
The Adhesive Properties of Coherent and Semicoherent NiAl/V Interfaces Within the Peierls-Nabarro Model

Abstract
:1. Introduction
2. Details of the Calculations
3. Bulk Properties
4. Surface Properties
5. Coherent Interfaces
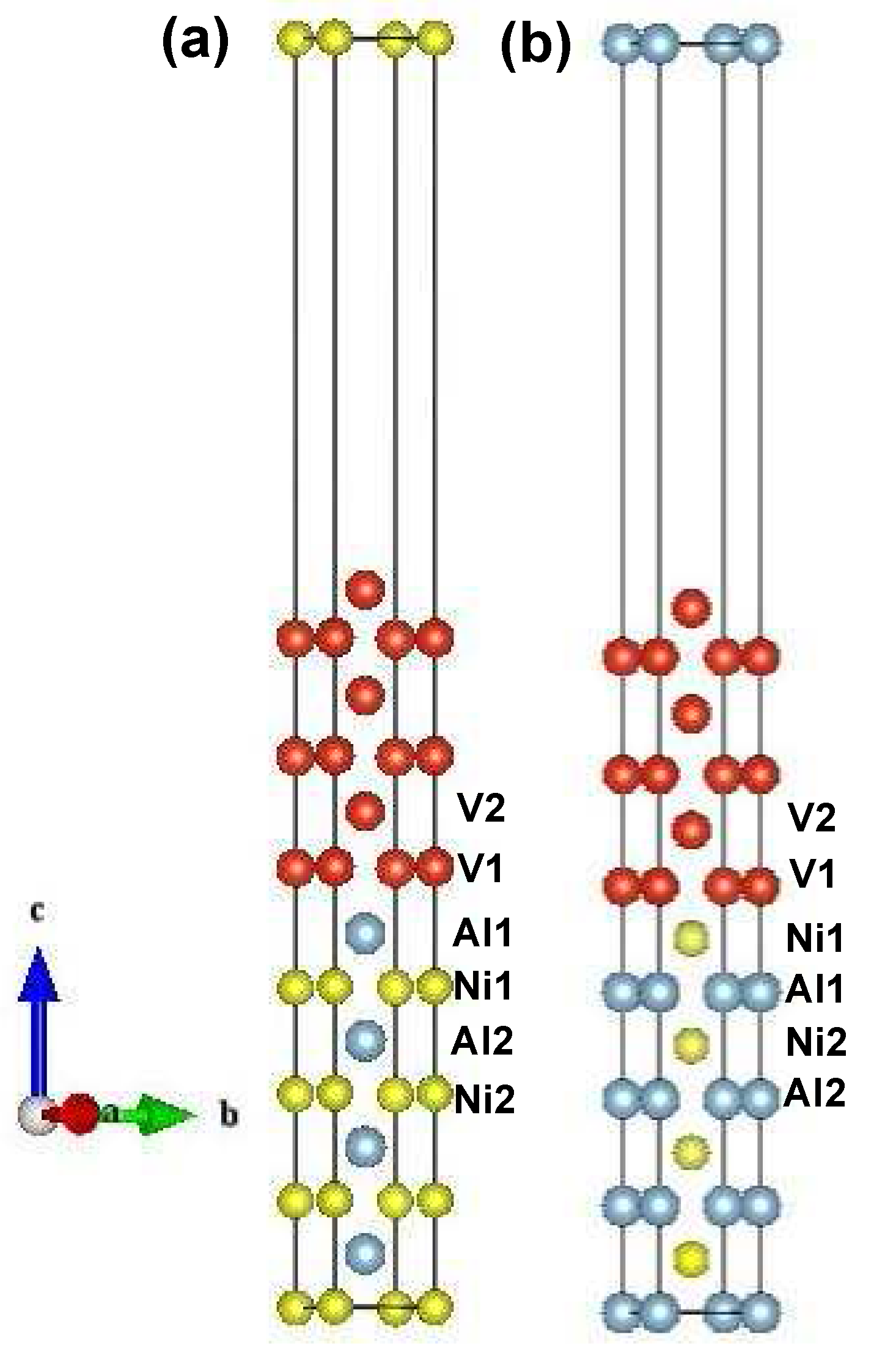
5.1. Interface Geometry
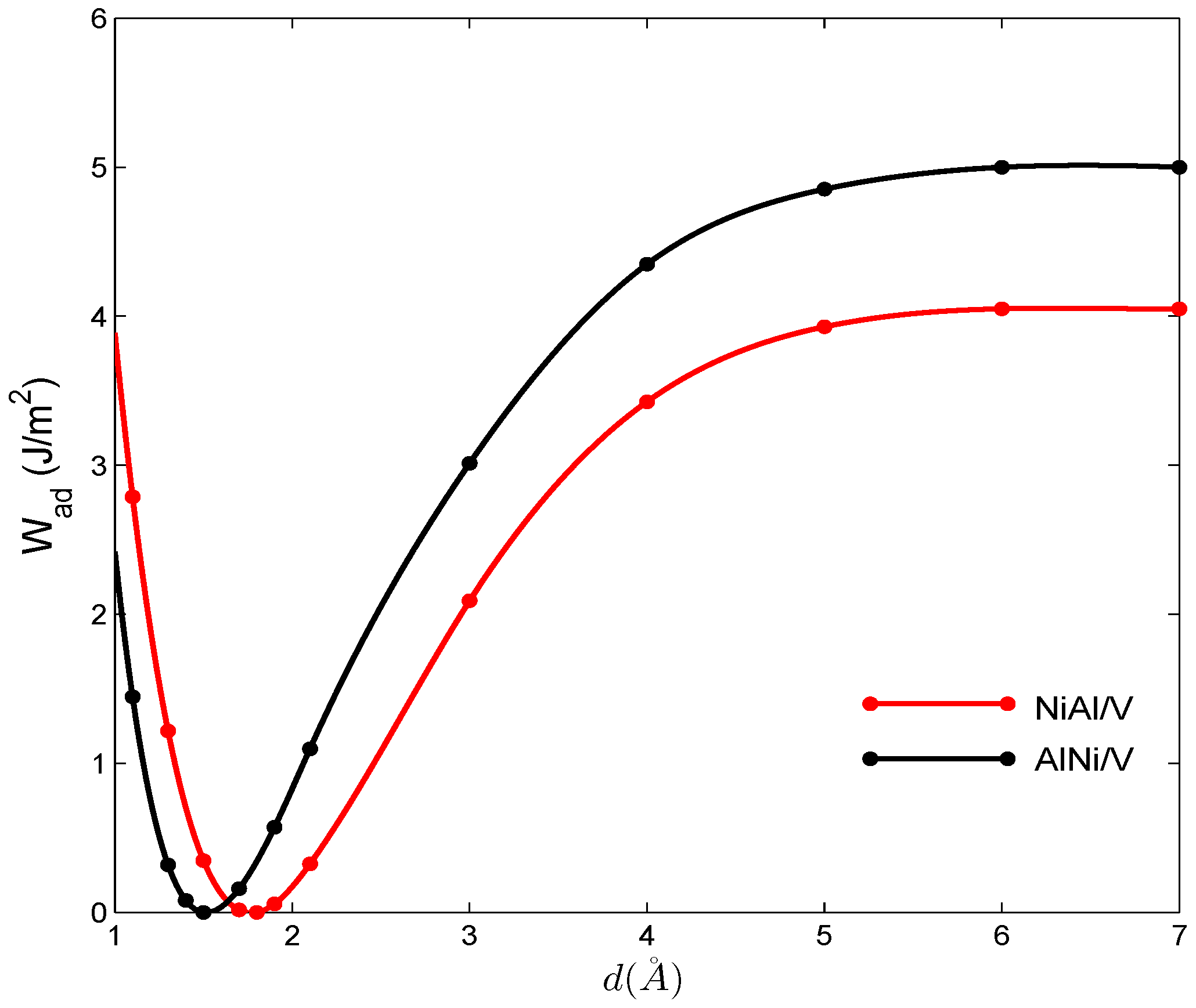
5.2. Work of Adhesion
5.3. Interface Energy
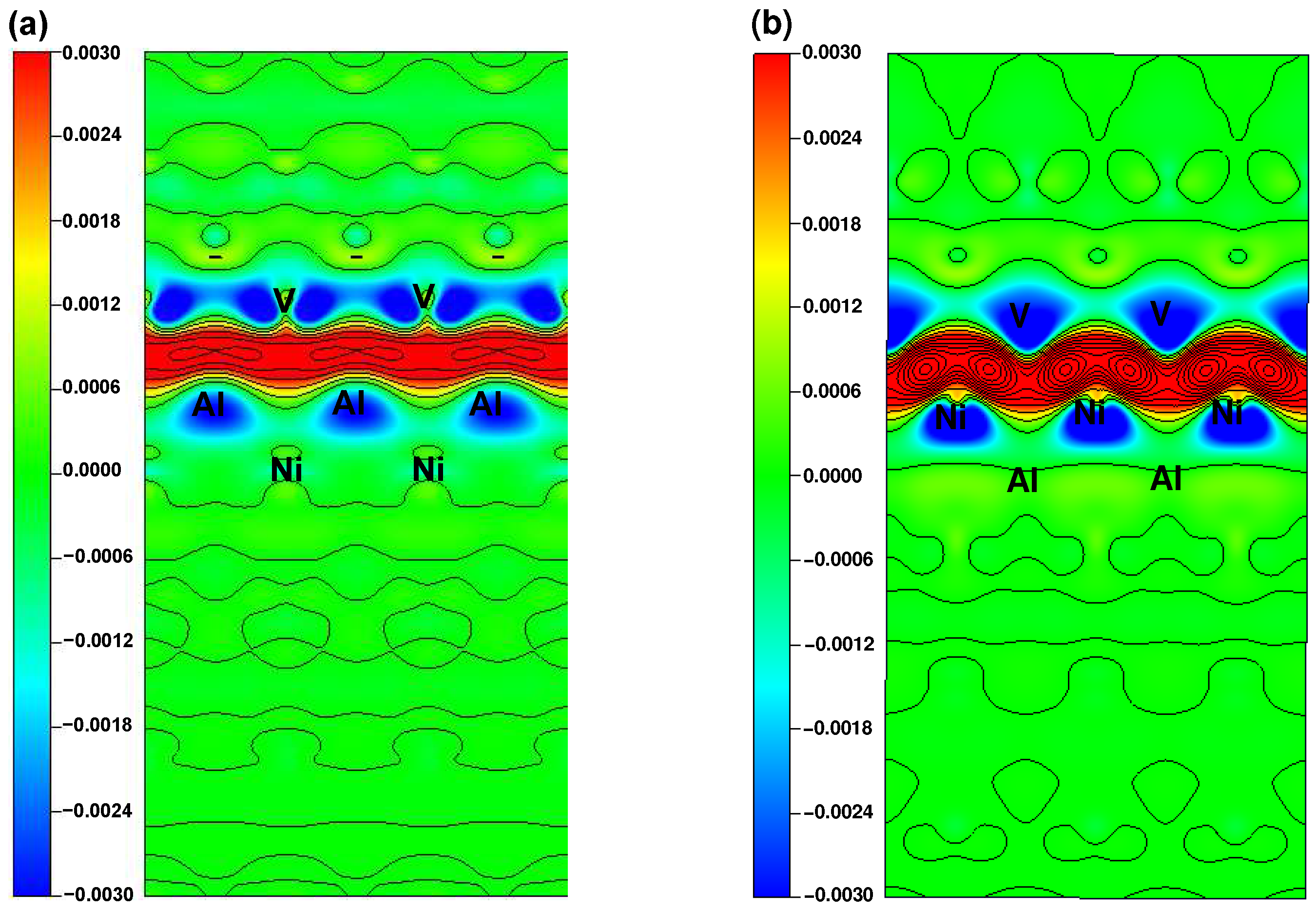
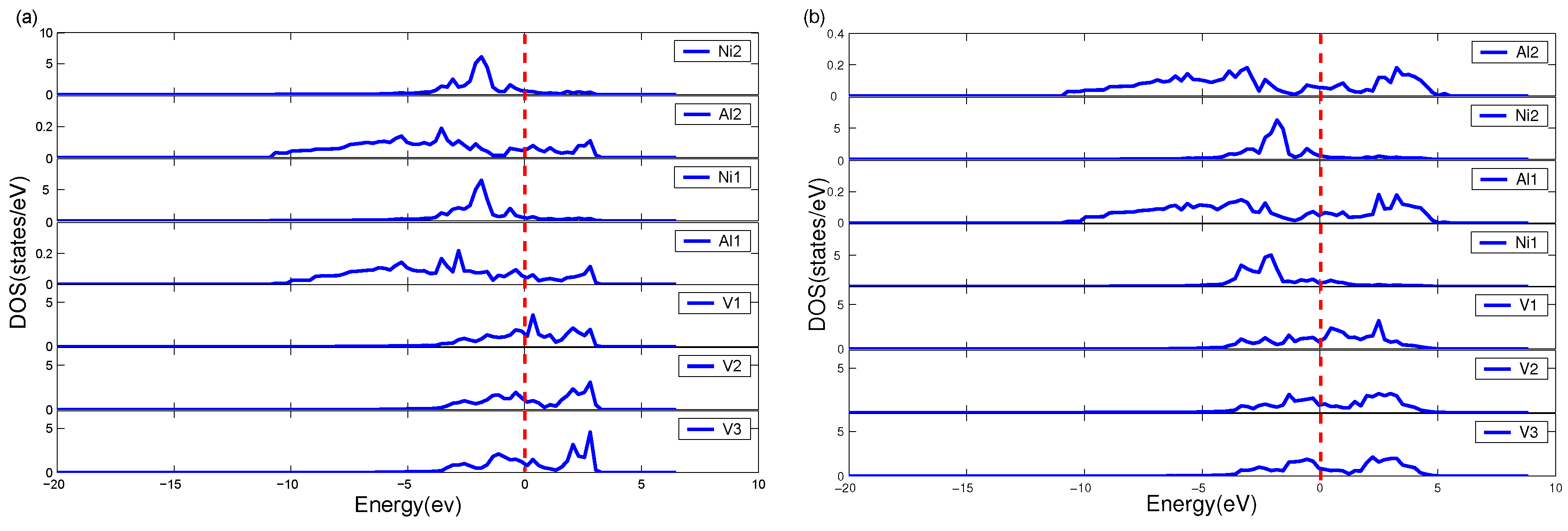
5.4. Electronic Characteristics
6. Semicoherent Interfaces
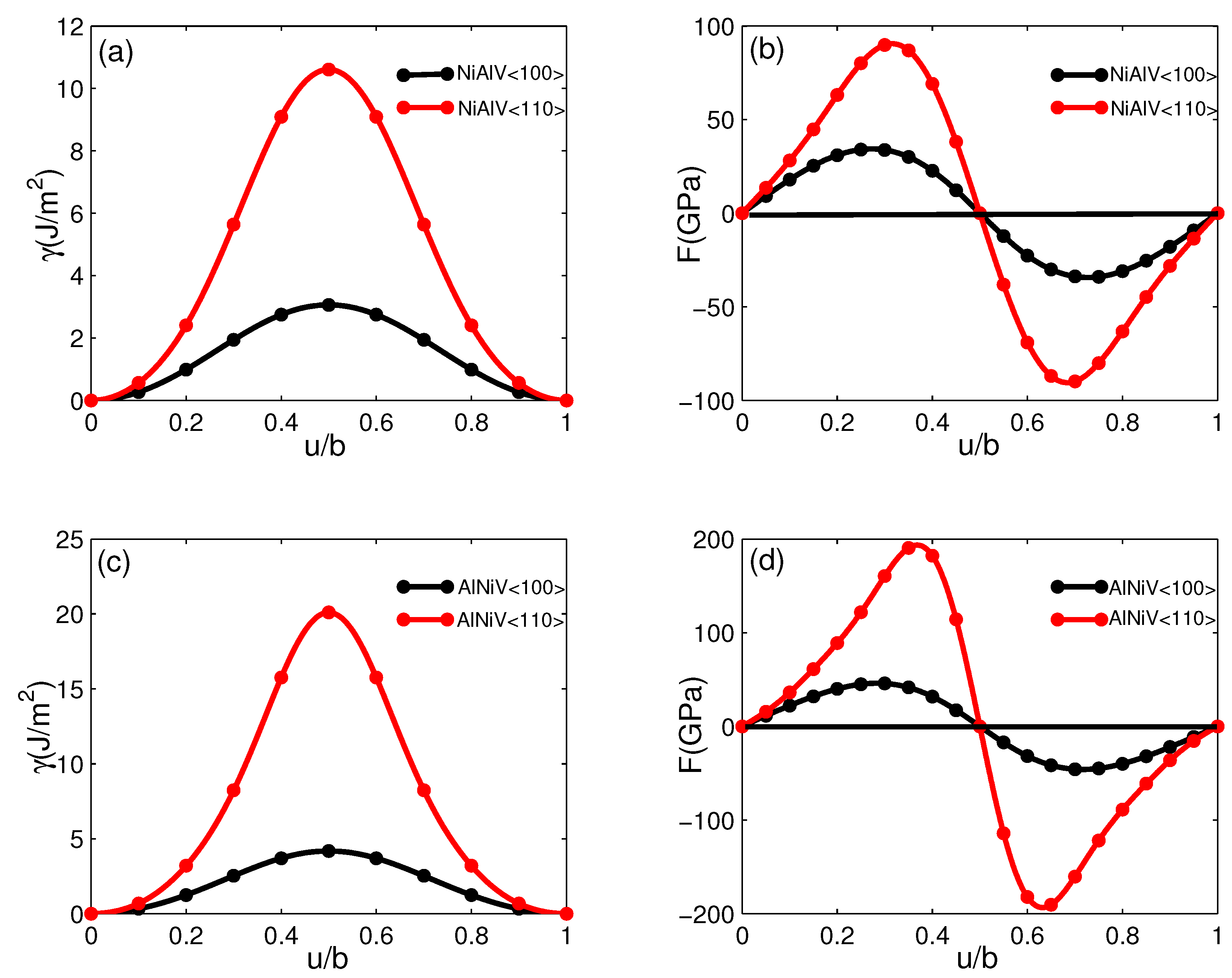
6.1. Generalized Stacking Fault Energy of the NiAlV and AlNiV Interfaces
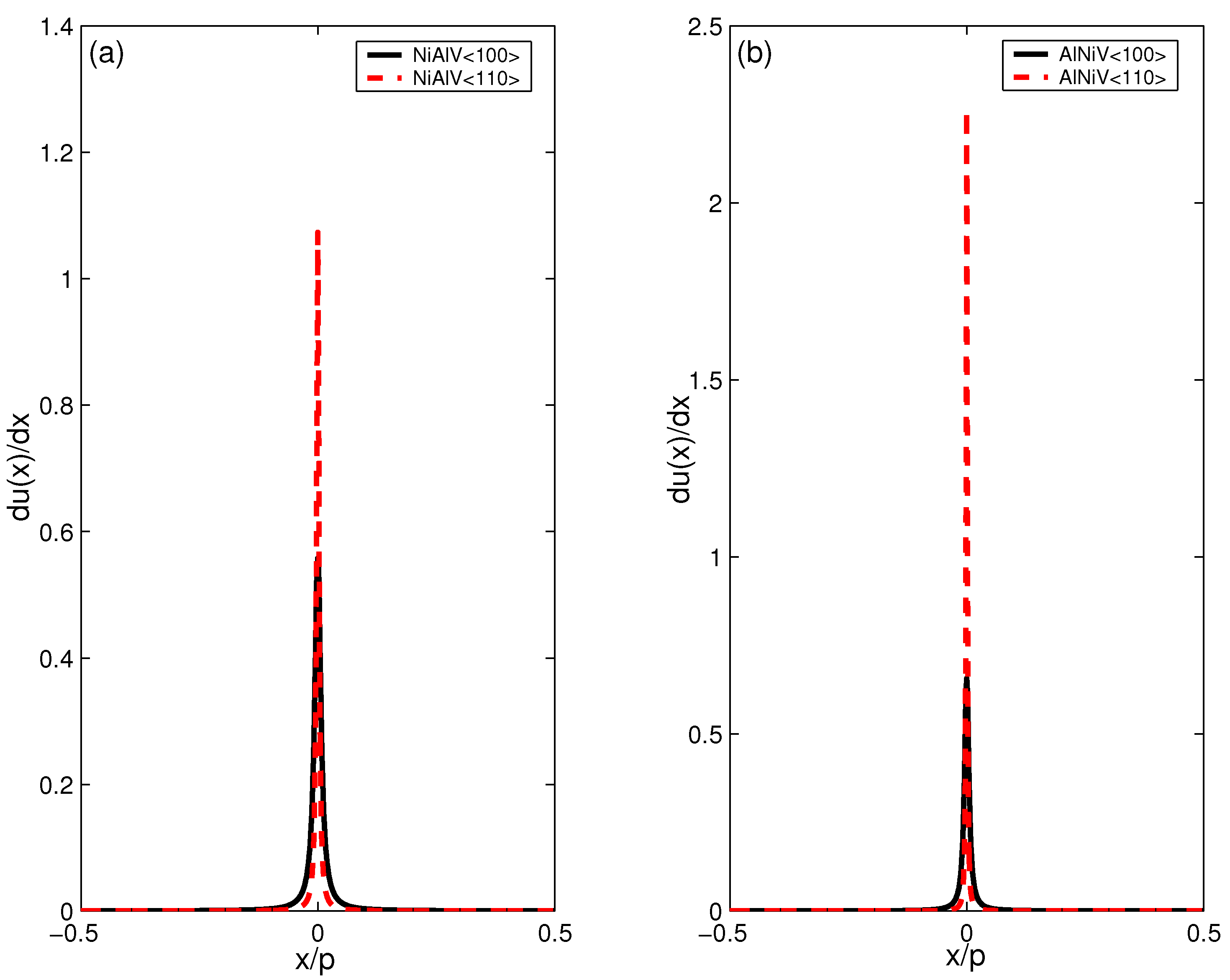
6.2. Core Structure and Energy of Misfit Dislocation
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baker, I. A review of the mechanical properties of B2 compounds. Mater. Sci. Eng. A 1995, 192, 1–13. [Google Scholar] [CrossRef]
- Sivakumar, R.; Mordike, B.L. High temperature coatings for gas turbine blades: A review. Surf. Coat. Technol. 1989, 37, 139–160. [Google Scholar] [CrossRef]
- Medvedeva, N.I.; Mryasov, O.N.; Gornostyrev, Y.N.; Novikov, D.L.; Freeman, A.J. First-principles total-energy calculations for planar shear and cleavage decohesion processes in B2-ordered NiAl and FeAl. Phys. Rev. B 1996, 54, 13506. [Google Scholar] [CrossRef]
- Grahle, P.; Arzt, E. Microstructural development in dispersion strengthened NiAl produced by mechanical alloying and secondary recrystallization. Acta Mater. 1997, 45, 201–211. [Google Scholar] [CrossRef]
- Miracle, D.B. Overview No. 104 The physical and mechanical properties of NiAl. Acta Metall. Mater. 1993, 41, 649–684. [Google Scholar] [CrossRef]
- Amouyal, Y.; Rabkin, E.; Mishin, Y. Correlation between grain boundary energy and geometry in Ni-rich NiAl. Acta Mater. 2005, 53, 3795–3805. [Google Scholar] [CrossRef]
- Joslin, S.M.; Chen, X.F.; Oliver, B.F.; Noebe, R.D. Fracture behavior of directionally solidified NiAl-Mo and NiAl-V eutectics. Mater. Sci. Eng. A 1995, 196, 9–18. [Google Scholar] [CrossRef]
