

Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Crystallization and X-ray Structural Analysis
2.3. Computations
3. Results and Discussion
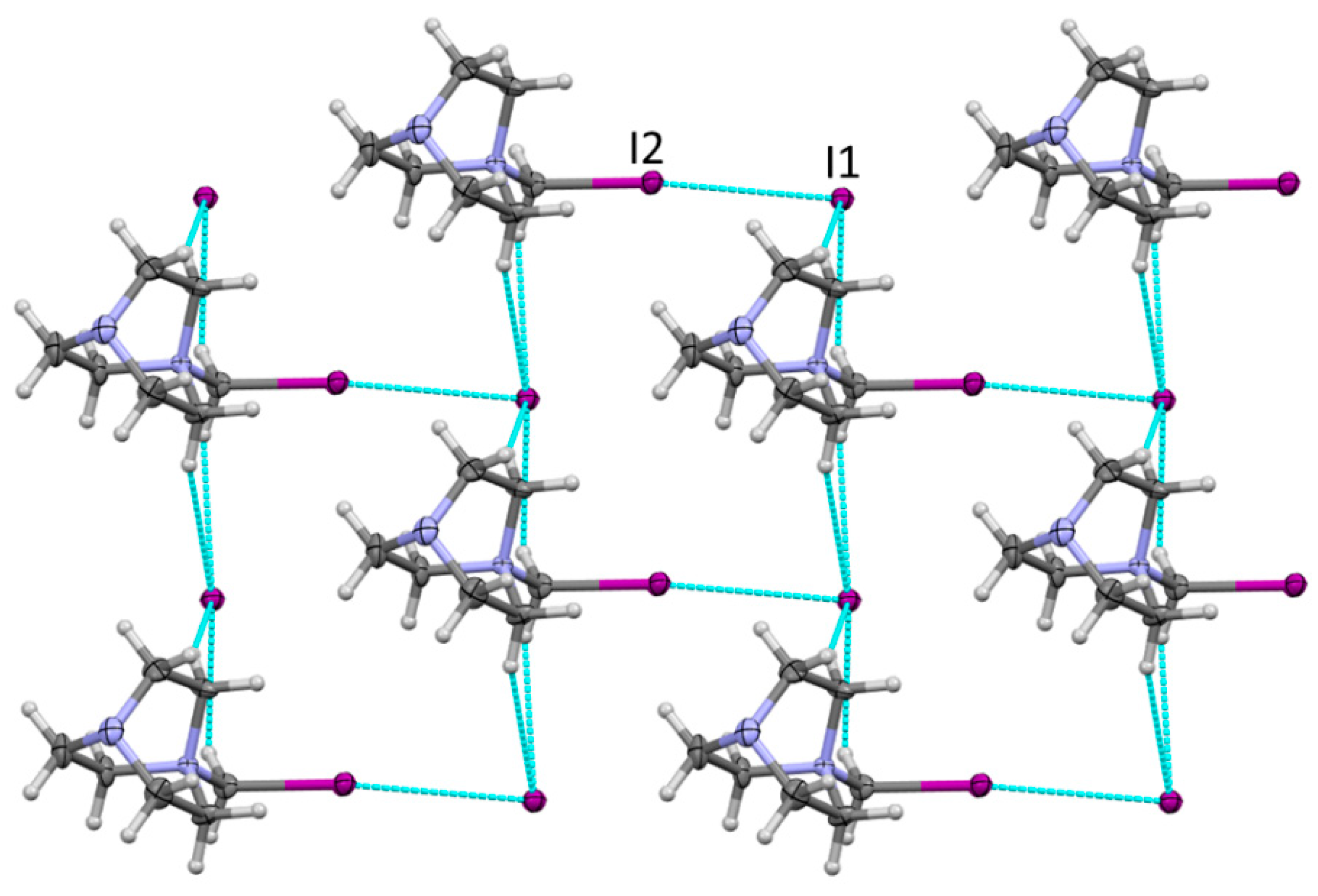
3.1. Preparation and X-ray Structural Characterization of the Quaternary Ammonium Salts
3.2. Computational Analysis of XB in Cation/Anion, Neutral/Anion, and Neutral/Neutral Associations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Metrangolo, P.; Resnati, G.; Poilati, T.; Biella, S. Halogen bonding in crystal engineering. Struct. Bond. 2008, 126, 105–136. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen bonding in supramolecular chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, C.; Hines, R.; Zeller, M.; Rosokha, S.V. Continuum of covalent to intermolecular bonding in the halogen-bonded complexes of 1,4-diazabicyclo[2.2.2]octane with bromine-containing electrophiles. Chem. Commun. 2018, 54, 8060–8063. [Google Scholar] [CrossRef] [PubMed]
- Loy, C.; Zeller, M.; Rosokha, S.V. Halogen bonding in the complexes of brominated electrophiles with chloride anions: From a weak supramolecular interaction to a covalent Br–Cl bond. Crystals 2020, 10, 1075. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Electrostatics and polarization in σ- and π-hole noncovalent interactions: An overview. ChemPhysChem 2020, 21, 579–588. [Google Scholar] [CrossRef]
- Holthoff, J.M.; Weiss, R.; Rosokha, S.V.; Huber, S.M. Anti-electrostatic halogen bonding between Ions of Like Charges. Chem. Eur. J. 2021, 27, 16530–16542. [Google Scholar] [CrossRef]
- Loy, C.; Holthoff, J.M.; Weiss, R.; Huber, S.M.; Rosokha, S.V. Anti-electrostatic” halogen bonding in solution. Chem. Sci. 2021, 12, 8246–8251. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Willett, R.D.; Twamley, B. The role of charge assisted arylhalogen-halide ion interactions in the structures of the dibromopyridinium halide salts. J. Mol. Struct. 2009, 918, 116–122. [Google Scholar] [CrossRef]
- Cametti, M.; Raatikainen, K.; Metrangolo, P.; Pilati, T.; Terraneo, G.; Resnati, G. 2-Iodo-imidazolium receptor binds oxoanions via charge-assisted halogen bonding. Org. Biomol. Chem. 2012, 10, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Kassl, C.J.; Swenson, D.C.; Pigge, F.C. Charge-assisted halogen bonding in bromo- and iodophenylpyridinium chlorides. Cryst. Growth Des. 2015, 15, 4571–4580. [Google Scholar] [CrossRef]
- Zapata, F.; Caballero, A.; White, N.G.; Claridge, T.D.W.; Costa, P.J.; Felix, V.; Beer, P.D. Fluorescent charge-assisted halogen-bonding macrocyclic halo-imidazolium receptors for anion recognition and sensing in aqueous media. J. Am. Chem. Soc. 2012, 134, 11533–11541. [Google Scholar] [CrossRef]
- Fotovic, L.; Stilinovic, V. Halogenide anions as halogen and hydrogen bond acceptors in iodopyridinium halogenides. CrystEngComm 2020, 22, 4039–4046. [Google Scholar] [CrossRef]
- Sutar, R.L.; Engelage, E.; Stoll, R.; Huber, S.M. Bidentate chiral bis(imidazolium)-based halogen-bond donors: Synthesis and applications in enantioselective recognition and catalysis. Angew. Chem. Int. Ed. 2020, 59, 6806–6810. [Google Scholar] [CrossRef]
