

A Comprehensive First-Principles Investigation of SnTiO3 Perovskite for Optoelectronic and Thermoelectric Applications

 , , ,
, , ,
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
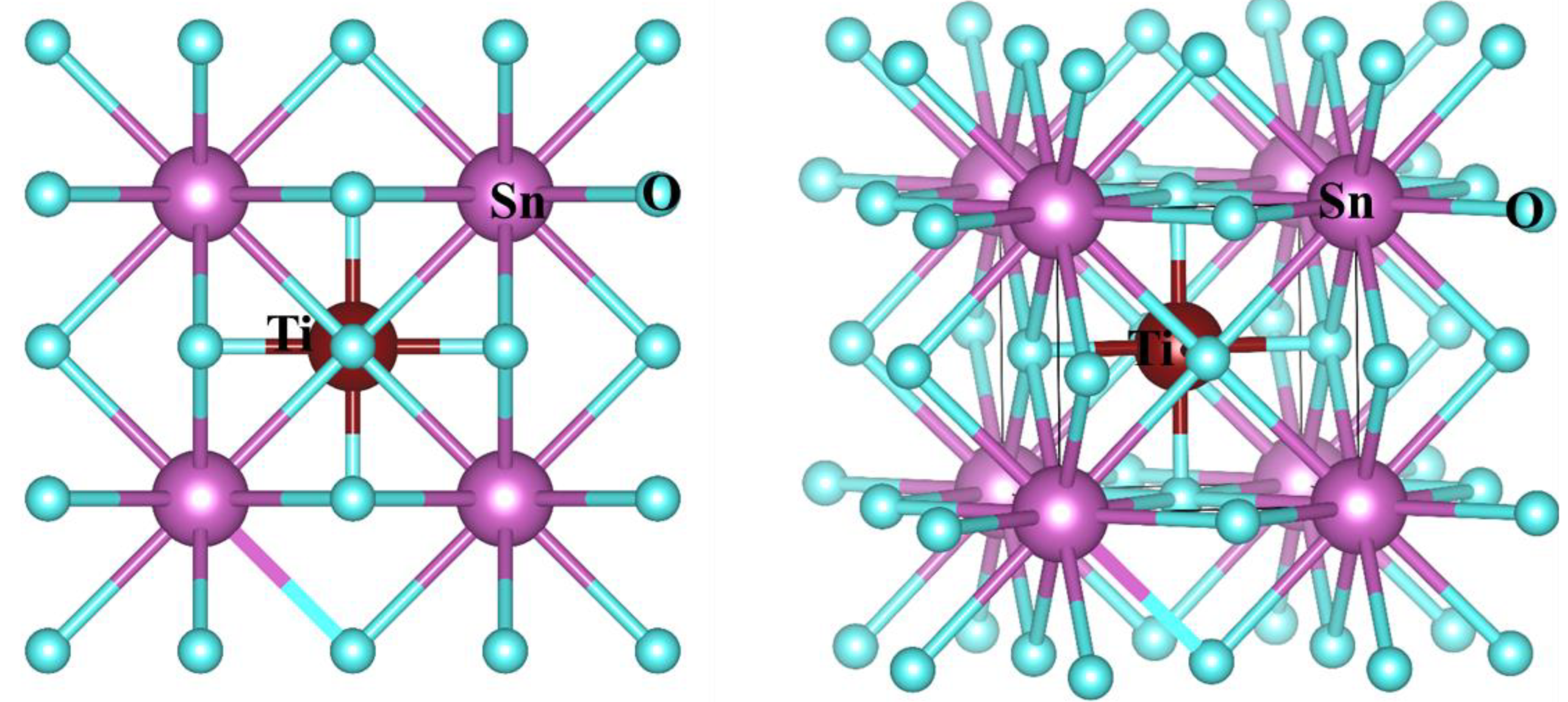
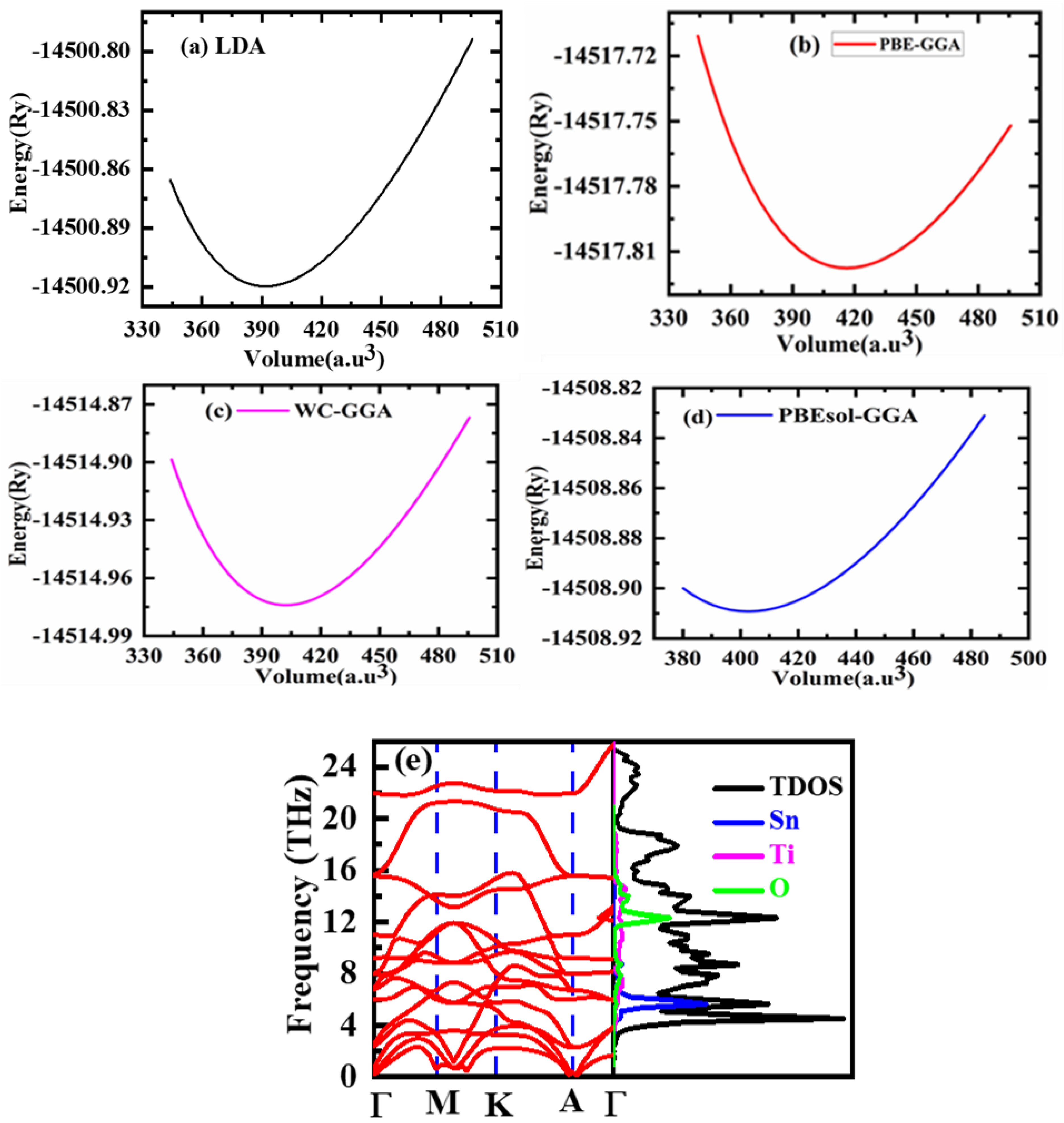
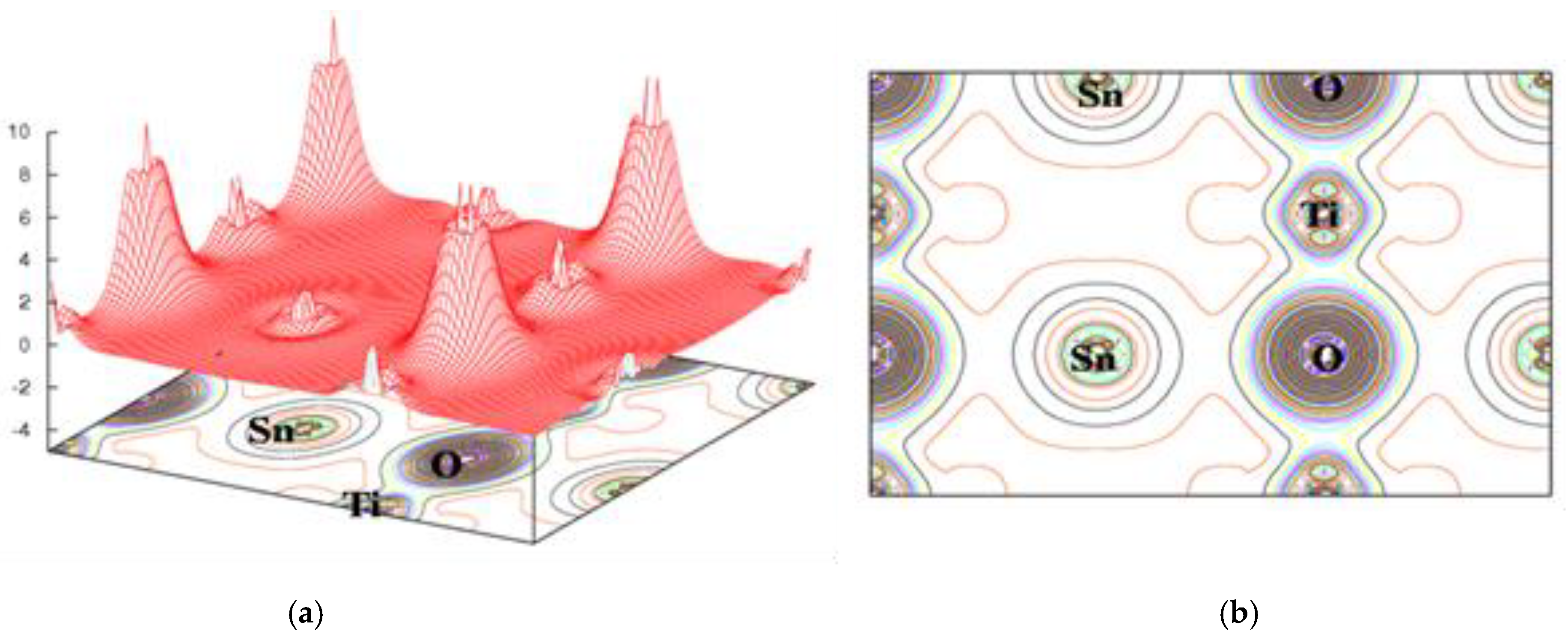
3.1. Structural Properties
3.2. Elastic Properties
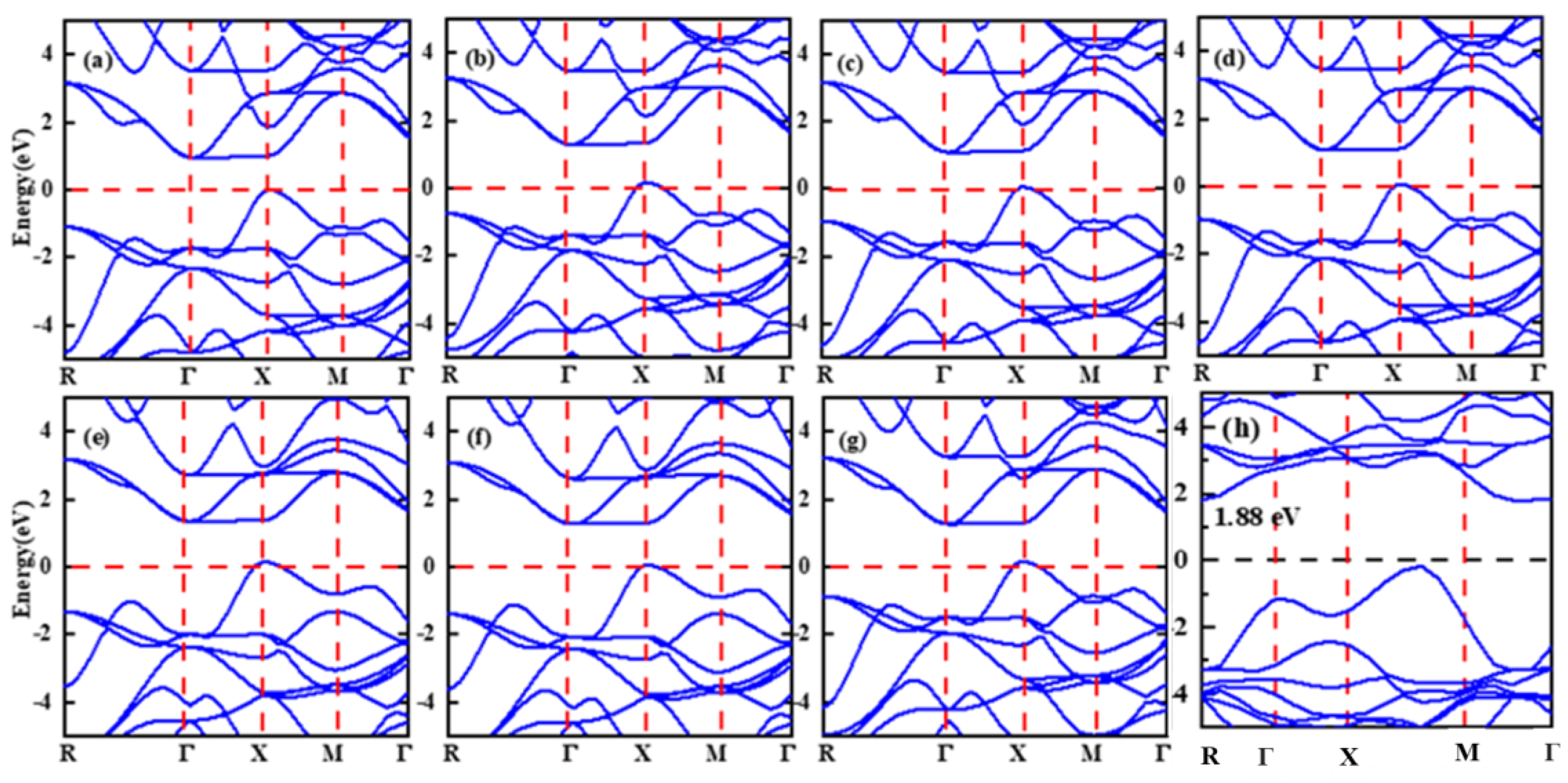
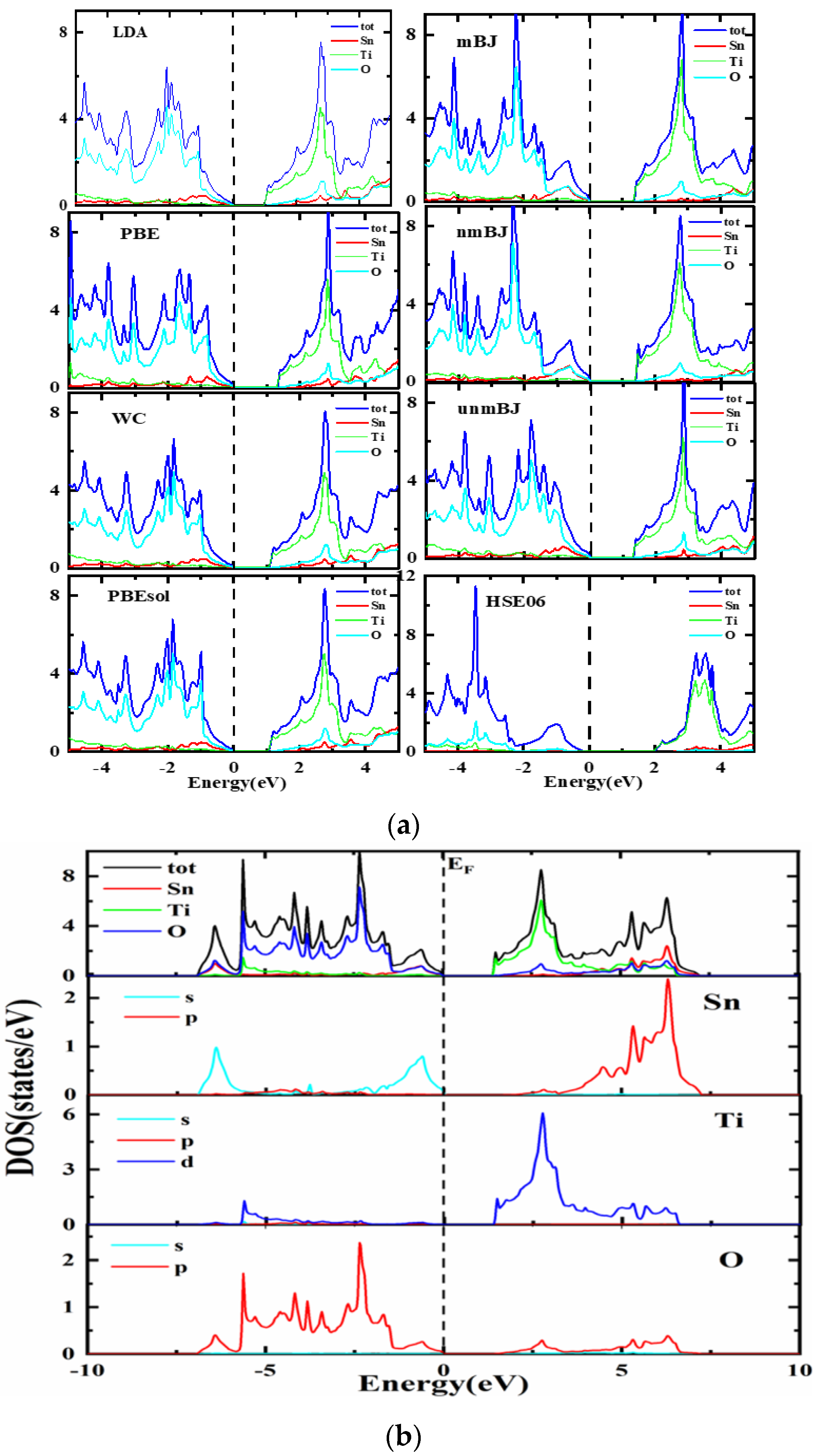
3.3. Electronic Structure
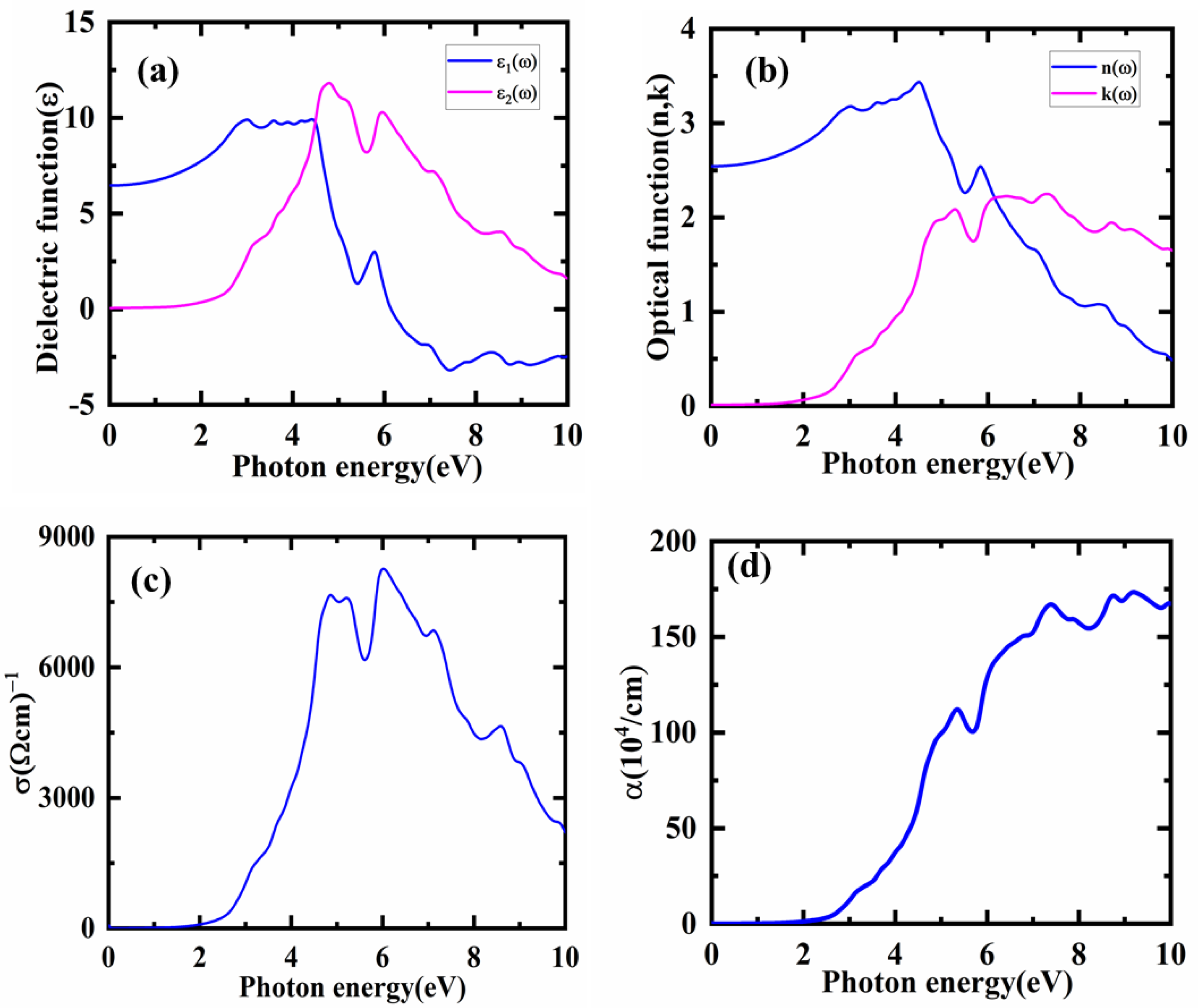
3.4. Optical Properties
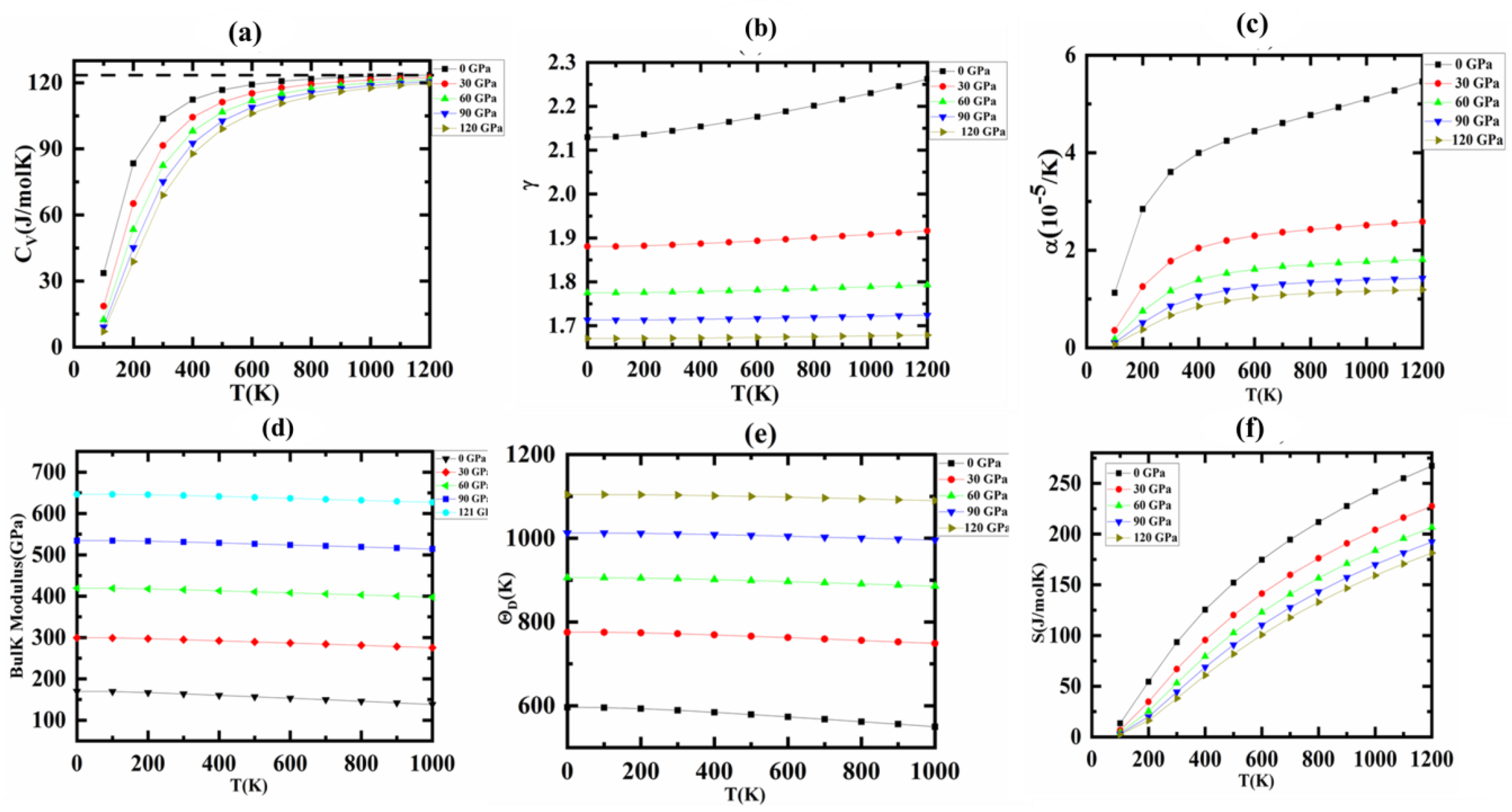
3.5. Thermodynamic Properties
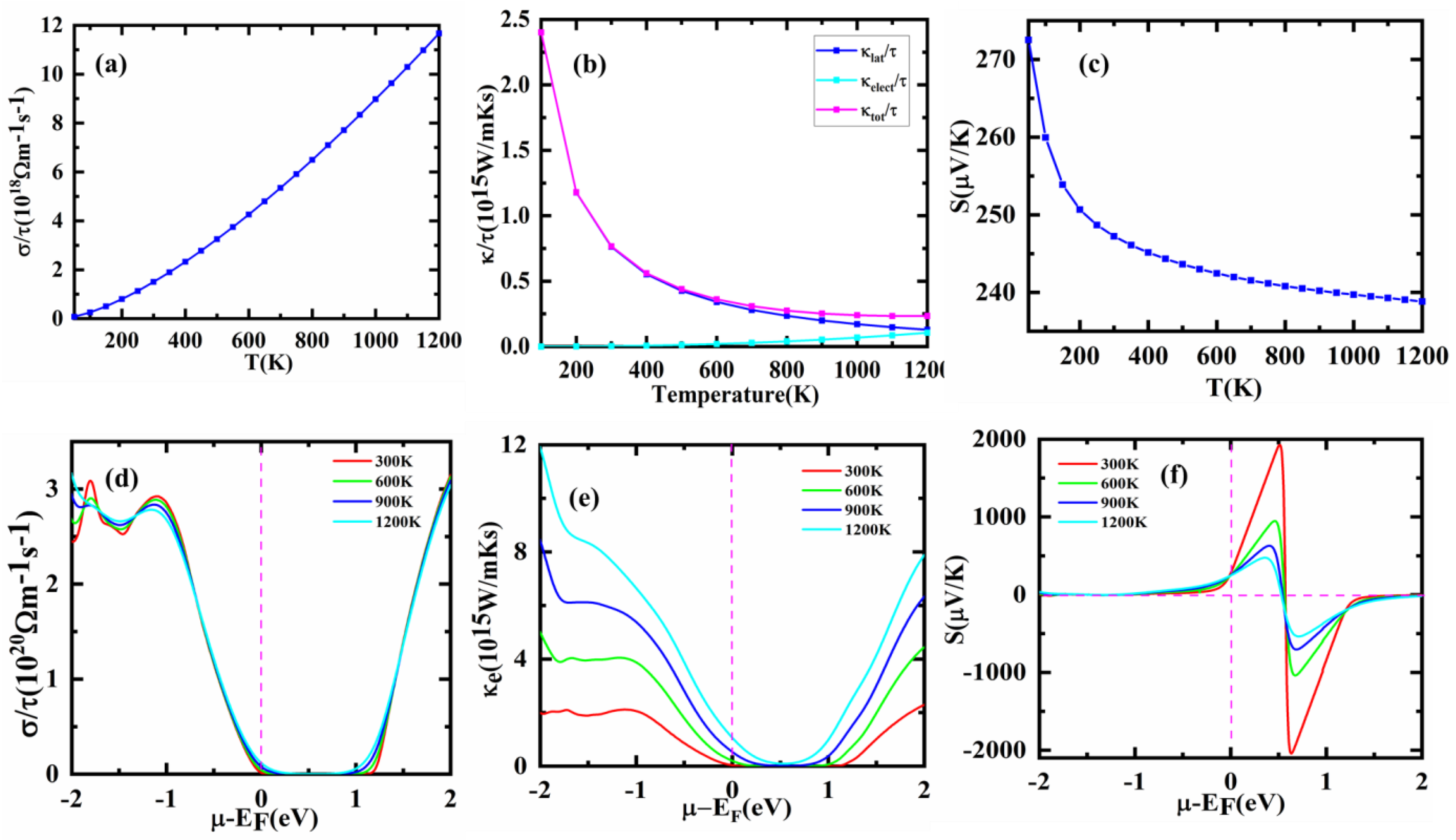
3.6. Thermoelectric Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Im, J.-H.; Lee, C.-R.; Lee, J.-W.; Park, S.-W.; Park, N.-G. 6.5% Efficient Perovskite Quantum-Dot-Sensitized Solar Cell. Nanoscale 2011, 3, 4088–4093. [Google Scholar] [CrossRef] [Green Version]
- Manzoor, M.; Behera, D.; Sharma, R.; Iqbal, M.W.; Mukherjee, S.K.; Khenata, R.; Alarfaji, S.S.; Alzahrani, H.A. Investigation of the Structural, Mechanical, Optoelectronic and, Thermoelectric Characteristics of Cubic GeTiO3: An Ab Initio Study. Mater. Today Commun. 2023, 34, 105053. [Google Scholar] [CrossRef]
- Moon, S.-J.; Itzhaik, Y.; Yum, J.-H.; Zakeeruddin, S.M.; Hodes, G.; Grätzel, M. Sb2S3-Based Mesoscopic Solar Cell Using an Organic Hole Conductor. J. Phys. Chem. Lett. 2010, 1, 1524–1527. [Google Scholar] [CrossRef]
- Taib, M.F.M.; Yaakob, M.K.; Badrudin, F.W.; Kudin, T.I.T.; Hassan, O.H.; Yahya, M.Z.A. First Principles Calculation of Tetragonal (P4 Mm) Pb-Free Ferroelectric Oxide of SnTiO3. Ferroelectrics 2014, 459, 134–142. [Google Scholar] [CrossRef]
- Akkerman, Q.A.; Gandini, M.; Di Stasio, F.; Rastogi, P.; Palazon, F.; Bertoni, G.; Ball, J.M.; Prato, M.; Petrozza, A.; Manna, L. Strongly Emissive Perovskite Nanocrystal Inks for High-Voltage Solar Cells. Nat. Energy 2016, 2, 16194. [Google Scholar] [CrossRef]
- Eperon, G.E.; Paternò, G.M.; Sutton, R.J.; Zampetti, A.; Haghighirad, A.A.; Cacialli, F.; Snaith, H.J. Inorganic Caesium Lead Iodide Perovskite Solar Cells. J. Mater. Chem. A 2015, 3, 19688–19695. [Google Scholar] [CrossRef]
- Behera, D.; Mukherjee, S.K. Structural, Elastic, Electronic and Thermoelectric Properties of K2GeBr6: A First Principle Approach. Mater. Today Proc. 2023, in press. [CrossRef]