- Hassel, A.W.; Smith, A.J.; Milenkovic, S. Nanostructures from directionally solidified NiAl-W eutectics. Electrochim. Acta 2006, 52, 1799–1804. [Google Scholar] [CrossRef]
- Milenkovic, S.; Caram, R. Microstructure of the microalloyed NiAl-V eutectics. Mater. Lett. 2002, 55, 126–131. [Google Scholar] [CrossRef]
- Subramanian, P.R.; Mendiratta, M.G.; Miracle, D.B. Microstructures and mechanical behavior of NiAl-Mo and NiAl-Mo-Ti two-phase alloys. Metall. Mater. Trans. A 1994, 25, 2769–2781. [Google Scholar] [CrossRef]
- Yang, J.M.; Jeng, S.M.; Bain, K.; Amato, R.A. Microstructure and mechanical behavior of in-situ directional solidified NiAl/Cr (Mo) eutectic composite. Acta Mater. 1997, 45, 295–308. [Google Scholar] [CrossRef]
- Milenkovic, S.; Coelho, A.A.; Caram, R. Directional solidification processing of eutectic alloys in the Ni-Al-V system. J. Cryst. Growth 2000, 211, 485–490. [Google Scholar] [CrossRef]
- Hua, K.; Guo, J.; Ren, Z.; Gao, Q.; Yang, R. Effect of Nb on the microstructure and mechanical properties of cast NiAl-Cr (Mo) eutectic alloy. J. Mater. Sci. Technol. 2006, 22, 164–168. [Google Scholar]
- Cui, C.Y.; Guo, J.T.; Qi, Y.H.; Ye, H.Q. Deformation behavior and microstructure of DS NiAl/Cr (Mo) alloy containing Hf. Intermetallics 2002, 10, 1001–1009. [Google Scholar] [CrossRef]
- Whittenberger, J.D.; Noebe, R.D.; Joslin, S.M.; Oliver, B.F. Elevated temperature compressive slow strain rate properties of several directionally solidified NiAl-(Nb,Mo) alloys. Intermetallics 1999, 7, 627–633. [Google Scholar] [CrossRef]
- Zeumert, B.; Sauthoff, G. Intermetallic NiAl-Ta alloys with strengthening Laves phase for high-temperature applications. I. Basic properties. Intermetallics 1997, 5, 563–577. [Google Scholar] [CrossRef]
- Milenkovic, S.; Caram, R. Mechanical properties and fracture behavior of directionally solidified NiAl-V eutectic composites. Metall. Mater. Trans. A 2015, 46, 557–565. [Google Scholar] [CrossRef]