- Fotovic, L.; Stilinovic, V. Halogen Bonding in N-Alkyl-3-halogenopyridinium Salts. Crystals 2021, 11, 1240. [Google Scholar] [CrossRef]
- Fotovic, L.; Bedekovic, N.; Stilinovic, V. Evaluation of Halogenopyridinium Cations as Halogen Bond Donors. Cryst. Growth Des. 2021, 21, 6889–6901. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, G.; Xu, Z.; Chen, Z.; Wang, J.; Shi, J.; Zhu, W. Halogen bonding in differently charged complexes: Basic profile, essential interaction terms and intrinsic σ-Hole. Phys. Chem. Chem. Phys. 2019, 21, 15106–15119. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Z.; Lu, Y.; Xu, Z.; Wu, W.; Zhu, W.; Peng, C.; Liu, H. Halogen bonding interactions in ion pairs versus conventional charge-assisted and neutral halogen bonds: A theoretical study based on imidazolium species. RSC Adv. 2015, 5, 74284–74294. [Google Scholar] [CrossRef]
- Posavec, L.; Nemec, V.; Stilinović, V.; Cinčić, D. Halogen and hydrogen bond motifs in ionic cocrystals derived from 3-halopyridinium halogenides and perfluorinated iodobenzenes. Cryst. Growth Des. 2021, 21, 6044–6050. [Google Scholar] [CrossRef]
- Pfrunder, M.C.; Micallef, A.S.; Rintoul, L.; Arnold, D.P.; Davy, K.J.P.; McMurtrie, J. Exploitation of the Menshutkin reaction for the controlled assembly of halogen bonded architectures incorporating 1,2-diiodotetrafluorobenzene and 1,3,5-triiodotrifluorobenzene. Cryst. Growth Des. 2012, 12, 714–724. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Kinzhalov, M.A.; Novikov, A.S.; Ananyev, I.V.; Romanova, A.A.; Boyarskiy, V.P.; Haukka, M.; Kukushkin, V.Y. H2C(X)–X···X– (X = Cl, Br) Halogen bonding of dihalomethanes. Cryst. Growth Des. 2017, 17, 1353–1362. [Google Scholar] [CrossRef]
- Gustafsson, B.; Håkansson, M.; Jagner, S. Copper(I)-mediated quaternisation of 1,4-diazabicyclo[2.2.2]octane (DABCO). Crystal structure of bis{(1-chloromethyl-4-aza-1-azoniabi cyclo [2.2.2]octane)-μ-chloro-chlorocopper(I)}. Inorg. Chim. Acta 2005, 358, 1309–1312. [Google Scholar] [CrossRef]
- Hua, X.-N.; Liao, W.-Q.; Tang, Y.-Y.; Li, P.-F.; Shi, P.-P.; Zhao, D.; Xiong, R.-G. A Room-temperature hybrid lead iodide perovskite ferroelectric. J. Am. Chem. Soc. 2018, 140, 12296–12302. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liao, W.-Q.; Ai, Y.; Li, J.; Deng, S.; Hou, Y.; Tang, Y.-Y. Precise molecular design toward organic-inorganic zinc chloride abx3 ferroelectrics. J. Am. Chem. Soc. 2020, 142, 6236–6243. [Google Scholar] [CrossRef]
- March, J. Advanced Organic Chemistry. In Reaction Mechanisms and Structure, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1992. [Google Scholar]
- Adeniyi, E.; Grounds, O.; Stephens, Z.; Zeller, M.; Rosokha, S.V. Thermodynamics and spectroscopy of halogen- and hydrogen-bonded complexes of haloforms with aromatic and aliphatic amines. Molecules 2022, 27, 6124. [Google Scholar] [CrossRef]
