


Prediction of Carbon Dioxide and Methane Adsorption on UiO-66 Metal–Organic Framework via Molecular Simulation



Abstract
:1. Introduction
2. Methods
Molecular Models and Solid Lattice

{kind=link}
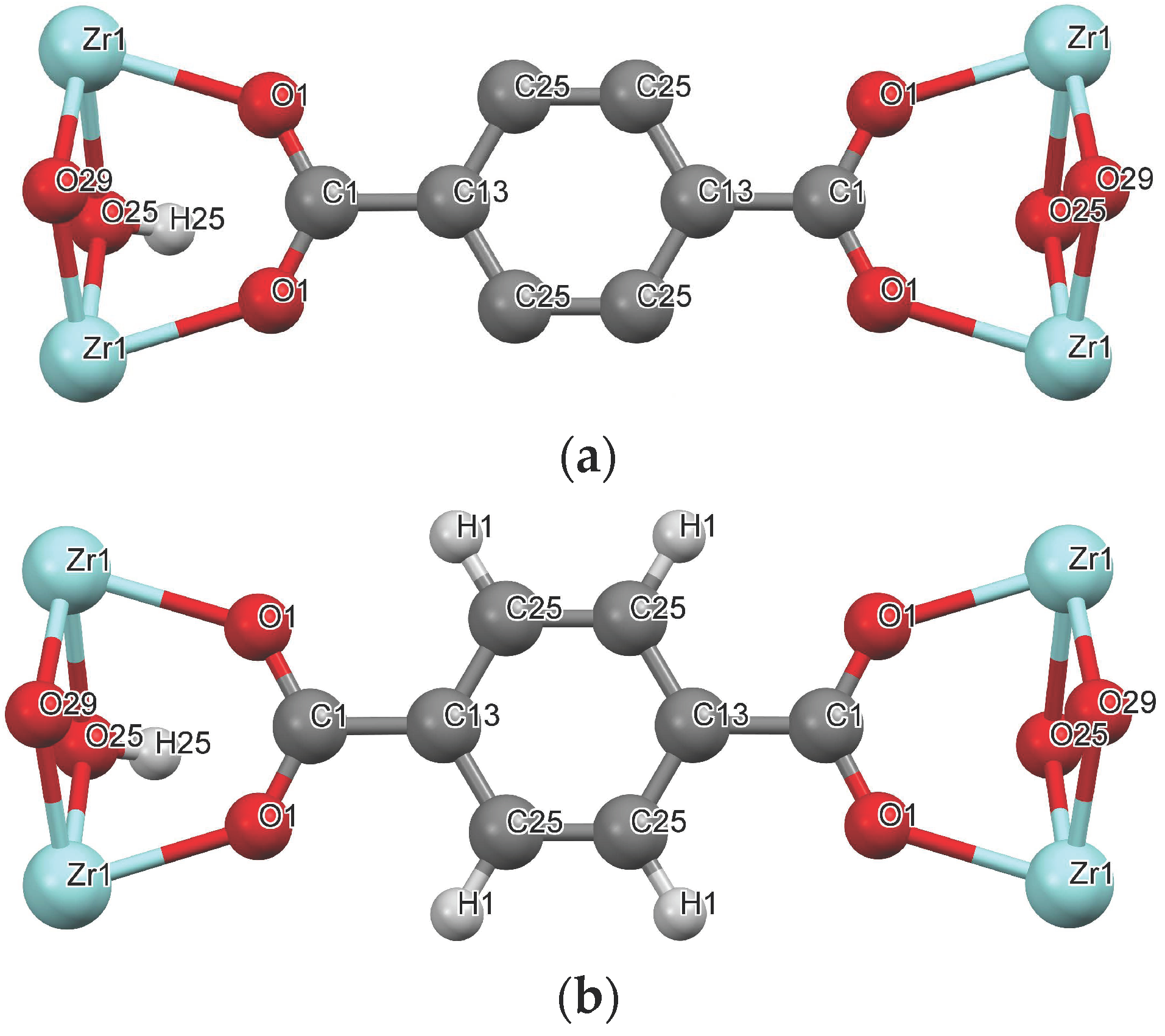{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | σ (Å) | ε/kB (K) | q (e) | Ref. |
|---|---|---|---|---|
| Methane | ||||
| CH4 | 3.73 | 148 | - | [42] |
| Carbon dioxide | ||||
| C_CO2 | 2.80 | 27.0 | +0.70 | [43] |
| O_CO2 | 3.05 | 79.0 | −0.35 | [43] |
3. Results and Discussion
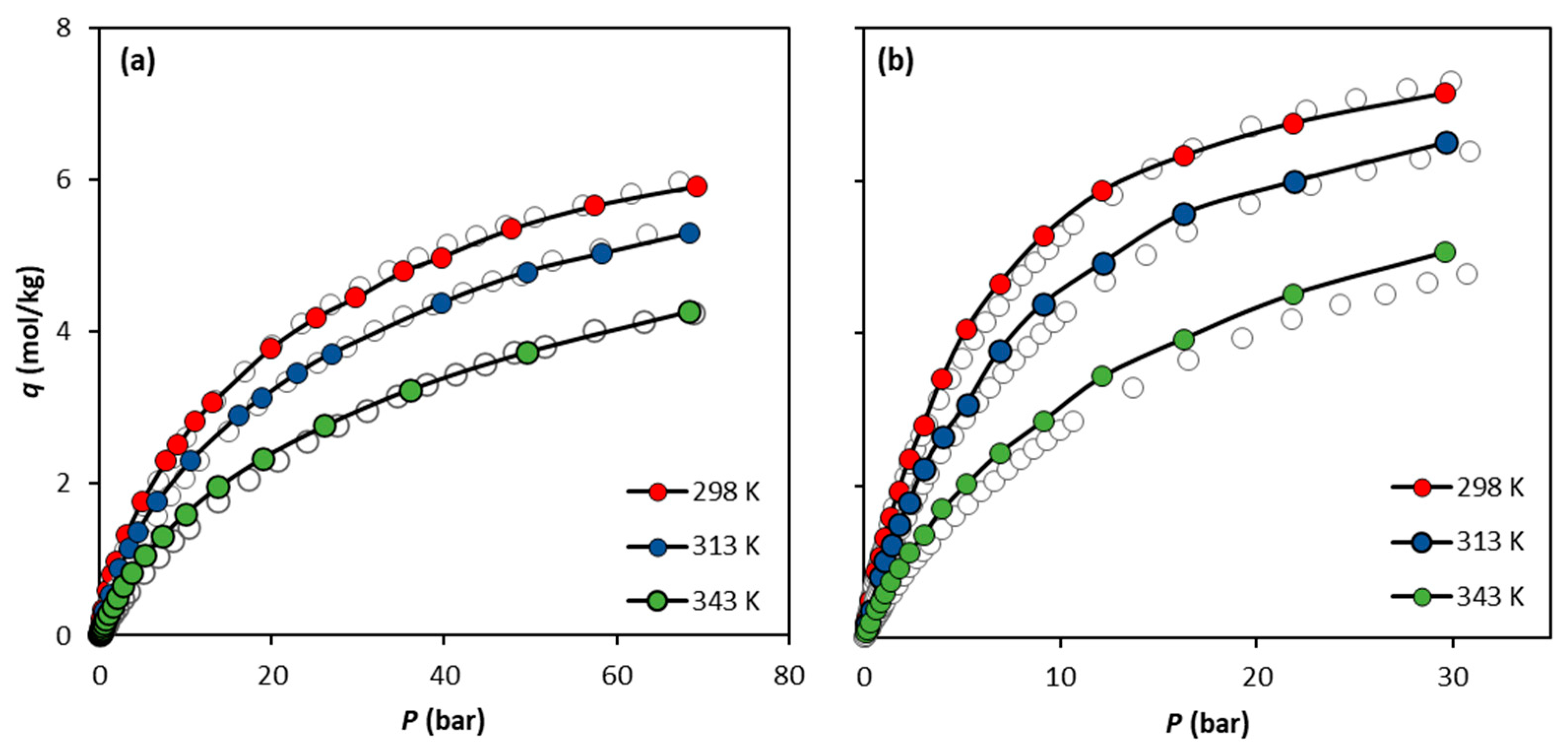
3.1. Single-Component Adsorption Equilibria
3.2. Adsorption Energetics
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Millward, A.R.; Yaghi, O.M. Metal-Organic Frameworks with Exceptionally High Capacity for Storage of Carbon Dioxide at Room Temperature. J. Am. Chem. Soc. 2005, 127, 17998–17999. [Google Scholar] [CrossRef] [PubMed]
- Rowsell, J.L.C.; Yaghi, O.M. Metal–Organic Frameworks: A New Class of Porous Materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Meek, S.T.; Greathouse, J.A.; Allendorf, M.D. Metal-Organic Frameworks: A Rapidly Growing Class of Versatile Nanoporous Materials. Adv. Mater. 2011, 23, 249–267. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.; Schubert, M.; Teich, F.; Puetter, H.; Schierle-Arndt, K.; Pastre, J. Metal-Organic Frameworks-Prospective Industrial Applications. J. Mater. Chem. 2006, 16, 626–636. [Google Scholar] [CrossRef]
- Schneemann, A.; Bon, V.; Schwedler, I.; Senkovska, I.; Kaskel, S.; Fischer, R.A. Flexible metal-organic frameworks. Chem. Soc. Rev. 2014, 43, 6062–6096. [Google Scholar] [CrossRef]
- Chen, X.; Li, M.; Lin, M.; Lu, C.; Kumar, A.; Pan, Y.; Liu, J.; Peng, Y. Current and promising applications of Hf(iv)-based MOFs in clinical cancer therapy. J. Mater. Chem. B 2023, 11, 5693–5714. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, Z.; Huang, S.; Ye, K.; Jiang, Y.; Liu, J.; Liu, J.; Lu, X.; Li, B. A metal-organic framework-based immunomodulatory nanoplatform for anti-atherosclerosis treatment. J. Control. Release 2023, 354, 615–625. [Google Scholar] [CrossRef]
