
New Acetylenic Amine Derivatives of 5,8-Quinolinediones: Synthesis, Crystal Structure and Antiproliferative Activity

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
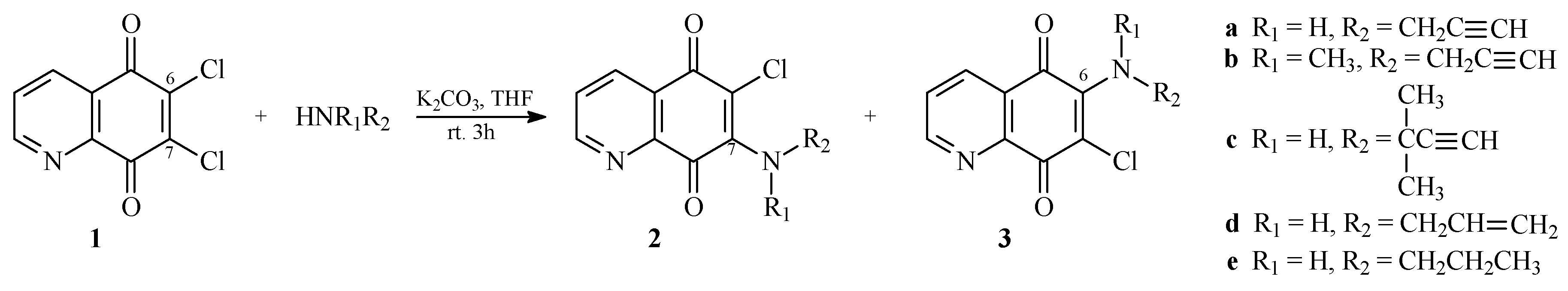
2.1. Chemistry
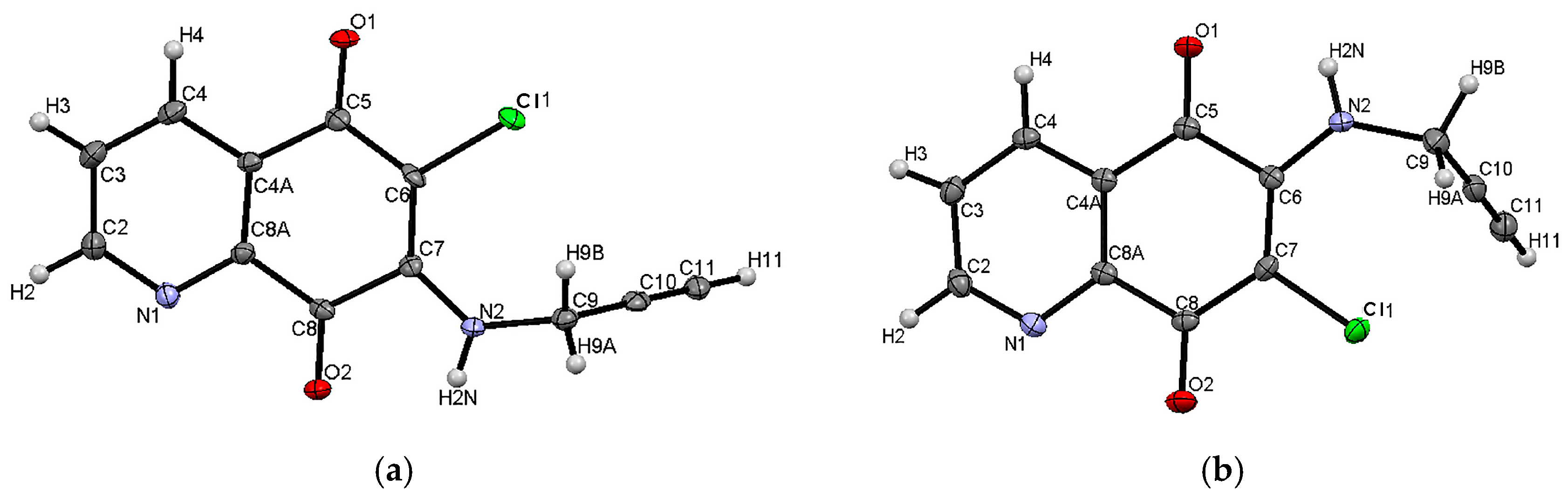
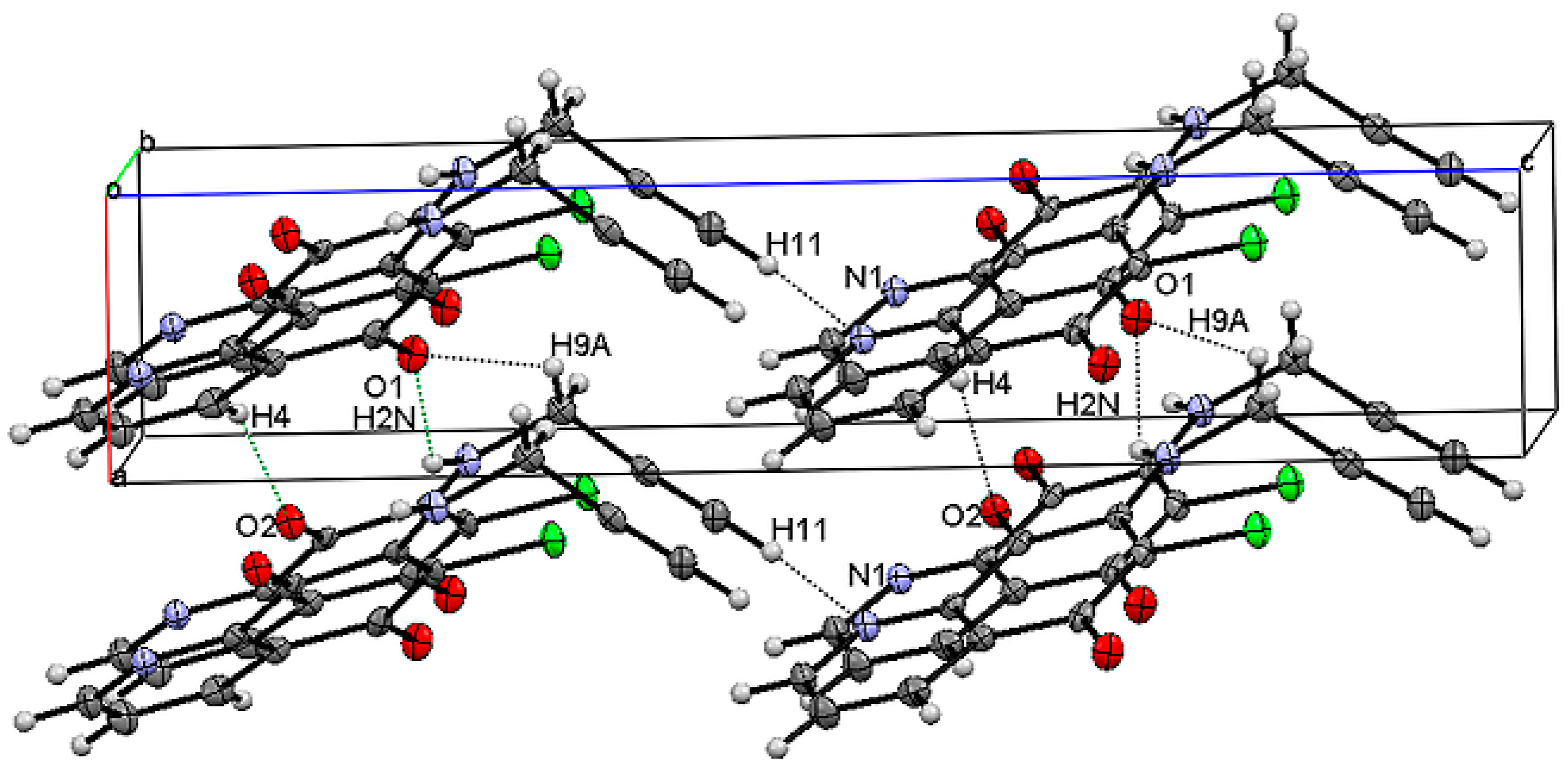
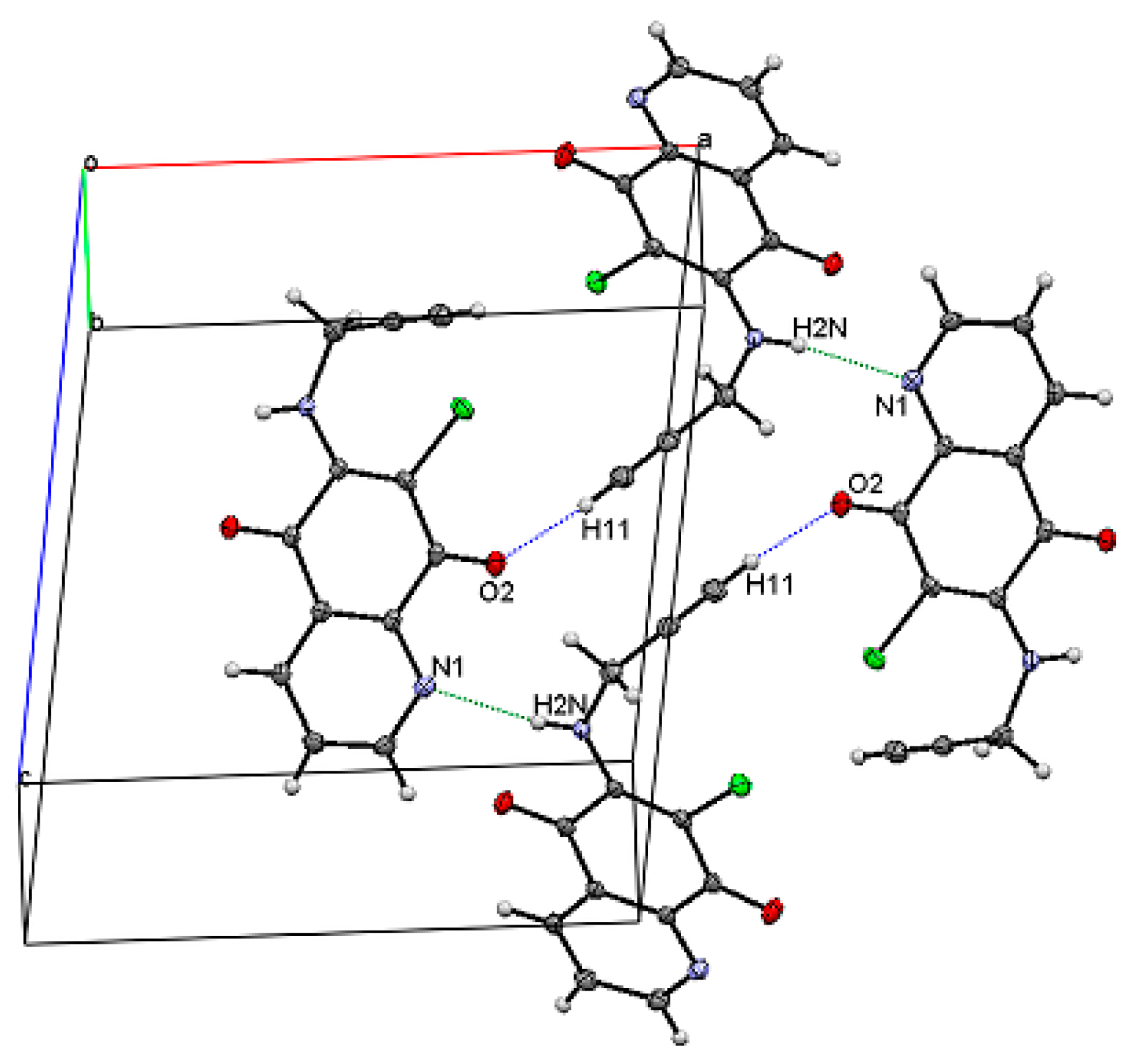
2.2. Crystal Structure and Formation of Hydrogen Bonds
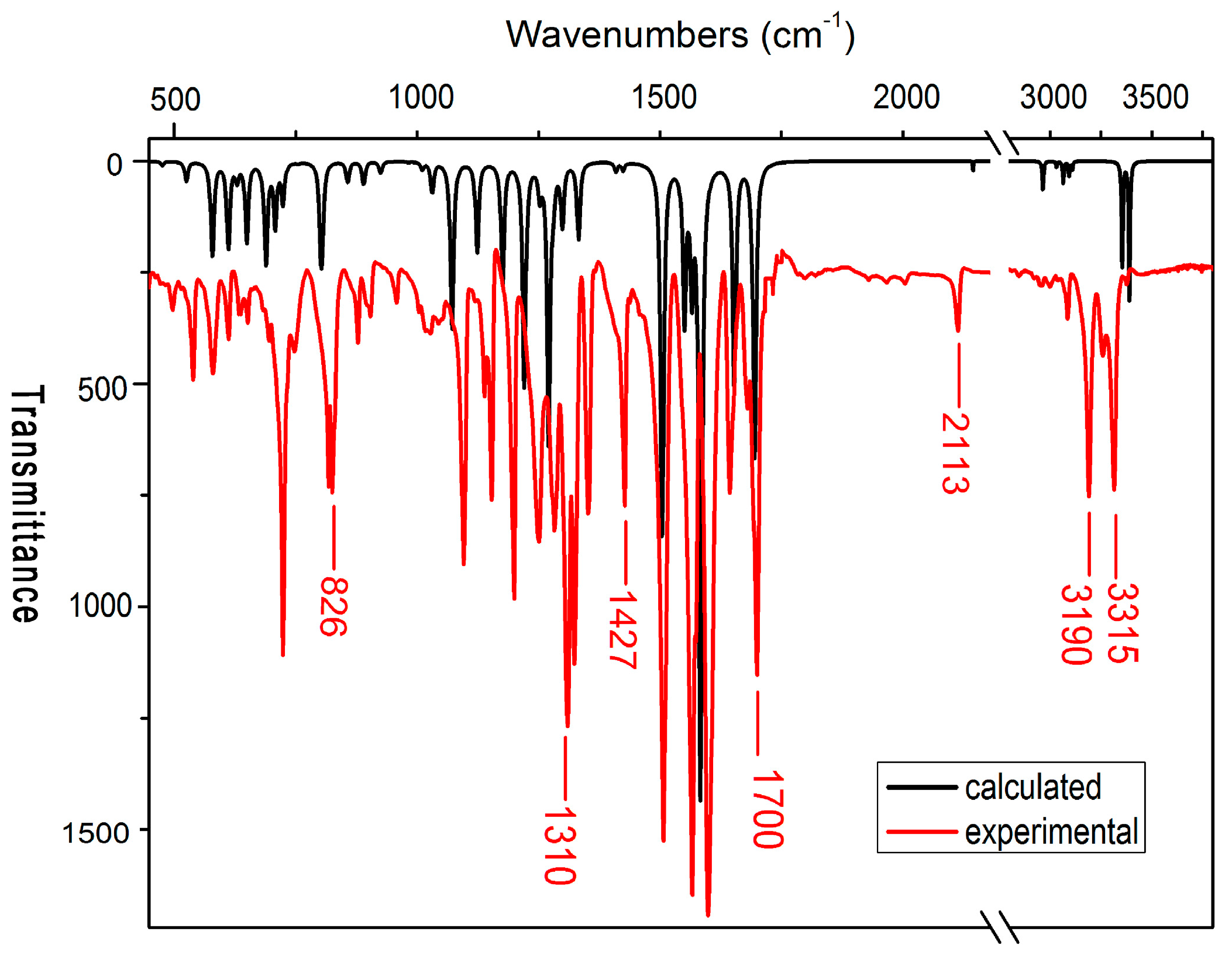
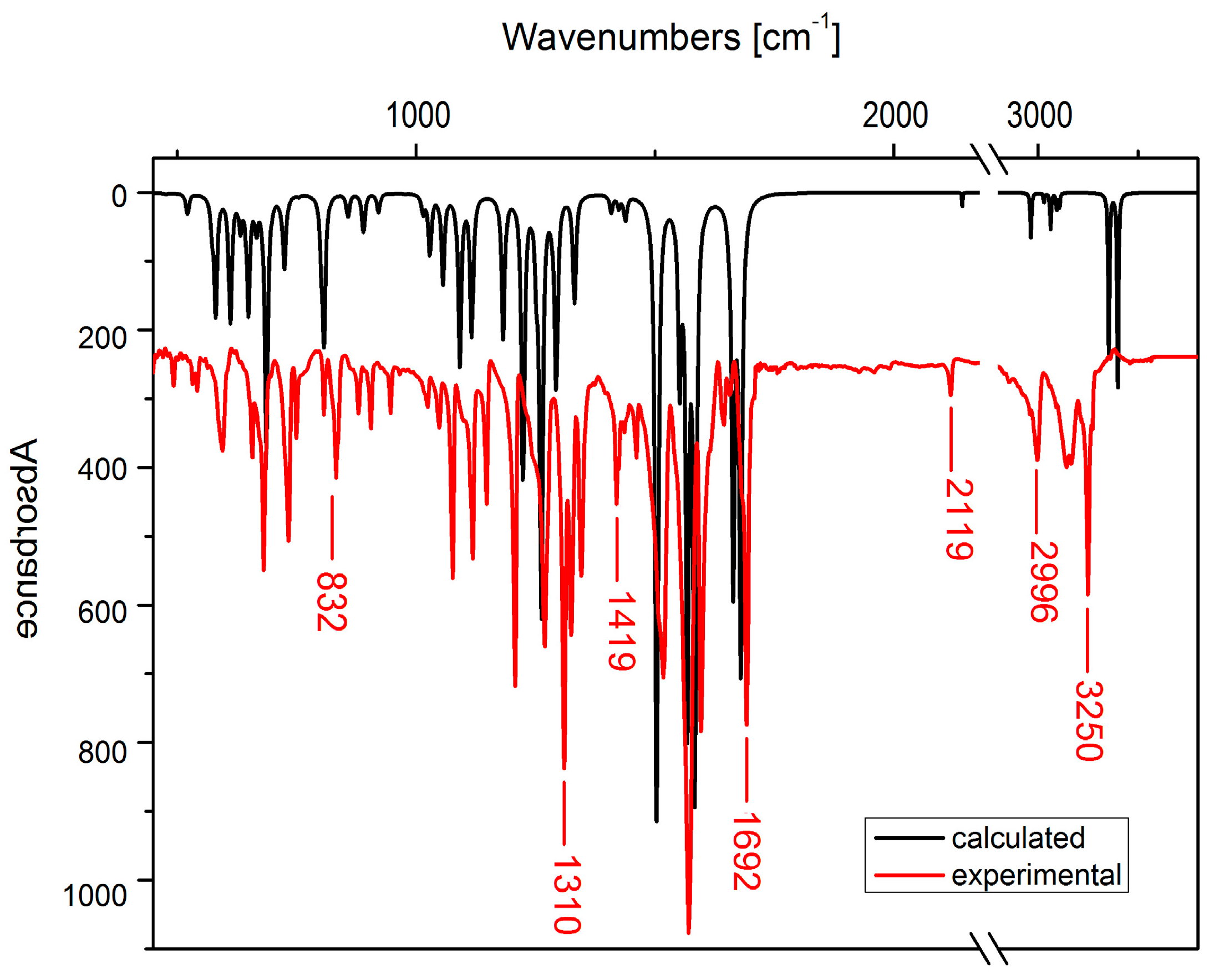
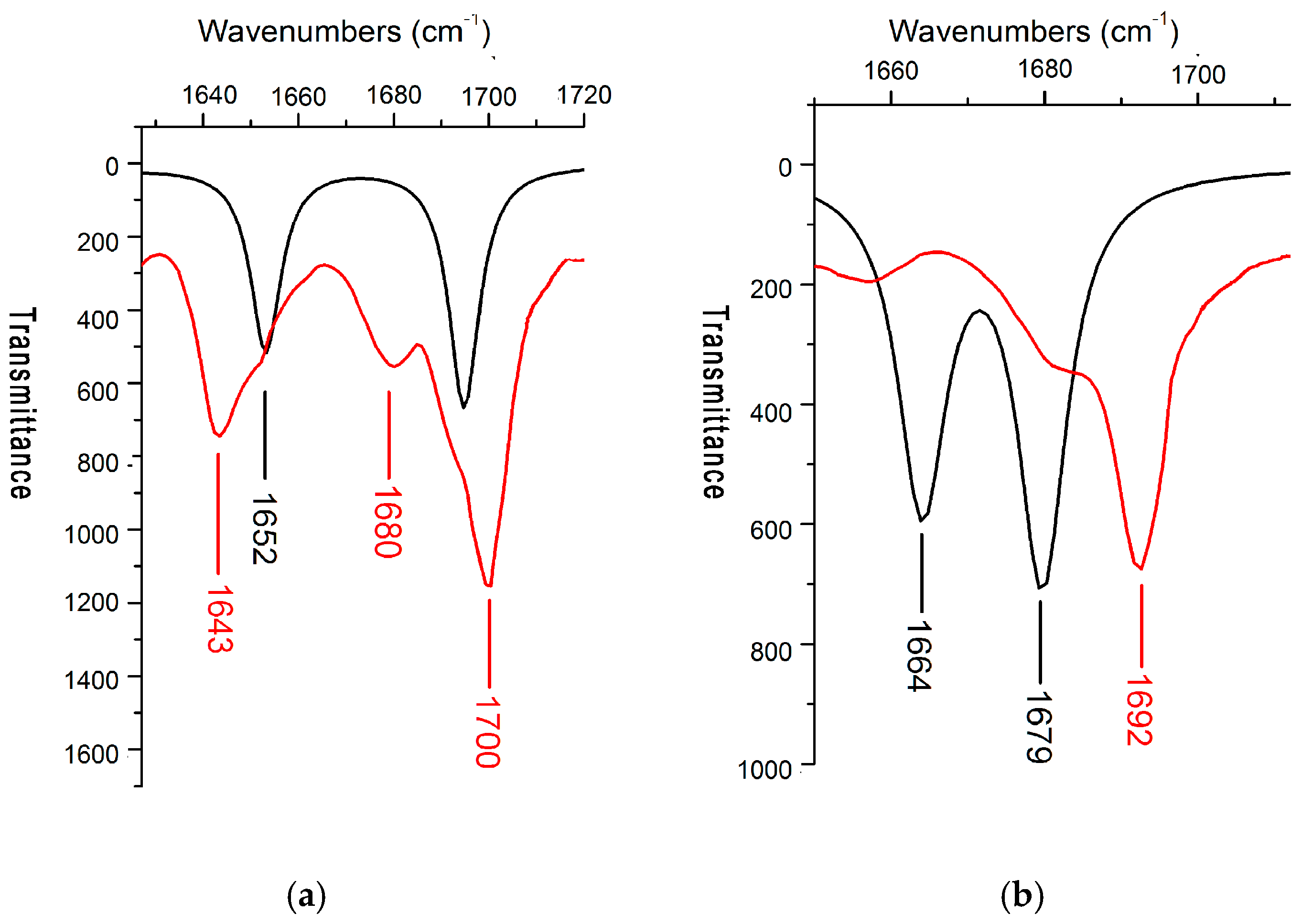
2.3. IR Spectra
2.4. Antiproliferative Activity
3. Materials and Methods
3.1. General Techniques
3.2. Chemistry
3.3. X-ray Diffraction
3.4. Density Functional Theory (DFT) Analysis
3.5. Antiproliferative Assay In Vitro
3.5.1. Cell Culture