- Blackstock, S.C.; Lorand, J.P.; Kochi, J.K. Charge-transfer interactions of amines with tetrahalomethanes. x-ray crystal structures of the donor-acceptor complexes of quinuclidine and diazabicyclo [2.2.2]Octane with carbon tetrabromide. J. Org. Chem. 1987, 52, 1451–1460. [Google Scholar] [CrossRef]
- Bruker. Apex3 v2019.1-0, SAINT V8.40A.; Bruker AXS Inc.: Madison, WI, USA, 2013/2014. [Google Scholar]
- SHELXTL Suite of Programs. Version 6.14, 2000-2003, Bruker Advanced X-ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G. Crystal Structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Hübschle, C.; Sheldrick, G.; Dittrich, B. ShelXle: A Qt Graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design an assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Xu, Z.; Zhu, W. Interaction Nature and Computational Methods for Halogen Bonding: A Perspective. J. Chem. Inf. Model. 2020, 60, 2683–2696. [Google Scholar] [CrossRef]
- Bauza, A.; Alkorta, I.; Frontera, A.; Elguero, J. On the reliability of pure and hybrid DFT methods for the evaluation of halogen, chalcogen, and pnicogen bonds involving anionic and neutral electron donors. J. Chem. Theory Comput. 2013, 9, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Is the use of diffuse functions essential for the properly description of noncovalent interactions involving anions? J. Phys. Chem. A 2013, 117, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Popelier, P.L.A. The QTAIM perspective of chemical bonding. In The Chemical Bond; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; p. 271. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Chernyshov, I.Y.; Ananyev, I.V.; Pidko, E.A. Revisiting van der Waals radii: From comprehensive structural analysis to knowledge-based classification of interatomic contacts. Chemphyschem 2020, 21, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.; Holl, S. Interaction in molecular crystals. Part 168. σ-donor/acceptor complexes {HCI3···XJ} (X− = Cl−, Br−, I−) of triiodomethane in tetraphenylphosphonium halides. Z. Naturforsch. B 2014, 56, 152–163. [Google Scholar] [CrossRef]
- du Mont, W.-W.; Stenzel, V.; Jeske, J.; Jones, P.G.; Sebald, A.; Pohl, S.; Saak, W.; Baetcher, M. Destructive or cooperative attack of iodide anions on alkyltriiodophosphonium cations: Elimination of iodine in solution and layer structures in the solid state. Inorg. Chem. 1994, 33, 1502–1505. [Google Scholar] [CrossRef]
- Watson, B.; Grounds, O.; Borley, W.; Rosokha, S.V. Resolving halogen vs hydrogen bonding dichotomy in solutions: Intermolecular complexes of trihalomethanes with halide and pseudohalide anions. Phys. Chem. Chem. Phys. 2018, 2, 21999–22007. [Google Scholar] [CrossRef]
- Mertsalov, D.F.; Gomila, R.M.; Zaytsev, V.P.; Grigoriev, M.S.; Nikitina, E.V.; Zubkov, F.I.; Frontera, A. On the importance of halogen bonding interactions in two x-ray structures containing all four (F, Cl, Br, I) halogen atoms. Crystals 2021, 11, 1406. [Google Scholar] [CrossRef]