- Ma, D.; Li, Z.; Zhu, J.; Zhou, Y.; Chen, L.; Mai, X.; Liufu, M.; Wu, Y.; Li, Y. Inverse and highly selective separation of CO2/C2H2 on a thulium–organic framework. J. Mater. Chem. A 2020, 8, 11933–11937. [Google Scholar] [CrossRef]
- Ahmadijokani, F.; Molavi, H.; Rezakazemi, M.; Tajahmadi, S.; Bahi, A.; Ko, F.; Aminabhavi, T.M.; Li, J.-R.; Arjmand, M. UiO-66 metal–organic frameworks in water treatment: A critical review. Prog. Mater. Sci. 2022, 125, 100904. [Google Scholar] [CrossRef]
- Wu, H.; Yildirim, T.; Zhou, W. Exceptional Mechanical Stability of Highly Porous Zirconium Metal–Organic Framework UiO-66 and Its Important Implications. J. Phys. Chem. Lett. 2013, 4, 925–930. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.; Vitillo, J.G.; Uddin, M.J.; Bonino, F.; Lamberti, C.; Groppo, E.; Lillerud, K.-P.; Bordiga, S. Functionalization of UiO-66 Metal−Organic Framework and Highly Cross-Linked Polystyrene with Cr(CO)3: In Situ Formation, Stability, and Photoreactivity. Chem. Mater. 2010, 22, 4602–4611. [Google Scholar] [CrossRef]
- Winarta, J.; Shan, B.; McIntyre, S.M.; Ye, L.; Wang, C.; Liu, J.; Mu, B. A Decade of UiO-66 Research: A Historic Review of Dynamic Structure, Synthesis Mechanisms, and Characterization Techniques of an Archetypal Metal–Organic Framework. Cryst. Growth Des. 2020, 20, 1347–1362. [Google Scholar] [CrossRef]
- Jasuja, H.; Walton, K.S. Experimental Study of CO2, CH4, and Water Vapor Adsorption on a Dimethyl-Functionalized Ui0-66 Framework. J. Phys. Chem. C 2013, 117, 7062–7068. [Google Scholar] [CrossRef]
- Cavka, J.H.; Grande, C.A.; Mondino, G.; Blom, R. High Pressure Adsorption of CO2 and CH4 on Zr-MOFs. Ind. Eng. Chem. Res. 2014, 53, 15500–15507. [Google Scholar] [CrossRef]
- Bárcia, P.S.; Guimarães, D.; Mendes, P.A.P.; Silva, J.A.C.; Guillerm, V.; Chevreau, H.; Serre, C.; Rodrigues, A.E. Reverse Shape Selectivity in the Adsorption of Hexane and Xylene Isomers in MOF UiO-66. Microporous Mesoporous Mater. 2011, 139, 67–73. [Google Scholar] [CrossRef]