- Choudhury, S.; Agular, J.A.; Fluss, M.J.; Hsiung, L.L.; Misra, A.; Uberuaga, B.P. Non-uniform solute segregation at semi-coherent metal/oxide interfaces. Sci. Rep. 2015, 5, 13086. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Morgan, D.; Uberuaga, B.P. Massive interfacial reconstruction at misfit dislocations in metal/oxide interfaces. Sci. Rep. 2014, 5, 6533. [Google Scholar] [CrossRef] [PubMed]
- Benedek, R.; Alavi, A.; Seidman, D.N.; Yang, L.H.; Muller, D.A.; Woodward, C. First principles simulation of a ceramic metal interface with misfit. Phys. Rev. Lett. 2000, 84, 3362. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Hu, Q.M.; Punkkinen, M.P.J.; Johansson, B.; Vitos, L. First-principles study of fcc-Ag/bcc-Fe interfaces. Phys. Rev. B 2013, 87, 22104. [Google Scholar] [CrossRef]
- Benedek, R.; Seidman, D.N.; Minkoff, M.; Yang, L.H.; Alavi, A. Atomic and electronic structure and interatomic potentials at a polar ceramic/metal interface: {222} MgO/Cu. Phys. Rev. B 1999, 60, 16094. [Google Scholar] [CrossRef]
- Arya, A.; Carter, E.A. Structure, bonding, and adhesion at the TiC (100)/Fe (110) interface from first principles. J. Chem. Phys. 2003, 118, 8982–8996. [Google Scholar] [CrossRef]
- Mu˜oz, M.C.; Gallego, S.; Beltr´n, J.I.; Cerd´, J. Adhesion at metal-ZrO2 interfaces. Sur. Sci. Rep. 2006, 61, 303–344. [Google Scholar]
- Shang, J.X.; Guan, K.; Wang, F.H. Atomic structure and adhesion of the Nb(001)/α-Nb5Si3(001) interface: A first-principles study. J. Phys. Condens. Matter 2010, 22, 085004. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Cho, M.; Zhou, M. Ab initio study of the fracture energy of LiFePO4/FePO4 interfaces. J. Power Sorces 2013, 243, 706–714. [Google Scholar] [CrossRef]
- Li, J.; Qi, Y.N.; Zhang, M.; Zhou, Y.; Li, X. First-principle study of adhesion, wetting and bonding on Al/Al3V (001) interface. Surf. Sci. 2014, 624, 1–7. [Google Scholar] [CrossRef]
- Dong, N.; Zhang, C.; Liu, H.; Fan, G.; Fang, X.D.; Han, P. Effects of different alloying additives X (X= Si, Al, V, Ti, Mo, W, Nb, Y) on the adhesive behavior of Fe/Cr2O3 interfaces: A first-principles study. Comput. Mater. Sci. 2015, 109, 293–299. [Google Scholar] [CrossRef]
