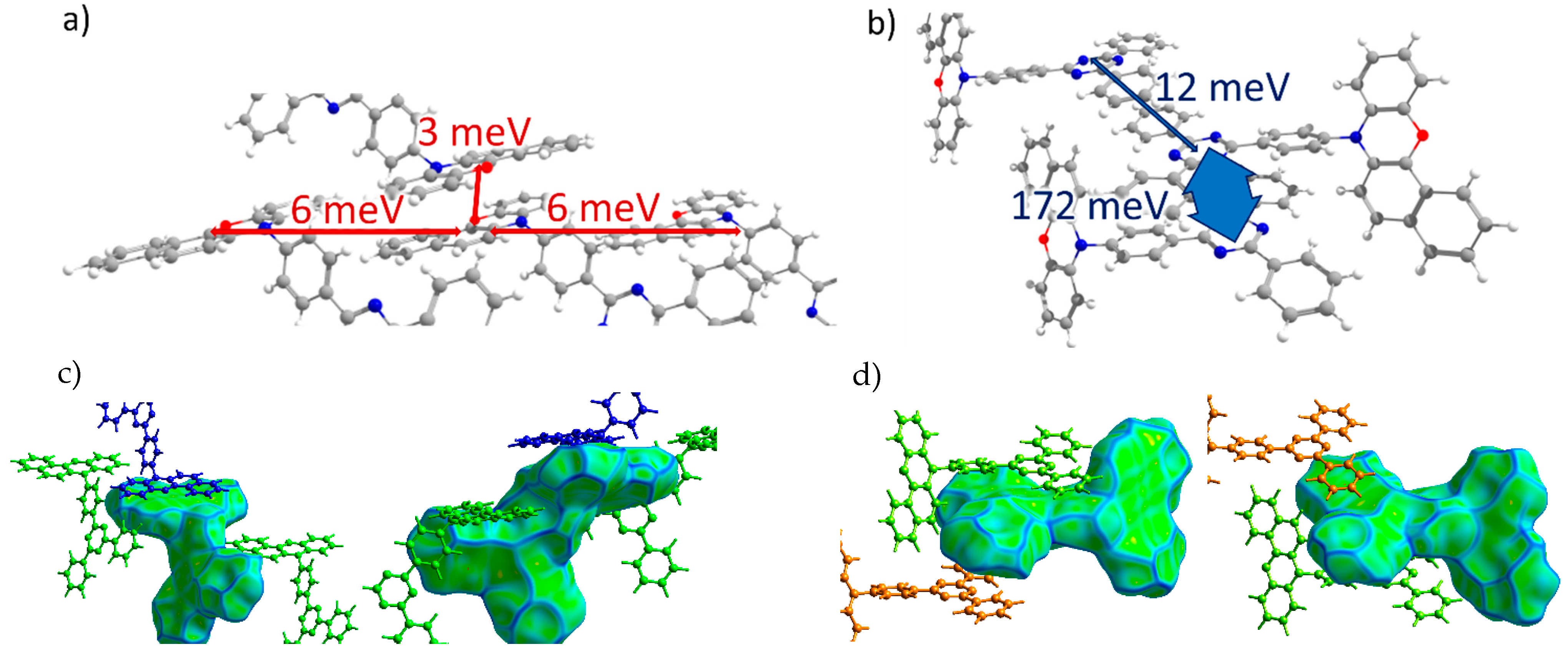
3.3.1. Transfer Integrals
3.3.2. Reorganization Energies
3.3.3. Charge Mobilities
3.4. Outlook
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Huang, Y.; Hsiang, E.-L.; Deng, M.-Y.; Wu, S.-T. Mini-LED, Micro-LED and OLED displays: Present status and future perspectives. Light Sci. Appl. 2020, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Fu, X.; Shin, D.-H.; So, F. Recent Advances in OLED Optical Design. Adv. Funct. Mater. 2019, 29, 1808803. [Google Scholar] [CrossRef]
- Lee, S.M.; Kwon, J.H.; Kwon, S.; Choi, K.C. A Review of Flexible OLEDs Toward Highly Durable Unusual Displays. IEEE Trans. Electron. Devices 2017, 64, 1922–1931. [Google Scholar] [CrossRef]
- Hsiang, E.-L.; Yang, Z.; Yang, Q.; Lan, Y.-F.; Wu, S.-T. Prospects and challenges of mini-LED, OLED, and micro-LED displays. J. Soc. Inf. Disp. 2021, 29, 446–465. [Google Scholar] [CrossRef]
- Cai, X.; Su, S.-J. Marching Toward Highly Efficient, Pure-Blue, and Stable Thermally Activated Delayed Fluorescent Organic Light-Emitting Diodes. Adv. Funct. Mater. 2018, 28, 1802558. [Google Scholar] [CrossRef]
- Cai, X.; Qiao, Z.; Li, M.; Wu, X.; He, Y.; Jiang, X.; Cao, Y.; Su, S.-J. Purely Organic Crystals Exhibit Bright Thermally Activated Delayed Fluorescence. Angew. Chem. Int. Ed. 2019, 58, 13522–13531. [Google Scholar] [CrossRef]
- Gužauskas, M.; Narbutaitis, E.; Volyniuk, D.; Baryshnikov, G.V.; Minaev, B.F.; Ågren, H.; Chao, Y.-C.; Chang, C.-C.; Rutkis, M.; Grazulevicius, J.V. Polymorph acceptor-based triads with photoinduced TADF for UV sensing. Chem. Eng. J. 2021, 425, 131549. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, H.; Wang, S.; Li, Z.; Ye, K.; Zhang, J.; Liu, Y.; Peng, Q.; Wang, Y. Supramolecular Structure-Dependent Thermally-Activated Delayed Fluorescence (TADF) Properties of Organic Polymorphs. J. Phys. Chem. C 2016, 120, 19759–19767. [Google Scholar] [CrossRef]
- Yang, W.; Yang, Y.; Cao, X.; Liu, Y.; Chen, Z.; Huang, Z.; Gong, S.; Yang, C. On-off switchable thermally activated delayed fluorescence controlled by multiple channels: Understanding the mechanism behind distinctive polymorph-dependent optical properties. Chem. Eng. J. 2021, 415, 128909. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Li, C.; Wang, Y. Suppressing Efficiency Roll-Off of TADF Based OLEDs by Constructing Emitting Layer with Dual Delayed Fluorescence. Front. Chem. 2019, 7, 302. [Google Scholar] [CrossRef]