- Kandiah, M.; Nilsen, M.H.; Usseglio, S.; Jakobsen, S.; Olsbye, U.; Tilset, M.; Larabi, C.; Quadrelli, E.A.; Bonino, F.; Lillerud, K.P. Synthesis and Stability of Tagged UiO-66 Zr-MOFs. Chem. Mater. 2010, 22, 6632–6640. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Wiersum, A.D.; Jobic, H.; Guillerm, V.; Serre, C.; Llewellyn, P.L.; Maurin, G. Understanding the Thermodynamic and Kinetic Behavior of the CO2/CH4 Gas Mixture within the Porous Zirconium Terephthalate UiO-66(Zr): A Joint Experimental and Modeling Approach. J. Phys. Chem. C 2011, 115, 13768–13774. [Google Scholar] [CrossRef]
- Moreira, M.A.; Santos, J.C.; Ferreira, A.F.P.; Loureiro, J.M.; Ragon, F.; Horcajada, P.; Shim, K.-E.; Hwang, Y.-K.; Lee, U.H.; Chang, J.-S.; et al. Reverse Shape Selectivity in the Liquid-Phase Adsorption of Xylene Isomers in Zirconium Terephthalate MOF UiO-66. Langmuir 2012, 28, 5715–5723. [Google Scholar] [CrossRef]
- Edubilli, S.; Gumma, S. A systematic evaluation of UiO-66 metal organic framework for CO2/N2 separation. Sep. Purif. Technol. 2019, 224, 85–94. [Google Scholar] [CrossRef]
- Yang, Q.; Guillerm, V.; Ragon, F.; Wiersum, A.D.; Llewellyn, P.L.; Zhong, C.; Devic, T.; Serre, C.; Maurin, G. CH4 storage and CO2 capture in highly porous zirconium oxide based metal-organic frameworks. Chem. Commun. 2012, 48, 9831–9833. [Google Scholar] [CrossRef] [PubMed]
- Lennox, M.J.; Düren, T. Understanding the Kinetic and Thermodynamic Origins of Xylene Separation in UiO-66(Zr) via Molecular Simulation. J. Phys. Chem. C 2016, 120, 18651–18658. [Google Scholar] [CrossRef]
- Granato, M.A.; Martins, V.D.; Ferreira, A.F.P.; Rodrigues, A.E. Adsorption of xylene isomers in MOF UiO-66 by molecular simulation. Microporous Mesoporous Mater. 2014, 190, 165–170. [Google Scholar] [CrossRef]
- Lyu, J.; Liu, H.; Zeng, Z.; Zhang, J.; Xiao, Z.; Bai, P.; Guo, X. Metal–Organic Framework UiO-66 as an Efficient Adsorbent for Boron Removal from Aqueous Solution. Ind. Eng. Chem. Res. 2017, 56, 2565–2572. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Chen, J.P.; Li, K. Superior removal of arsenic from water with zirconium metal-organic framework UiO-66. Sci. Rep. 2015, 5, 16613. [Google Scholar] [CrossRef] [PubMed]