- Junkaew, A.; Ham, B.; Zhang, R.X. Arro´yave, Investigation of interfaces in Mg/Nb multilayer thin films. Comput. Mater. Sci. 2015, 108, 212–225. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, Y.H.; Wang, H.; Guo, J.M.; Singh, A.; Fu, C.Y. Interfacial stability, electronic structure and bond characteristics of Pt3Zr(111)/Pt (111) interfaces: A first-principles study. Comput. Mater. Sci. 2016, 111, 74–78. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.-K.; Chen, L.-Q.; Wolverton, C. First-principles calculations of β′′-Mg5Si6/α-Al interfaces. Acta Mater. 2007, 55, 5934–5937. [Google Scholar]
- Finns, M.W. The theory of metal-ceramic interfaces. J. Phys. Condens. Matter 1996, 8, 5811–5836. [Google Scholar] [CrossRef]
- Joos, B.; Duesbery, M.S. The Peierls stress of dislocations: An analytic formula. Phys. Rev. Lett. 1997, 78, 266. [Google Scholar] [CrossRef]
- Schoeck, G. The core structure, recombination energy and Peierls energy for dislocations in Al. Philos. Mag. A 2001, 81, 1161–1176. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, T.C.; Wang, C.Y. Peierls-Nabarro model of interfacial misfit dislocation: An analytic solution. Phys. Rev. B 1999, 59, 8232. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, T.C. The modified Peierls-Nabarro model of interfacial misfit dislocation. Acta Mater. 1999, 47, 3063–3068. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y. The two-dimensional Peierls-Nabarro model for interfacial misfit dislocation networks of cubic lattice. Eur. Phys. J. B 2007, 55, 355–362. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y. The multiscale model combining elastic theory with ab initio calculations for metal-ceramic interfaces. Mod. Phys. Lett. B 2008, 22, 3135–3143. [Google Scholar] [CrossRef]
- Johansson, S.A.E.; Christensen, M.; Wahnstrom, G. Interface energy of semicoherent metal-ceramic interfaces. Phys. Rev. Lett. 2005, 95, 226108. [Google Scholar] [CrossRef] [PubMed]