3.5.2. Analysis of Antiproliferative Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bringmann, G.; Reichert, Y.; Kane, V.V. The total synthesis of streptonigrin and related antitumor antibiotic natural products. Tetrahedron 2004, 60, 3539–3574. [Google Scholar] [CrossRef]
- Boger, D.L.; Yasuda, M.; Mitscher, L.A.; Drake, S.D.; Kitos, P.A.; Thompson, S.C. Streptonigrin and lavendamycin partial structures. Probes for the minimum, potent pharmacophore of streptonigrin, lavendamycin, and synthetic quinoline-5,8-diones. J. Med. Chem. 1987, 30, 1913–1928. [Google Scholar] [CrossRef]
- Fryatt, T.; Goroski, D.T.; Nilson, Z.D.; Moody, C.J.; Beall, H. Novel quinolinequinone antitumor agents: Structure-metabolism studies with NAD(P)H:quinone oxidoreductase (NQO1). Bioorg. Med. Chem. Lett. 1999, 9, 2195–2198. [Google Scholar] [CrossRef]
- Leteurtre, F.; Kohlhagen, G.; Pommier, Y. Streptonigrin induced topoisomerase II sites exhibit base preferences in the middle of the enzym stragger. Biochem. Biophys. Res. Commun. 1994, 203, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Park, S.-Y.; Lee, H.-J.; Suh, M.-E.; Schollmeyer, D.; Lee, C.-O. Synthesis and cytotoxicity of 6,11-dihydro-pyrido- and 6,11-dihydro-benzo[2,3-b]phenazine-6,11-dione derivatives. Bioorg. Med. Chem. 2003, 11, 1709–1714. [Google Scholar] [CrossRef]
- Alfadhli, A.; Mack, A.; Harper, L.; Berk, S.; Ritchie, C.; Barklis, E. Analysis of quinolinequinone reactivity, cytotoxicity, and anti-HIV-1 properties. Bioorg. Med. Chem. 2016, 24, 5618–5625. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.-K.; Song, A.L.; Hong, J.A. Synthesis and antifungal evaluation of 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates. Bioorg. Med. Chem. Lett. 2013, 23, 2065–2068. [Google Scholar] [CrossRef] [PubMed]