- Han, J.; Huang, Z.; Miao, J.; Qiu, Y.; Xie, Z.; Yang, C. Narrowband blue emission with insensitivity to the doping concentration from an oxygen-bridged triarylboron-based TADF emitter: Nondoped OLEDs with a high external quantum efficiency up to 21.4%. Chem. Sci. 2022, 13, 3402–3408. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, D.; Wang, P.; Huang, X.; Chen, H.; Zhang, Y.; Zhang, D.; Jiang, W.; Sun, Y.; Duan, L. Exceeding 30 % External Quantum Efficiency in Non-doped OLEDs Utilizing Solution Processable TADF Emitters with High Horizontal Dipole Orientation via Anchoring Strategy. Angew. Chem. Int. Ed. 2022, 61, e202212861. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-Z.; Wu, H.; Wang, K.; Yu, J.; Ou, X.-M.; Zhang, X.-H. Recent progress in thermally activated delayed fluorescence emitters for nondoped organic light-emitting diodes. Chem. Sci. 2022, 13, 3625–3651. [Google Scholar] [CrossRef] [PubMed]
- Danyliv, Y.; Ivaniuk, K.; Danyliv, I.; Bezvikonnyi, O.; Volyniuk, D.; Galyna, S.; Lazauskas, A.; Skhirtladze, L.; Ågren, H.; Stakhira, P.; et al. Carbazole-σ-sulfobenzimide derivative exhibiting mechanochromic thermally activated delayed fluorescence as emitter for flexible OLEDs: Theoretical and experimental insights. Dyes Pigm. 2022, 208, 110841. [Google Scholar] [CrossRef]
- Ran, Y.; Yang, G.; Liu, Y.; Han, W.; Gao, G.; Su, R.; Bin, Z.; You, J. A methyl-shield strategy enables efficient blue thermally activated delayed fluorescence hosts for high-performance fluorescent OLEDs. Mater. Horiz. 2021, 8, 2025–2031. [Google Scholar] [CrossRef]
- Bucinskas, A.; Bezvikonnyi, O.; Gudeika, D.; Volyniuk, D.; Grazulevicius, J.V. Methoxycarbazolyl-disubstituted dibenzofuranes as holes- and electrons-transporting hosts for phosphorescent and TADF-based OLEDs. Dyes Pigm. 2020, 172, 107781. [Google Scholar] [CrossRef]
- Bezvikonnyi, O.; Gudeika, D.; Volyniuk, D.; Rutkis, M.; Grazulevicius, J.V. Diphenylsulfone-based hosts for electroluminescent devices: Effect of donor substituents. Dyes Pigm. 2020, 175, 108104. [Google Scholar] [CrossRef]
- Liu, C.; Huang, K.; Park, W.-T.; Li, M.; Yang, T.; Liu, X.; Liang, L.; Minari, T.; Noh, Y.-Y. A unified understanding of charge transport in organic semiconductors: The importance of attenuated delocalization for the carriers. Mater. Horiz. 2017, 4, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Menard, E.; Podzorov, V.; Hur, S.-H.; Gaur, A.; Gershenson, M.E.; Rogers, J.A. High-Performance n- and p-Type Single-Crystal Organic Transistors with Free-Space Gate Dielectrics. Adv. Mater. 2004, 16, 2097–2101. [Google Scholar] [CrossRef]
- Qin, Z.; Gao, C.; Dong, H.; Hu, W. Organic Semiconductor Single-Crystal Light-Emitting Transistors. Adv. Opt. Mater. 2022, in press. [CrossRef]
- Fedorenko, R.S.; Kuevda, A.V.; Trukhanov, V.A.; Konstantinov, V.G.; Sosorev, A.Y.; Sonina, A.A.; Kazantsev, M.S.; Surin, N.M.; Grigorian, S.; Borshchev, O.V.; et al. Luminescent High-Mobility 2D Organic Semiconductor Single Crystals. Adv. Electron. Mater. 2022, 8, 2101281. [Google Scholar] [CrossRef]