- Barreto, J.; Xavier, M.D.G.; Ribeiro, R.P.P.L.; Martins, D.; Esteves, I.A.A.C.; Branco, M.; Tirolien, T.; Mota, J.P.B.; Bonfait, G. Neon Adsorption on HKUST-1 and UiO-66 Metal–Organic Frameworks over Wide Pressure and Temperature Ranges. J. Chem. Eng. Data 2019, 64, 5407–5414. [Google Scholar] [CrossRef]
- Garibay, S.J.; Cohen, S.M. Isoreticular synthesis and modification of frameworks with the UiO-66 topology. Chem. Commun. 2010, 46, 7700–7702. [Google Scholar] [CrossRef]
- Xian, S.; Wu, Y.; Wu, J.; Wang, X.; Xiao, J. Enhanced Dynamic CO2 Adsorption Capacity and CO2/CH4 Selectivity on Polyethylenimine-Impregnated UiO-66. Ind. Eng. Chem. Res. 2015, 54, 11151–11158. [Google Scholar] [CrossRef]
- Ribeiro, R.P.P.L.; Camacho, B.C.R.; Lyubchyk, A.; Esteves, I.A.A.C.; Cruz, F.J.A.L.; Mota, J.P.B. Experimental and Computational Study of Ethane and Ethylene Adsorption in the MIL-53(Al) Metal Organic Framework. Microporous Mesoporous Mater. 2016, 230, 154–165. [Google Scholar] [CrossRef]
- Ribeiro, R.P.P.L.; Mota, J.P.B. Surface Area and Porosity of Co3(ndc)3(dabco) Metal–Organic Framework and Its Methane Storage Capacity: A Combined Experimental and Simulation Study. J. Phys. Chem. C 2021, 125, 2411–2423. [Google Scholar] [CrossRef]
- Rogacka, J.; Seremak, A.; Luna-Triguero, A.; Formalik, F.; Matito-Martos, I.; Firlej, L.; Calero, S.; Kuchta, B. High-throughput screening of metal—Organic frameworks for CO2 and CH4 separation in the presence of water. Chem. Eng. J. 2021, 403, 126392. [Google Scholar] [CrossRef]
- Becker, T.M.; Heinen, J.; Dubbeldam, D.; Lin, L.-C.; Vlugt, T.J.H. Polarizable Force Fields for CO2 and CH4 Adsorption in M-MOF-74. J. Phys. Chem. C 2017, 121, 4659–4673. [Google Scholar] [CrossRef] [PubMed]
- Skoulidas, A.I. Molecular Dynamics Simulations of Gas Diffusion in Metal−Organic Frameworks: Argon in CuBTC. J. Am. Chem. Soc. 2004, 126, 1356–1357. [Google Scholar] [CrossRef] [PubMed]
- Boyd, P.G.; Moosavi, S.M.; Witman, M.; Smit, B. Force-Field Prediction of Materials Properties in Metal-Organic Frameworks. J. Phys. Chem. Lett. 2017, 8, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhong, C. Molecular Simulation of Carbon Dioxide/Methane/Hydrogen Mixture Adsorption in Metal−Organic Frameworks. J. Phys. Chem. B 2006, 110, 17776–17783. [Google Scholar] [CrossRef]