- Prado-Gonjal, J.; Lopez, C.A.; Pinacca, R.M.; Serrano-Sánchez, F.; Nemes, N.M.; Dura, O.J.; Martínez, J.L.; Fernández-Díaz, M.T.; Alonso, J.A. Correlation between Crystal Structure and Thermoelectric Properties of Sr1− XTi0. 9Nb0. 1O3− δ Ceramics. Crystals 2020, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk Properties and Electronic Structure of SrTiO3, BaTiO3, PbTiO3 Perovskites: An Ab Initio HF/DFT Study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Cohen, R.E.; Krakauer, H. Electronic Structure Studies of the Differences in Ferroelectric Behavior of BaTiO3 and PbTiO3. Ferroelectrics 1992, 136, 65–83. [Google Scholar] [CrossRef]
- Sághi-Szabó, G.; Cohen, R.E.; Krakauer, H. First-Principles Study of Piezoelectricity in PbTiO3. Phys. Rev. Lett. 1998, 80, 4321. [Google Scholar] [CrossRef]
- Saghi-Szabo, G.; Cohen, R.E.; Krakauer, H. First-Principles Study of Piezoelectricity in Tetragonal PbTiO3 and PbZr 1/2 Ti 1/2 O 3. Phys. Rev. B 1999, 59, 12771. [Google Scholar] [CrossRef]
- Rabe, K.M.; Ghosez, P. Ferroelectricity in PbTiO3 Thin Films: A First Principles Approach. J. Electroceram. 2000, 4, 379–383. [Google Scholar] [CrossRef]
- Meyer, B.; Padilla, J.; Vanderbilt, D. Theory of PbTiO3, BaTiO3, and SrTiO3 Surfaces. Faraday Discuss. 1999, 114, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Neaton, J.B.; Zheng, H.; Nagarajan, V.; Ogale, S.B.; Liu, B.; Viehland, D.; Vaithyanathan, V.; Schlom, D.G.; Waghmare, U. V Epitaxial BiFeO3 Multiferroic Thin Film Heterostructures. Science 2003, 299, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.Y.; Ricinschi, D.; Kanashima, T.; Noda, M.; Okuyama, M. Giant Ferroelectric Polarization beyond 150 ΜC/Cm2 in BiFeO3 Thin Film. Jpn. J. Appl. Phys. 2004, 43, L647. [Google Scholar] [CrossRef]
- Konishi, Y.; Ohsawa, M.; Yonezawa, Y.; Tanimura, Y.; Chikyow, T.; Wakisaka, T.; Koinuma, H.; Miyamoto, A.; Kubo, M.; Sasata, K. Possible Ferroelectricity in SnTiO3 by First-Principles Calculations. MRS Online Proc. Libr. (OPL) 2002, 748. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, J.-T.; Grin, Y.; Wang, X.-J.; Tang, M.-B.; Man, Z.-Y.; Chen, H.-H.; Yang, X.-X. A New Type of Thermoelectric Material, Eu Zn2Sb2. J. Chem. Phys. 2008, 129, 164713. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Rondinelli, J.M.; Nakhmanson, S.M. First-Principles Study of Misfit Strain-Stabilized Ferroelectric SnTiO3. Phys. Rev. B 2011, 84, 245126. [Google Scholar] [CrossRef] [Green Version]
- Hautier, G.; Jain, A.; Ong, S.P. From the Computer to the Laboratory: Materials Discovery and Design Using First-Principles Calculations. J. Mater. Sci. 2012, 47, 7317–7340. [Google Scholar] [CrossRef] [Green Version]
- Hautier, G.; Fischer, C.C.; Jain, A.; Mueller, T.; Ceder, G. Finding Nature’s Missing Ternary Oxide Compounds Using Machine Learning and Density Functional Theory. Chem. Mater. 2010, 22, 3762–3767. [Google Scholar] [CrossRef]