- Fors, D.H.R.; Johansson, S.A.E.; Petisme, M.V.G.; Wahnstrom, G. Theoretical investigation of moderate misfit and interface energetics in the Fe/VN system. Comput. Mater. Sci. 2010, 50, 550–559. [Google Scholar] [CrossRef]
- Medvedeva, N.I.; Gornostyrev, Y.N.; Kontsevoi, O.Y.; Freeman, A.J. Ab-initio study of interfacial strength and misfit dislocations in eutectic composites: NiAl/Mo. Acta Mater. 2004, 52, 675–682. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J.; Hafner, J. Ab initio force constant approach to phonon dispersion relations of diamond and graphite. Phys. Rev. B 1995, 32, 729. [Google Scholar] [CrossRef]
- Bl˙chl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Lazar, P.; Podloucky, R. Ab initio study of the mechanical properties of NiAl microalloyed by X= Cr, Mo, Ti, Ga. Phys. Rev. B 2006, 73, 104114. [Google Scholar] [CrossRef]
- Ponomareva, A.V.; Isaev, E.I.; Vekilov, Y.K.; Abrikosov, I.A. Site preference and effect of alloying on elastic properties of ternary B2 NiAl-based alloys. Phys. Rev. B 2012, 85, 144117. [Google Scholar] [CrossRef]
- Rai, R.C.; Hemkar, M.P. Crystal equilibrium and lattice dynamics of chromium and vanadium. J. Phys. F Met. Phys. 1978, 8, 45. [Google Scholar] [CrossRef]
- Bolef, D.I. Elastic constants of single crystals of the bcc transition elements V, Nb, and Ta. J. Appl. Phys. 1961, 32, 100. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.C.; Zheng, W.T.; Jiang, Q. NiAl(110)/Cr(110) interface: A density functional theory study. Phys. Rev. B 2006, 73, 205421. [Google Scholar] [CrossRef]
- Fiorentini, V.; Methfessel, M. Extracting convergent surface energies from slab calculations. J. Phys. Condens. Matter 1996, 8, 6525. [Google Scholar] [CrossRef]
- Zhang, J.M.; Ma, F.; Xu, K.W. Calculation of the surface energy of bcc metals by using the modified embedded-atom method. Surf. Interface Anal. 2003, 34, 662–666. [Google Scholar] [CrossRef]
- Neugebauer, J.; Scheffler, M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al (111). Phys. Rev. B 1992, 46, 16067. [Google Scholar] [CrossRef]