- Ahmad, V.; Sobus, J.; Bencheikh, F.; Mamada, M.; Adachi, C.; Lo, S.-C.; Namdas, E.B. High EQE and High Brightness Solution-Processed TADF Light-Emitting Transistors and OLEDs. Adv. Opt. Mater. 2020, 8, 2000554. [Google Scholar] [CrossRef]
- Sobus, J.; Bencheikh, F.; Mamada, M.; Wawrzinek, R.; Ribierre, J.-C.; Adachi, C.; Lo, S.-C.; Namdas, E.B. High Performance p- and n-Type Light-Emitting Field-Effect Transistors Employing Thermally Activated Delayed Fluorescence. Adv. Funct. Mater. 2018, 28, 1800340. [Google Scholar] [CrossRef]
- Choi, H.H.; Cho, K.; Frisbie, C.D.; Sirringhaus, H.; Podzorov, V. Critical assessment of charge mobility extraction in FETs. Nat. Mater. 2018, 17, 2–7. [Google Scholar] [CrossRef]
- Yavuz, I.; Lopez, S.A.; Lin, J.B.; Houk, K.N. Quantitative prediction of morphology and electron transport in crystal and disordered organic semiconductors. J. Mater. Chem. C 2016, 4, 11238–11243. [Google Scholar] [CrossRef]
- Sokolov, A.N.; Atahan-Evrenk, S.; Mondal, R.; Akkerman, H.B.; Sánchez-Carrera, R.S.; Granados-Focil, S.; Schrier, J.; Mannsfeld, S.C.B.; Zoombelt, A.P.; Bao, Z.; et al. From computational discovery to experimental characterization of a high hole mobility organic crystal. Nat. Commun. 2011, 2, 437. [Google Scholar] [CrossRef] [Green Version]
- Schober, C.; Reuter, K.; Oberhofer, H. Virtual Screening for High Carrier Mobility in Organic Semiconductors. J. Phys. Chem. Lett. 2016, 7, 3973–3977. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta—Bioenerg. 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Baumeier, B.; Kirkpatrick, J.; Andrienko, D. Density-functional based determination of intermolecular charge transfer properties for large-scale morphologies. Phys. Chem. Chem. Phys. 2010, 12, 11103–11113. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, J. An approximate method for calculating transfer integrals based on the ZINDO Hamiltonian. Int. J. Quantum Chem. 2008, 108, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Kobayashi, N.; Hosoi, S.; Koshitani, N.; Murakami, D.; Shirasawa, R.; Kudo, Y.; Hobara, D.; Tokita, Y.; Itabashi, M. Hopping and band mobilities of pentacene, rubrene, and 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene (C8-BTBT) from first principle calculations. J. Chem. Phys. 2013, 139, 014707. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.-L. Charge Transport in Organic Semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Chapter 41—Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [Green Version]
- Sure, R.; Grimme, S. Corrected small basis set Hartree-Fock method for large systems. J. Comput. Chem. 2013, 34, 1672–1685. [Google Scholar] [CrossRef]
- Chemcraft. Graphical Software for Quantum Chemical Computations. Available online: https://www.chemcraftprog.com/ (accessed on 15 November 2022).