- Diehl, L.; Bette, S.; Pielnhofer, F.; Betzler, S.; Moudrakovski, I.; Ozin, G.A.; Dinnebier, R.; Lotsch, B. V Structure-Directing Lone Pairs: Synthesis and Structural Characterization of SnTiO3. Chem. Mater. 2018, 30, 8932–8938. [Google Scholar] [CrossRef]
- Pielnhofer, F.; Diehl, L.; Jiménez-Solano, A.; Bussmann-Holder, A.; Schön, J.C.; Lotsch, B. V Examination of Possible High-Pressure Candidates of SnTiO3: The Search for Novel Ferroelectric Materials. APL Mater. 2021, 9, 21103. [Google Scholar] [CrossRef]
- de Lazaro, S.; Longo, E.; Sambrano, J.R.; Beltrán, A. Structural and Electronic Properties of PbTiO3 Slabs: A DFT Periodic Study. Surf. Sci. 2004, 552, 149–159. [Google Scholar] [CrossRef]
- Matar, S.F.; Baraille, I.; Subramanian, M.A. First Principles Studies of SnTiO3 Perovskite as Potential Environmentally Benign Ferroelectric Material. Chem. Phys. 2009, 355, 43–49. [Google Scholar] [CrossRef]
- Zhao, H.; Kimura, H.; Cheng, Z.; Wang, X.; Yao, Q.; Osada, M.; Li, B.; Nishida, T. A New Multiferroic Heterostructure of YMnO3/SnTiO3+ X. Scr. Mater. 2011, 65, 618–621. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Density Functional Theory (DFT). Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Blaha, P.; Schwarz, K.; Madsen, G.K.H.; Kvasnicka, D.; Luitz, J. Wien2k. Augment. Plane Wave+ Local Orbitals Program Calc. Cryst. Prop. 2001, 60, 1–302. [Google Scholar]
- Hybertsen, M.S.; Louie, S.G. First-Principles Theory of Quasiparticles: Calculation of Band Gaps in Semiconductors and Insulators. Phys. Rev. Lett. 1985, 55, 1418. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Cohen, R.E. More Accurate Generalized Gradient Approximation for Solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Behera, D.; Abraham, J.A.; Sharma, R.; Mukerjee, S.K.; Jain, E. First Principles Study of New D0 Half-Metallic Ferromagnetism in CsBaC Ternary Half-Heusler Alloy. J. Supercond. Nov. Magn. 2022, 35, 3431–3437. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Sharma, R.; Iqbal, M.W.; Mukherjee, S.K. First Principle Insight on Structural, Opto-Electronic and Transport Properties of Novel Zintl-Phase AMg2Bi2 (A = Sr, Ba). J. Solid State Chem. 2023, 320, 123860. [Google Scholar] [CrossRef]
- Hafner, J. Materials Simulations Using VASP—A Quantum Perspective to Materials Science. Comput. Phys. Commun. 2007, 177, 6–13. [Google Scholar] [CrossRef]
- Dahbi, S.; Tahiri, N.; El Bounagui, O.; Ez-Zahraouy, H. Electronic, Optical, and Thermoelectric Properties of Perovskite BaTiO3 Compound under the Effect of Compressive Strain. Chem. Phys. 2021, 544, 111105. [Google Scholar] [CrossRef]