- Martín-Calvo, A.; García-Pérez, E.; Manuel Castillo, J.; Calero, S. Molecular simulations for adsorption and separation of natural gas in IRMOF-1 and Cu-BTC metal-organic frameworks. Phys. Chem. Chem. Phys. 2008, 10, 7085–7091. [Google Scholar] [CrossRef]
- Sladekova, K.; Campbell, C.; Grant, C.; Fletcher, A.J.; Gomes, J.R.B.; Jorge, M. The effect of atomic point charges on adsorption isotherms of CO2 and water in metal organic frameworks. Adsorption 2020, 26, 663–685. [Google Scholar] [CrossRef]
- Demir, H.; Walton, K.S.; Sholl, D.S. Computational Screening of Functionalized UiO-66 Materials for Selective Contaminant Removal from Air. J. Phys. Chem. C 2017, 121, 20396–20406. [Google Scholar] [CrossRef]
- Planchais, A.; Devautour-Vinot, S.; Salles, F.; Ragon, F.; Devic, T.; Serre, C.; Maurin, G. A Joint Experimental/Computational Exploration of the Dynamics of Confined Water/Zr-Based MOFs Systems. J. Phys. Chem. C 2014, 118, 14441–14448. [Google Scholar] [CrossRef]
- Ghosh, P.; Colón, Y.J.; Snurr, R.Q. Water adsorption in UiO-66: The importance of defects. Chem. Commun. 2014, 50, 11329–11331. [Google Scholar] [CrossRef]
- Ramsahye, N.A.; Maurin, G.; Bourrelly, S.; Llewellyn, P.; Loiseau, T.; Ferey, G. Charge distribution in metal organic framework materials: Transferability to a preliminary molecular simulation study of the CO2 adsorption in the MIL-53 (Al) system. Phys. Chem. Chem. Phys. 2007, 9, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Potoff, J.J.; Siepmann, J.I. Vapor–liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 2001, 47, 1676–1682. [Google Scholar] [CrossRef]
- Wick, C.D.; Siepman, J.I.; Klotz, W.L.; Schure, M.R. Temperature effects on the retention of n-alkanes and arenes in helium-squalane gas-liquid chromatography. Experiment and molecular simulation. J. Chromatogr. A 2002, 954, 181–190. [Google Scholar] [CrossRef]