- Kadela, M.; Jastrzębska, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Kusz, J.; Książek, M.; Boryczka, S. Synthesis, structure and cytotoxic activity of mono- and dialkoxy derivatives of 5,8-quinolinedione. Molecules 2016, 21, 156. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Pawełczak, B.; Bębenek, E.; Chrobak, E.; Latocha, M.; Książek, M.; Kusz, J.; Boryczka, S. Alkynyloxy derivatives of 5,8-quinolinedione: Synthesis, in vitro cytotoxicity studies and computational molecular modeling with NAD(P)H:quinone oxidoreductase 1. Eur. J. Med. Chem. 2017, 126, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.K.; Park, H.J.; Lee, S.K.; Lee, C.-O.; Choo, H.-Y. Synthesis, cytotoxicity and DNA topoisomerase II inhibitory activity of benzofuroquinolinediones. Bioorg. Med. Chem. 2007, 15, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.-K.; Jeong, H.J.; Lee, S.K.; You, H.-J.; Chio, K.U.; Shim, J.-Y.; Heo, Y.-H.; Lee, C.-O. Effects of 6-arylamine-5,8-quinolinediones and 6-chloro 7-arylo-5,8-isoquinolinediones on NAD(P)H:quinone oxidoreductase (NQO1) activity and their cytotoxic potential. Arch. Pharm. Res. 2001, 24, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Mulchin, B.J.; Newton, C.G.; Baty, J.W.; Grasso, C.H.; Martin, W.J.; Walton, M.C.; Dangerfield, E.M.; Plunkett, C.H.; Berridge, M.V.; Harper, J.L.; et al. The anti-cancer, anti-inflammatory and tuberculostatic activities of a series of 6,7-substituted-5,8-quinolinequinones. Bioorg. Med. Chem. 2010, 18, 3238–3251. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.Y.; Choi, H.Y.; Shin, K.J.; Yoo, K.H.; Chi, D.Y.; Kim, D.J. The regioselectivity in the reaction of 6,7-dihaloquinoline-5,8-diones with amine nucleophiles in various solvents. Tetrahedron Lett. 2000, 41, 7475–7480. [Google Scholar] [CrossRef]