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 15 November 2022).
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef]
- Mas-Torrent, M.; Hadley, P.; Bromley, S.T.; Ribas, X.; Tarrés, J.; Mas, M.; Molins, E.; Veciana, J.; Rovira, C. Correlation between Crystal Structure and Mobility in Organic Field-Effect Transistors Based on Single Crystals of Tetrathiafulvalene Derivatives. J. Am. Chem. Soc. 2004, 126, 8546–8553. [Google Scholar] [CrossRef] [PubMed]
- Sosorev, A.Y. Simple charge transport model for efficient search of high-mobility organic semiconductor crystals. Mater. Des. 2020, 192, 108730. [Google Scholar] [CrossRef]
- Andruleviciene, V.; Leitonas, K.; Volyniuk, D.; Sini, G.; Grazulevicius, J.V.; Getautis, V. TADF versus TTA emission mechanisms in acridan and carbazole-substituted dibenzo[a,c]phenazines: Towards triplet harvesting emitters and hosts. Chem. Eng. J. 2021, 417, 127902. [Google Scholar] [CrossRef]
- Masimukku, N.; Gudeika, D.; Volyniuk, D.; Bezvikonnyi, O.; Simokaitiene, J.; Matulis, V.; Lyakhov, D.; Azovskyi, V.; Gražulevičius, J.V. Bipolar 1,8-naphthalimides showing high electron mobility and red AIE-active TADF for OLED applications. Phys. Chem. Chem. Phys. 2022, 24, 5070–5082. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, J.; Zhu, X.; Guo, J.; Zhao, Z.; Tang, B.Z. Versatile Aggregation-Enhanced Delayed Fluorescence Luminogens Functioning as Emitters and Hosts for High-Performance Organic Light-Emitting Diodes. CCS Chem. 2021, 3, 230–240. [Google Scholar] [CrossRef]
- Liu, H.; Liu, H.; Fan, J.; Guo, J.; Zeng, J.; Qiu, F.; Zhao, Z.; Tang, B.Z. An Effective Design Strategy for Robust Aggregation-Induced Delayed Fluorescence Luminogens to Improve Efficiency Stability of Nondoped and Doped OLEDs. Adv. Opt. Mater. 2020, 8, 2001027. [Google Scholar] [CrossRef]
- Watt, M.; Hardebeck, L.K.E.; Kirkpatrick, C.C.; Lewis, M. Face-to-Face Arene−Arene Binding Energies: Dominated by Dispersion but Predicted by Electrostatic and Dispersion/Polarizability Substituent Constants. J. Am. Chem. Soc. 2011, 133, 3854–3862. [Google Scholar] [CrossRef]
- Trukhanov, V.A.; Dominskiy, D.I.; Parashchuk, O.D.; Feldman, E.V.; Surin, N.M.; Svidchenko, E.A.; Skorotetcky, M.S.; Borshchev, O.V.; Paraschuk, D.Y.; Sosorev, A.Y. Impact of N-substitution on structural, electronic, optical, and vibrational properties of a thiophene–phenylene co-oligomer. RSC Adv. 2020, 10, 28128–28138. [Google Scholar] [CrossRef]
- Sosorev, A.; Dominskiy, D.; Chernyshov, I.; Efremov, R. Tuning of Molecular Electrostatic Potential Enables Efficient Charge Transport in Crystalline Azaacenes: A Computational Study. Int. J. Mol. Sci. 2020, 21, 5654. [Google Scholar] [CrossRef]