- Johnston, K.; Huang, X.; Neaton, J.B.; Rabe, K.M. First-Principles Study of Symmetry Lowering and Polarization in Ba TiO3/SrTiO3 Superlattices with in-Plane Expansion. Phys. Rev. B 2005, 71, 100103. [Google Scholar] [CrossRef] [Green Version]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef] [Green Version]
- Pielnhofer, F.; Diehl, L.; Jiménez-Solano, A.; Bußmann-Holder, A.; Christian, J.; Schön, B.V.L. 3. Examining Experimentally Accessible Structural Candidates of SnTiO3: The Search for Novel Ferroelectric Materials. In SnTiO3–A Lone Pair Model System for Studying Structure-Property Relationships in Photocatalysis, Ferroelectricity and Beyond; Ludwig-Maximilians-Universität München: Munich, Germany, 2020; Volume 44. [Google Scholar]
- Taib, M.F.M.; Yaakob, M.K.; Chandra, A.; Arof, A.K.M.; Yahya, M.Z.A. Effect of Pressure on Structural, Electronic and Elastic Properties of Cubic (Pm3m) SnTiO3 Using First Principle Calculation. Adv. Mater. Res. 2012, 501, 342–346. [Google Scholar]
- Wang, J.J.; Meng, F.Y.; Ma, X.Q.; Xu, M.X.; Chen, L.Q. Lattice, Elastic, Polarization, and Electrostrictive Properties of BaTiO3 from First-Principles. J. Appl. Phys. 2010, 108, 34107. [Google Scholar] [CrossRef] [Green Version]
- Taib, M.F.M.; Yaakob, M.K.; Hassan, O.H.; Chandra, A.; Arof, A.K.; Yahya, M.Z.A. First Principles Calculation on Structural and Lattice Dynamic of SnTiO3 and SnZrO3. Ceram. Int. 2013, 39, S297–S300. [Google Scholar] [CrossRef]
- Taib, M.F.M.; Yaakob, M.K.; Hassan, O.H.; Yahya, M.Z.A. Structural, Electronic, and Lattice Dynamics of PbTiO3, SnTiO3, and SnZrO3: A Comparative First-Principles Study. Integr. Ferroelectr. 2013, 142, 119–127. [Google Scholar] [CrossRef]
- Alam, N.N.; Malik, N.A.; Hussin, N.H.; Ali, A.M.M.; Hassan, O.H.; Yahya, M.Z.A.; Taib, M.F.M. First-Principles Study on Electronic Properties, Phase Stability and Strain Properties of Cubic (Pm3m) and Tetragonal (P4mm) ATiO3 (A = Pb, Sn). Int. J. Nanoelectron. Mater. 2020, 13. [Google Scholar]
- Behera, D.; Manzoor, M.; Mukherjee, S.K. Incorporation of Te in Enhancing Thermoelectric Response of AeAg2SeTe (Ae = Sr, Ba) Compounds: A DFT Insight. Comput. Condens. Matter 2022, 33, e00757. [Google Scholar] [CrossRef]
- Gao, S.; Broux, T.; Fujii, S.; Tassel, C.; Yamamoto, K.; Xiao, Y.; Oikawa, I.; Takamura, H.; Ubukata, H.; Watanabe, Y. Hydride-Based Antiperovskites with Soft Anionic Sublattices as Fast Alkali Ionic Conductors. Nat. Commun. 2021, 12, 201. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Maharana, M.; Iqbal, M.W.; Zahid, T.; Lakra, S.; Mukherjee, S.K.; Alarfaji, S.S. Structural, Electronic, Optical, and Thermoelectric Response of Zintl Phase AAg2S2 (A = Sr/Ba) Compounds for Renewable Energy Applications. Phys. B Condens. Matter 2023, 649, 414446. [Google Scholar] [CrossRef]