- Kamath, G.; Cao, F.; Potoff, J.J. An Improved Force Field for the Prediction of the Vapor−Liquid Equilibria for Carboxylic Acids. J. Phys. Chem. B 2004, 108, 14130–14136. [Google Scholar] [CrossRef]
- Stubbs, J.M.; Potoff, J.J.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 6. United-Atom Description for Ethers, Glycols, Ketones, and Aldehydes. J. Phys. Chem. B 2004, 108, 17596–17605. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Rai, N.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 10. Explicit-Hydrogen Description of Substituted Benzenes and Polycyclic Aromatic Compounds. J. Phys. Chem. B 2013, 117, 273–288. [Google Scholar] [CrossRef]
- Shearer, G.C.; Chavan, S.; Bordiga, S.; Svelle, S.; Olsbye, U.; Lillerud, K.P. Defect Engineering: Tuning the Porosity and Composition of the Metal–Organic Framework UiO-66 via Modulated Synthesis. Chem. Mater. 2016, 28, 3749–3761. [Google Scholar] [CrossRef]
- Thornton, A.W.; Babarao, R.; Jain, A.; Trousselet, F.; Coudert, F.X. Defects in metal–organic frameworks: A compromise between adsorption and stability? Dalton Trans. 2016, 45, 4352–4359. [Google Scholar] [CrossRef]
- Wu, H.; Chua, Y.S.; Krungleviciute, V.; Tyagi, M.; Chen, P.; Yildirim, T.; Zhou, W. Unusual and Highly Tunable Missing-Linker Defects in Zirconium Metal–Organic Framework UiO-66 and Their Important Effects on Gas Adsorption. J. Am. Chem. Soc. 2013, 135, 10525–10532. [Google Scholar] [CrossRef] [PubMed]
- Vandenbrande, S.; Verstraelen, T.; Gutiérrez-Sevillano, J.J.; Waroquier, M.; Van Speybroeck, V. Methane Adsorption in Zr-Based MOFs: Comparison and Critical Evaluation of Force Fields. J. Phys. Chem. C 2017, 121, 25309–25322. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.; Parsonage, N.G. Computer Simulation and the Statistical Mechanics of Adsorption; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Vlugt, T.J.H.; García-Pérez, E.; Dubbeldam, D.; Ban, S.; Calero, S. Computing the Heat of Adsorption using Molecular Simulations: The Effect of Strong Coulombic Interactions. J. Chem. Theory Comput. 2008, 4, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Babarao, R.; Hu, Z.; Jiang, J.; Chempath, S.; Sandler, S.I. Storage and Separation of CO2 and CH4 in Silicalite, C168 Schwarzite, and IRMOF-1: A Comparative Study from Monte Carlo Simulation. Langmuir 2007, 23, 659–666. [Google Scholar] [CrossRef]