- Miller, D.K.; Loy, C.; Rosokha, S.V. Examining a transition from supramolecular halogen bonding to covalent bonds: Topological analysis of electron densities and energies in the complexes of bromosubstituted electrophiles. ACS Omega 2021, 6, 23588–23597. [Google Scholar] [CrossRef]
- Bertolotti, F.; Shishkina, A.V.; Forni, A.; Gervasio, G.; Stash, A.I.; Tsirelson, V.G. Intermolecular bonding features in solid iodine. Cryst. Growth Des. 2014, 14, 3587–3595. [Google Scholar] [CrossRef]
- Isse, A.A.; Lin, C.Y.; Coote, M.L.; Gennaro, A. Estimation of standard reduction potentials of halogen atoms and alkyl halides. J. Phys. Chem. B 2011, 115, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Kennepohl, P. Catalytic activation via pi-backbonding in halogen bonds. Faraday Discuss. 2023, 244, 241–251. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | Contact | d(X···Y), Å | ∠(C-X-Y), deg | RXY a |
|---|---|---|---|---|
| 1 | C-I2+···I1− | 3.670 | 160.6 | 0.93 |
| 2 | C-I3···Cl2− | 3.317 | 171.2 | 0.89 |
| C-I1···Cl2− | 3.276 | 171.4 | 0.88 | |
| C-Cl1+···Cl2− | 3.361 | 175.7 | 0.96 | |
| C-I1···I2-C | 3.637 | 170.6 | 0.91 | |
| 3 | C-I2···I3− | 3.581 | 167.7 | 0.90 |
| C-I1···I3− | 3.652 | 163.4 | 0.92 | |
| 4 | C-I1···I3− | 3.644 | 176.5 | 0.92 |
| C-I2···I3− | 3.648 | 160.2 | 0.92 |
| Complex a | ρ(r) | ∇2ρ(r) | H(r) | G(r) | V(r) | Eint b | Eint c |
|---|---|---|---|---|---|---|---|
| DABCO-CH2I+···I− | 0.0123 | 0.0285 | 0.0006 | 0.0065 | −0.0059 | −2.7 | −2.5 |
| QN-CH2Cl+···Cl− | 0.0094 | 0.0316 | 0.0014 | 0.0065 | −0.0051 | −2.7 | −2.2 |
| CHI3···I− | 0.0129 | 0.0297 | 0.0006 | 0.0069 | −0.0063 | −2.9 | −2.7 |
| CHI3···Cl− | 0.0157 | 0.0447 | 0.0011 | 0.0101 | −0.0090 | −4.3 | −3.9 |
| CHI3···CHI3 | 0.0122 | 0.0311 | 0.0008 | 0.0070 | −0.0063 | −3.0 | −2.7 |
| Gas Phase | Dichloromethane | Acetonitrile | ||||
|---|---|---|---|---|---|---|
| Complex | dX···Y, Å | ΔE, kcal/mol | dX···Y, Å | ΔE, kcal/mol | dX···Y, Å | ΔE, kcal/mol |
| DABCO-CH2I+···I− | 3.006 | −78.6 | 3.506 | −10.4 | 3.596 | −5.2 |
| QN-CH2Cl+···Cl− | N/A a | −95.3 | 3.216 | −6.7 | 3.324 | −2.6 |
| CHI3···I− | 3.166 | −18.3 | 3.513 | −4.6 | 3.557 | −3.9 |
| CHI3···Cl− | 2.733 | −28.0 | 3.057 | −6.8 | 3.108 | −5.7 |
| CHI3···CHI3 | 3.757 | −2.0 | 3.790 | −1.9 | 3.754 | −1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grounds, O.; Zeller, M.; Rosokha, S.V. Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines. Crystals 2024, 14, 124. https://doi.org/10.3390/cryst14020124
Grounds O, Zeller M, Rosokha SV. Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines. Crystals. 2024; 14(2):124. https://doi.org/10.3390/cryst14020124
Chicago/Turabian StyleGrounds, Olivia, Matthias Zeller, and Sergiy V. Rosokha. 2024. "Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines" Crystals 14, no. 2: 124. https://doi.org/10.3390/cryst14020124
APA StyleGrounds, O., Zeller, M., & Rosokha, S. V. (2024). Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines. Crystals, 14(2), 124. https://doi.org/10.3390/cryst14020124