- Jamal, M.; Bilal, M.; Ahmad, I.; Jalali-Asadabadi, S. IRelast Package. J. Alloy. Compd. 2018, 735, 569–579. [Google Scholar] [CrossRef]
- Grimvall, G. Thermophysical Properties of Materials; Elsevier: Amsterdam, The Netherlands, 1999; ISBN 0080542867. [Google Scholar]
- Barsch, G.R. Adiabatic, Isothermal, and Intermediate Pressure Derivatives of the Elastic Constants for Cubic Symmetry. I. Basic Formulae. Phys. Status Solidi 1967, 19, 129–138. [Google Scholar] [CrossRef]
- Behera, D.; Dixit, A.; Kumari, K.; Srivastava, A.; Sharma, R.; Mukherjee, S.K.; Khenata, R.; Boumaza, A.; Bin-Omran, S. Structural, Elastic, Mechanical, and Thermodynamic Characteristic of NaReO3 and KReO3 Perovskite Oxides from First Principles Study. Eur. Phys. J. Plus 2022, 137, 1345. [Google Scholar] [CrossRef]
- Bahera, D.; Dixit, A.; Nahak, B.; Srivastava, A.; Dubey, S.; Sharma, R.; Mishra, A.K.; Mukeerjee, S.K. Structural, Electronic, Elastic, Vibrational and Thermodynamic Properties of Antiperovskites Mg3NX (X = Ge, Sn): A DFT Study. Phys. Lett. A 2022, 453, 128478. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Piskunov, S.; Popov, A.I.; Purans, J.; Bocharov, D.; Jia, R. Systematic Trends in Hybrid-DFT Computations of BaTiO3/SrTiO3, PbTiO3/SrTiO3 and PbZrO3/SrZrO3 (001) Hetero Structures. Condens. Matter 2022, 7, 70. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Popov, A.I.; Bocharov, D.; Chekhovska, A.; Jia, R. Ab Initio Computations of O and AO as Well as ReO2, WO2 and BO2-Terminated ReO3, WO3, BaTiO3, SrTiO3 and BaZrO3 (001) Surfaces. Symmetry 2022, 14, 1050. [Google Scholar] [CrossRef]
- Manzoor, M.; Chowdhury, S.; Sharma, R.; Iqbal, M.W.; Mukherjee, S.K.; Alarfaji, S.S.; Alzahrani, H.A. Insight on the Lattice Dynamics, Thermodynamic and Thermoelectric Properties of CdYF3 Perovskite: A DFT Study. Comput. Theor. Chem. 2022, 1217, 113928. [Google Scholar] [CrossRef]
- Behera, D.; Mukherjee, S.K. Theoretical Investigation of the Lead-Free K2InBiX6 (X = Cl, Br) Double Perovskite Compounds Using First Principle Calculation. JETP Lett. 2022, 116, 537–546. [Google Scholar] [CrossRef]
- Taib, M.F.M.; Yaakob, M.K.; Badrudin, F.W.; Rasiman, M.S.A.; Kudin, T.I.T.; Hassan, O.H.; Yahya, M.Z.A. First-Principles Comparative Study of the Electronic and Optical Properties of Tetragonal (P4mm) ATiO3 (A = Pb, Sn, Ge). Integr. Ferroelectr. 2014, 155, 23–32. [Google Scholar] [CrossRef]