| Label | σ (Å) | ε/kB (K) | Mw (g/mol) | q (e) | Ref. |
|---|---|---|---|---|---|
| C25 | 3.74 | 48.00 | 13.02 | +0.006 | [44] |
| C1 | 3.90 | 41.00 | 12.01 | +0.625 | [45] |
| C13 | 3.88 | 21.00 | 12.01 | −0.002 | [44] |
| O1 | 2.80 | 55.00 | 16.00 | −0.582 | [46] |
| O25 | 3.02 | 93.00 | 17.01 | −1.179 | [46] |
| O29 | 2.80 | 55.00 | 16.00 | −0.741 | [46] |
| Zr | 2.78 | 34.72 | 91.22 | +2.008 | [47] |
| H25 | 0.00 | 0.00 | 1.01 | +0.495 | [39] |
| Label | σ (Å) | ε/kB (K) | Mw (g/mol) | q (e) | Ref. |
|---|---|---|---|---|---|
| C25 | 3.60 | 30.70 | 12.01 | −0.121 | [48] |
| C1 | 3.90 | 41.00 | 12.01 | +0.625 | [45] |
| C13 | 3.88 | 21.00 | 12.01 | −0.002 | [44] |
| O1 | 2.80 | 55.00 | 16.00 | −0.582 | [46] |
| O25 | 3.02 | 93.00 | 16.00 | −1.179 | [46] |
| O29 | 2.80 | 55.00 | 16.00 | −0.741 | [46] |
| Zr | 2.78 | 34.72 | 91.22 | +2.008 | [47] |
| H1 | 2.36 | 25.45 | 1.01 | +0.127 | [48] |
| H25 | 0.00 | 0.00 | 1.01 | +0.495 | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maia, J.M.M.; Ribeiro, R.P.P.L.; Mota, J.P.B. Prediction of Carbon Dioxide and Methane Adsorption on UiO-66 Metal–Organic Framework via Molecular Simulation. Crystals 2023, 13, 1523. https://doi.org/10.3390/cryst13101523
Maia JMM, Ribeiro RPPL, Mota JPB. Prediction of Carbon Dioxide and Methane Adsorption on UiO-66 Metal–Organic Framework via Molecular Simulation. Crystals. 2023; 13(10):1523. https://doi.org/10.3390/cryst13101523
Chicago/Turabian StyleMaia, João M. M., Rui P. P. L. Ribeiro, and José P. B. Mota. 2023. "Prediction of Carbon Dioxide and Methane Adsorption on UiO-66 Metal–Organic Framework via Molecular Simulation" Crystals 13, no. 10: 1523. https://doi.org/10.3390/cryst13101523
APA StyleMaia, J. M. M., Ribeiro, R. P. P. L., & Mota, J. P. B. (2023). Prediction of Carbon Dioxide and Methane Adsorption on UiO-66 Metal–Organic Framework via Molecular Simulation. Crystals, 13(10), 1523. https://doi.org/10.3390/cryst13101523