- Sosorev, A.Y.; Trukhanov, V.A.; Maslennikov, D.R.; Borshchev, O.V.; Polyakov, R.A.; Skorotetcky, M.S.; Surin, N.M.; Kazantsev, M.S.; Dominskiy, D.I.; Tafeenko, V.A.; et al. Fluorinated Thiophene-Phenylene Co-Oligomers for Optoelectronic Devices. ACS Appl. Mater. Interfaces 2020, 12, 9507–9519. [Google Scholar] [CrossRef]
- Qin, Z.; Gao, H.; Dong, H.; Hu, W. Organic Light-Emitting Transistors Entering a New Development Stage. Adv. Mater. 2021, 33, 2007149. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimer # | Jh, meV | Je, meV |
|---|---|---|
| oTE-DRZ (Crystal 1) | ||
| 1 | 111 | 0 |
| 2 | 82 | 0 |
| 3 | 0 | 15 |
| 4 | 0 | 15 |
| 5 | 6 | 1 |
| 6 | 3 | 7 |
| 7 | 11 | 1 |
| 8 | 10 | 0 |
| CPPD (Crystal 2) | ||
| 1 | 3 | 0 |
| 2 | 3 | 0 |
| 3 | 0 | 94 |
| 4 | 149 | 0 |
| 5 | 1 | 26 |
| 6 | 4 | 0 |
| PXZ-XO (Crystal 3) | ||
| 1 | 23 | 0 |
| 2 | 5 | 0 |
| 3 | 3 | 16 |
| 4 | 47 | 1 |
| 5 | 0 | 35 |
| 6 | 1 | 54 |
| TRZ-c-BPXZ (Crystal 4) | ||
| 1 | 6 | 2 |
| 2 | 6 | 2 |
| 3 | 0 | 12 |
| 4 | 0 | 172 |
| 5 | 3 | 1 |
| Compound | Hole Reorganization Energy | Electron Electron Energy |
|---|---|---|
| OTE-DRZ (Crystal 1) | 990 (540 a) | 440 (150 a) |
| CPPD (Crystal 2) | 220 | 660 |
| PXZ-XO (Crystal 3) | 227 | 255 |
| TRZ-c-BPXZ (Crystal 4) | 323 | 604 |
| Crystal | Hole Mobility, cm2/(V∙s) | Electron Mobility, cm2/(V∙s) |
|---|---|---|
| OTE-DRZ (Crystal 1) | 0.001 (0.08 a) | 0.009 (0.156 a) |
| CPPD (Crystal 2) | 0.03 | 0.002 |
| PXZ-XO (Crystal 3) | 0.39 | 0.34 |
| TRZ-c-BPXZ (Crystal 4) | 0.003 | 0.003 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sosorev, A.Y.; Dominskiy, D.I.; Dubinets, N.O. Charge Transport in Organic Semiconducting Crystals Exhibiting TADF: Insight from Quantum Chemical Calculations. Crystals 2023, 13, 55. https://doi.org/10.3390/cryst13010055
Sosorev AY, Dominskiy DI, Dubinets NO. Charge Transport in Organic Semiconducting Crystals Exhibiting TADF: Insight from Quantum Chemical Calculations. Crystals. 2023; 13(1):55. https://doi.org/10.3390/cryst13010055
Chicago/Turabian StyleSosorev, Andrey Y., Dmitry I. Dominskiy, and Nikita O. Dubinets. 2023. "Charge Transport in Organic Semiconducting Crystals Exhibiting TADF: Insight from Quantum Chemical Calculations" Crystals 13, no. 1: 55. https://doi.org/10.3390/cryst13010055
APA StyleSosorev, A. Y., Dominskiy, D. I., & Dubinets, N. O. (2023). Charge Transport in Organic Semiconducting Crystals Exhibiting TADF: Insight from Quantum Chemical Calculations. Crystals, 13(1), 55. https://doi.org/10.3390/cryst13010055