- Li, X.; Xia, C.; Wang, M.; Wu, Y.; Chen, D. First-Principles Investigation of Structural, Electronic and Elastic Properties of HFX (X = Os, Ir and Pt) Compounds. Metals 2017, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- Behera, D.; Sharma, R.; Ullah, H.; Waheed, H.S.; Mukherjee, S.K. Electronic, Optical, and Thermoelectric Investigations of Zintl Phase AAg2Se2 (A= Sr, Ba) Compounds: A First First-Principles Approach. J. Solid State Chem. 2022, 132, 123259. [Google Scholar] [CrossRef]
- Karki, B.B.; Wentzcovitch, R.M.; De Gironcoli, S.; Baroni, S. High-Pressure Lattice Dynamics and Thermoelasticity of MgO. Phys. Rev. B 2000, 61, 8793. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Iqbal, M.W.; Lakra, S.; Mukherjee, S.K. Revealing Excellent Electronic, Optical, and Thermoelectric Behavior of EU Based Euag2y2 (Y = S/Se): For Solar Cell Applications. Comput. Condens. Matter 2022, 32, e00723. [Google Scholar] [CrossRef]
- Andritsos, E.I.; Zarkadoula, E.; Phillips, A.E.; Dove, M.T.; Walker, C.J.; Brazhkin, V.V.; Trachenko, K. The Heat Capacity of Matter beyond the Dulong–Petit Value. J. Phys. Condens. Matter 2013, 25, 235401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajeeston, P.; Ravindran, P.; Fjellvåg, H. Prediction of Structural, Lattice Dynamical, and Mechanical Properties of CaB 2. RSC Adv. 2012, 2, 11687–11694. [Google Scholar] [CrossRef]
- Yaseen, M.; Butt, M.K.; Ashfaq, A.; Iqbal, J.; Almoneef, M.M.; Iqbal, M.; Murtaza, A.; Laref, A. Phase Transition and Thermoelectric Properties of Cubic KNbO3 under Pressure: DFT Approach. J. Mater. Res. Technol. 2021, 11, 2106–2113. [Google Scholar] [CrossRef]
- Behram, R.B.; Iqbal, M.A.; Alay-e-Abbas, S.M.; Sajjad, M.; Yaseen, M.; Arshad, M.I.; Murtaza, G. Theoretical Investigation of Mechanical, Optoelectronic and Thermoelectric Properties of BiGaO3 and BiInO3 Compounds. Mater. Sci. Semicond. Process. 2016, 41, 297–303. [Google Scholar] [CrossRef]
- Noor, N.A.; Hassan, M.; Rashid, M.; Alay-e-Abbas, S.M.; Laref, A. Systematic Study of Elastic, Electronic, Optical and Thermoelectric Properties of Cubic BiBO3 and BiAlO3 Compounds at Different Pressure by Using Ab-Initio Calculations. Mater. Res. Bull. 2018, 97, 436–443. [Google Scholar] [CrossRef]
- Ju, S.; Shiomi, J. Materials Informatics for Heat Transfer: Recent Progresses and Perspectives. Nanoscale Microscale Thermophys. Eng. 2019, 23, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Wood, C. Materials for Thermoelectric Energy Conversion. Rep. Prog. Phys. 1988, 51, 459. [Google Scholar] [CrossRef]
- Shah, S.H.; Khan, S.H.; Laref, A.; Murtaza, G. Optoelectronic and Transport Properties of LiBZ (B = Al, In, Ga and Z = Si, Ge, Sn) Semiconductors. J. Solid State Chem. 2018, 258, 800–808. [Google Scholar] [CrossRef]