- Liu, L.M.; Wang, S.Q.; Ye, H.Q. First-principles study of polar Al/TiN (111) interfaces. Acta Mater 2004, 52, 3681–3688. [Google Scholar] [CrossRef]
- Siegel, D.J.; Hector, L.G.; Adams, J.B. Adhesion, atomic structure, and bonding at the Al(111)/α-Al2O3 (0001) interface: A first principles study. Phys. Rev. B 2002, 65, 085415. [Google Scholar] [CrossRef]
- Brown, J.A.; Mishin, Y. Monte Carlo modeling of low-index surfaces in stoichiometric and Ni-rich NiAl. Phys. Rev. B 2003, 67, 195414. [Google Scholar] [CrossRef]
- Kriese, M.D.; Moody, N.R.; Gerberich, W.W. Effects of annealing and interlayers on the adhesion energy of copper thin films to SiO2/Si substrates. Acta Mater. 1998, 46, 6623–6630. [Google Scholar] [CrossRef]
- Raynolds, J.E.; Smith, J.R.; Zhao, G.L.; Srolovitz, D.J. Adhesion in NiAl-Cr from first principles. Phys. Rev. B 1996, 53, 13883. [Google Scholar] [CrossRef]
- Christensen, M.; Dudiy, S.; Wahnstr¨m, G. First-principles simulations of metal-ceramic interface adhesion: Co/WC versus Co/TiC. Phys. Rev. B 2002, 65, 045408. [Google Scholar] [CrossRef]
- Suo, Z.; Hutchinson, J.W. Interface crack between two elastic layers. Int. J. Fract. 1990, 43, 1–18. [Google Scholar] [CrossRef]
- Vitek, V. Intrinsic stacking faults in body-centred cubic crystals. Philos. Mag. 1968, 18, 773–786. [Google Scholar] [CrossRef]
- Wang, C.; Wang, C.Y. Density functional theory study of Ni/Ni3Al interface alloying with Re and Ru. Surf. Sci. 2008, 602, 2604–2609. [Google Scholar] [CrossRef]
- De Hosson1, J.T.M.; Groen, H.B.; Kooi, B.J.; Vitek, V. Metal-ceramic interfaces studied with high-resolution transmission electron microscopy. Acta Mater. 1999, 47, 4077–4092. [Google Scholar] [CrossRef]
- Vellinga, W.P.; De Hosson, J.T.M.; Vitek, V. Misfit dislocations: An atomistic and elastic continuum approach. Acta Mater. 1997, 45, 1525–1534. [Google Scholar] [CrossRef]
- Vellinga, W.P.; De Hosson, J.T.M. Atomic structure and orientation relation of interfaces between Ag and ZnO. Acta Mater. 1997, 45, 933–950. [Google Scholar] [CrossRef]