- Hoat, D.M. Electronic Structure and Thermoelectric Properties of Ta-Based Half-Heusler Compounds with 18 Valence Electrons. Comput. Mater. Sci. 2019, 159, 470–477. [Google Scholar] [CrossRef]
- Anissa, B.; Radouan, D.; Benaouda, B. Optical and Thermoelectric Response of RhTiSb Half-Heusler. Int. J. Mod. Phys. B 2019, 33, 1950247. [Google Scholar] [CrossRef]
- Abraham, J.A.; Behera, D.; Kumari, K.; Srivastava, A.; Sharma, R.; Mukherjee, S.K. A Comprehensive DFT Analysis on Structural, Electronic, Optical, Thermoelectric, SLME Properties of New Double Perovskite Oxide Pb2ScBiO6. Chem. Phys. Lett. 2022, 806, 139987. [Google Scholar] [CrossRef]
- Haq, B.U.; AlFaify, S.; Laref, A.; Ahmed, R.; Butt, F.K.; Chaudhry, A.R.; Rehman, S.U.; Mahmood, Q. Optoelectronic Properties of New Direct Bandgap Polymorphs of Single-Layered Germanium Sulfide. Ceram. Int. 2019, 45, 18073–18078. [Google Scholar] [CrossRef]
- Behera, D.; Mukherjee, S.K. Optoelectronics and Transport Phenomena in Rb2InBiX6 (X = Cl, Br) Compounds for Renewable Energy Applications: A DFT Insight. Chemistry 2022, 4, 1044–1059. [Google Scholar] [CrossRef]
- Noor, N.A.; Mahmood, Q.; Rashid, M.; Haq, B.U.; Laref, A.; Ahmad, S.A. Ab-Initio Study of Thermodynamic Stability, Thermoelectric and Optical Properties of Perovskites ATiO3 (A = Pb, Sn). J. Solid State Chem. 2018, 263, 115–122. [Google Scholar] [CrossRef]
- Zhu, H.; Zhao, T.; Zhang, B.; An, Z.; Mao, S.; Wang, G.; Han, X.; Lu, X.; Zhang, J.; Zhou, X. Entropy Engineered Cubic N-Type AgBiSe2 Alloy with High Thermoelectric Performance in Fully Extended Operating Temperature Range. Adv. Energy Mater. 2021, 11, 2003304. [Google Scholar] [CrossRef]
- Manzoor, M.; Bahera, D.; Sharma, R.; Tufail, F.; Iqbal, M.W.; Mukerjee, S.K. Investigated the Structural, Optoelectronic, Mechanical, and Thermoelectric Properties of Sr2BTaO6 (B = Sb, Bi) for Solar Cell Applications. Int. J. Energy Res. 2022, 46, 23698–23714. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | SnTiO3 (Cubic) | ||
|---|---|---|---|
| LDA | This study | Others’ Theory | Exp. |
| a(Å) | 3.8731 | 3.850 [41] | 3.960 [42] |
| V(Å)3 | 392.0831 | ||
| B(GPa) | 209.2639 | ||
| BP | 4.6452 | ||
| E0(Ry) | −14,500.9203 | ||
| PBE-GGA | |||
| a(Å) | 3.9511 | ||
| V(Å)3 | 416.2415 | ||
| B(GPa) | 175.1369 | ||
| B.P | 4.5807 | ||
| E0(Ry) | −14,517.819155 | ||
| WC-GGA | |||
| a(Å) | 3.9069 | 3.910 [43] | |
| V(Å)3 | 402.4362 | ||
| B(GPa) | 194.3111 | ||
| BP | 4.5816 | ||
| E0(Ry) | −14,514.870912 | ||
| PBEsol-GGA | |||
| a(Å) | 3.9078 | Ref. 3.916 [44] | |
| V(Å)3 | 402.7150 | ||
| B(GPa) | 191.0451 | ||
| BP | 4.2555 | ||
| E0(Ry) | −14,508.909256 | ||
| Formation energy(eV/atom) | −2.578 | ||