- Trampert, A.; Ernst, F.; Flynn, C.P.; Fischmeister, H.F.; Ruhle, M. High resolution transmission electron microscopy studies of the Ag/MgO interface. Acta Metall. Mater. 1992, 40, S227–S236. [Google Scholar] [CrossRef]
- Schnitker, J.; Srolovitz, D.J. Misfit effects in adhesion calculations. Model. Simul. Mater. Eng. 1998, 6, 153–164. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | a(Å) | (GPa) | (GPa) | (GPa) | G (GPa) | B (GPa) | E (GPa) | ν |
|---|---|---|---|---|---|---|---|---|
| NiAl | 2.89 | 205.1 | 136.1 | 116.9 | 83.9 | 159.1 | 214.2 | 0.40 |
| [47] | 2.89 | 203 | 140 | 113 | 80.4 | 161.0 | 206.8 | 0.41 |
| [48] | 2.89 | 233 | 121 | 114 | 85 | 159 | 218 | 0.34 |
| V | 3.01 | 253.7 | 134.7 | 19.9 | 35.7 | 174.3 | 100.4 | 0.35 |
| [49] | 3.04 | 228.0 | 118.8 | 42.6 | 47.4 | 155.2 | 129.0 | 0.34 |
| [50] | – | 228 | 119 | 42.6 | 47.4 | 155.3 | 129.0 | 0.34 |
| Systems | Symbol | Atomic Position | ||
|---|---|---|---|---|
| x | y | z | ||
| NiAl (Al-terminated) | Al1 | 0.0000 | 0.0000 | 0.0018 |
| Ni1 | 0.5000 | 0.5000 | 0.0743 | |
| Al2 | 0.0000 | 0.0000 | 0.1536 | |
| Ni2 | 0.5000 | 0.5000 | 0.2308 | |
| AlNi (Ni-terminated) | Ni1 | 0.0000 | 0.0000 | 0.0042 |
| Al1 | 0.5000 | 0.5000 | 0.0730 | |
| Ni2 | 0.0000 | 0.0000 | 0.1548 | |
| Al2 | 0.5000 | 0.5000 | 0.2308 | |
| V | V1 | 0.0000 | 0.0000 | 0.0108 |
| V2 | 0.5000 | 0.5000 | 0.0770 | |
| V3 | 0.0000 | 0.0000 | 0.1539 | |
| V4 | 0.5000 | 0.5000 | 0.2308 | |
| Systems | Interlayer | Atom Layers of Slab | ||||
|---|---|---|---|---|---|---|
| 3 | 5 | 7 | 9 | 11 | ||
| NiAl (Al terminated) | −1.4% | −6.6% | −5.7% | −6.4% | −7.0% | |
| – | 4.2% | 3.1% | 3.6% | 4.1% | ||
| – | – | 0.3% | 1.1% | 1.0% | ||
| – | – | – | −1.3% | −0.5% | ||
| – | – | – | – | −0.3% | ||
| AlNi (Ni terminated) | −5.3% | −10.0% | −10.7% | −10.3% | −10.9% | |
| – | 4.8% | 6.3% | 7.0% | 6.1% | ||
| – | – | −1.2% | −2.8% | −1.9% | ||
| – | – | – | 0.7% | 1.2% | ||
| – | – | – | – | 0.0% | ||
| V | −13.6% | −15.2% | −13.9% | −14.2% | −14.3% | |