| Ecoh(eV) | −17.600 | −37.088 [45] | |
| Bond length (Å) | Sn-Sn = 4.07 Sn-O = 2.818 Ti-O = 2.0378 | ||
| Material Property | SnTiO3 | Ref. [44] | Ref. [43] |
|---|---|---|---|
| C11 (GPa) | 220.36 | 314.69 | 356.79 |
| C12 (GPa) | 97.96 | 119.62 | 132.93 |
| C44 (GPa) | 100.2846 | 94.06 | 90.47 |
| Bulk modulus, B (GPa) | 138.764 | 184.64 | 207.55 |
| Shear modulus, G (GPa) | 82.265 | ||
| Young’s modulus, Y (GPa) | 206.072 | ||
| Poisson ratio, σ (GPa) | 0.252 | ||
| Pugh ratio, B/G (GPa) | 1.68 | ||
| Frantsevich ratio, G/B (GPa) | 0.59 | ||
| Shear anisotropy factor, A (GPa) | 1.63 | ||
| Cauchy pressure CP (GPa) | −2.32 | ||
| Transverse sound velocity (m/s) | 3772.96 | ||
| Longitudinal sound velocity (m/s) | 6556.84 | ||
| Average sound velocity (m/s) | 4189.92 | ||
| Debye Temperature ΘD (K) | 539.942 | ||
| Melting temperature (K) | 1855.3660 ± 300 K |
| Bandgaps | ||
|---|---|---|
| XC | SnTiO3 | |
| Present Work | Others’ Works | |
| LDA | 1.002 | |
| PBE-GGA | 1.373 | 1.164 eV [45] |
| WC-GGA | 1.080 | |
| PBEsol-GGA | 1.065 | 2.445 eV [44] |
| mBJ-GGA | 1.415 | |
| nmBJ-GGA | 1.451 | |
| unmBJ-GGA | 1.347 | |
| HSE | 1.88 | |
| Parameter | SnTiO3 | |
|---|---|---|
| Present Work | Others’ Works | |
| ε1(0) | 6.51 | 7.15 [4] |
| n(0) | 2.54 | 2.67 [4] |
| R(0)% | 0.18 | 0.21 [58] |
| Parameter | SnTiO3 | |
|---|---|---|
| Present Work | Others’ Works [76] | |
| Σ (Ωms)1 (1018) | 1.50 | 2.5 |
| Ktot (W/mKs) (1015) | 0.76 | 1.8 |
| S (µV/K) | 247 | 90 |
| P.F (W/K2ms1010) | 1.50 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behera, D.; Manzoor, M.; Sharma, R.; Salah, M.M.; Stich, I.; Mukherjee, S.K. A Comprehensive First-Principles Investigation of SnTiO3 Perovskite for Optoelectronic and Thermoelectric Applications. Crystals 2023, 13, 408. https://doi.org/10.3390/cryst13030408
Behera D, Manzoor M, Sharma R, Salah MM, Stich I, Mukherjee SK. A Comprehensive First-Principles Investigation of SnTiO3 Perovskite for Optoelectronic and Thermoelectric Applications. Crystals. 2023; 13(3):408. https://doi.org/10.3390/cryst13030408
Chicago/Turabian StyleBehera, Debidatta, Mumtaz Manzoor, Ramesh Sharma, Mostafa M. Salah, Ivan Stich, and Sanat Kumar Mukherjee. 2023. "A Comprehensive First-Principles Investigation of SnTiO3 Perovskite for Optoelectronic and Thermoelectric Applications" Crystals 13, no. 3: 408. https://doi.org/10.3390/cryst13030408
APA StyleBehera, D., Manzoor, M., Sharma, R., Salah, M. M., Stich, I., & Mukherjee, S. K. (2023). A Comprehensive First-Principles Investigation of SnTiO3 Perovskite for Optoelectronic and Thermoelectric Applications. Crystals, 13(3), 408. https://doi.org/10.3390/cryst13030408