| – | 0.7% | 0.2% | −0.7% | −0.2% | ||
| – | – | 0.3% | 1.9% | 2.0% | ||
| – | – | – | −1.8% | −3.0% | ||
| – | – | – | – | 0.7% | ||
| Number of Layers; n | ||||||
|---|---|---|---|---|---|---|
| Stoichiometric | Non-Stoichiometric | Stoichiometric | Non-Stoichiometric | |||
| 3 | – | – | – | |||
| 4 | 2.27 | – | 2.27 | – | 2.61 | |
| 5 | – | – | – | |||
| 6 | 2.31 | – | 2.31 | – | 2.36 | |
| 7 | – | – | – | |||
| 8 | 2.30 | – | 2.30 | – | 2.36 | |
| 9 | – | – | – | |||
| 10 | 2.30 | – | 2.30 | – | 2.36 | |
| 11 | – | – | – | |||
| Interfaces | Unrelaxed | Relaxed | |||||
|---|---|---|---|---|---|---|---|
| NiAlV | 1.769 | 4.09 | 1.775 | 4.03 | |||
| AlNiV | 1.515 | 5.01 | 1.506 | 4.85 | |||
| Misfit Dislocations | b | p | a | β | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NiAlV | 3.06 | 2.89 | 72.5 | 0.029 | 0.048 | 1.83 | 0.75 | 2.57 | ||
| NiAlV | 10.60 | 4.09 | 102.5 | 0.010 | 0.018 | 3.63 | 1.07 | 4.70 | ||
| AlNiV | 4.18 | 2.89 | 72.5 | 0.021 | 0.035 | 2.06 | 0.75 | 2.81 | ||
| AlNiV | 20.10 | 4.09 | 102.5 | 0.005 | 0.008 | 4.43 | 1.08 | 5.51 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linghu, Y.; Wu, X.; Wang, R.; Li, W.; Liu, Q. The Adhesive Properties of Coherent and Semicoherent NiAl/V Interfaces Within the Peierls-Nabarro Model. Crystals 2016, 6, 32. https://doi.org/10.3390/cryst6040032
Linghu Y, Wu X, Wang R, Li W, Liu Q. The Adhesive Properties of Coherent and Semicoherent NiAl/V Interfaces Within the Peierls-Nabarro Model. Crystals. 2016; 6(4):32. https://doi.org/10.3390/cryst6040032
Chicago/Turabian StyleLinghu, Yaoyao, Xiaozhi Wu, Rui Wang, Weiguo Li, and Qing Liu. 2016. "The Adhesive Properties of Coherent and Semicoherent NiAl/V Interfaces Within the Peierls-Nabarro Model" Crystals 6, no. 4: 32. https://doi.org/10.3390/cryst6040032
APA StyleLinghu, Y., Wu, X., Wang, R., Li, W., & Liu, Q. (2016). The Adhesive Properties of Coherent and Semicoherent NiAl/V Interfaces Within the Peierls-Nabarro Model. Crystals, 6(4), 32. https://doi.org/10.3390/cryst6040032