- Yin, H.; Kong, F.; Wang, S.; Yao, Z.-Y. Assembly of pentacyclic pyrido[4,3,2-mn]acridin-8-ones via a domino reaction initiated by Au(I)-catalyzed 6-endo-dig cycloisomerization of N-propargylaminoquinones. Tetrahedron Lett. 2012, 53, 7078–7082. [Google Scholar] [CrossRef]
- Mól, W.; Matyja, M.; Filip, B.; Wietrzyk, J.; Boryczka, S. Synthesis and antiproliferative activity in vitro of novel (2-butynyl)thioquinolines. Bioorg. Med. Chem. 2008, 16, 8136–8141. [Google Scholar] [CrossRef] [PubMed]
- Mól, W.; Naczyński, A.; Boryczka, S.; Wietrzyk, J.; Opolski, A. Synthesis and antiproliferative activity in vitro of diacetylenic thioquinolines. Pharmazie 2006, 61, 742–746. [Google Scholar] [PubMed]
- Boryczka, S.; Bębenek, E.; Wietrzyk, J.; Kempińska, K.; Jastrzębska, M.; Kusz, J.; Nowak, M. Synthesis, structure and cytotoxic activity of new acetylenic derivatives of betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Latocha, M.; Boryczka, S.; Kurczab, R. Synthesis, molecular docking study, and evaluation of the antiproliferative action of a new group of propargylthio- and propargylselenoquinolines. Med. Chem. Res. 2014, 23, 3468–3477. [Google Scholar] [CrossRef]
- Siddiq, A.; Dembitsky, V. Acetylenic anticancer agents. Anti-Cancer Agents Med. Chem. 2008, 8, 132–170. [Google Scholar] [CrossRef]
- Dembitsky, V.M.; Levitsky, D.O.; Gloriozovac, T.A.; Poroikovc, V.V. Acetylenic aquatic anticancer agents and related compounds. Nat. Prod. Commun. 2006, 1, 773–812. [Google Scholar]
- Kuklev, D.V.; Dembitsky, V.M. Epoxy acetylenic lipids: their analogues and derivatives. Prog. Lipid Res. 2014, 56, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Kuklev, D.V.; Domb, A.J.; Dembitsky, V.M. Bioactive acetylenic metabolites. Phytomedicine 2013, 20, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Tang, H.; Wu, P. Interpretation of carbonyl band splitting phenomenon of a novel thermotropic liquid crystalline polymer without conventional mesogens: Combination method of spectral analysis and molecular simulation. J. Phys. Chem. B 2010, 114, 3439–3448. [Google Scholar] [CrossRef] [PubMed]
- Socrates, G. Infrared Characteristic Group Frequencies; John Wiley & Sons Inc.: New York, NY, USA, 1994. [Google Scholar]
- Jastrzebska, M.; Boryczka, S.; Kadela, M.; Wrzalik, R.; Kusz, J.; Nowak, M. Synthesis, crystal structure and infrared spectra of new 6- and 7-propylamine-5,8-quinolinediones. J. Mol. Struct. 2014, 1067, 160–168. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond; Oxford University Press: London, UK, 2006. [Google Scholar]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. 2002, 41, 48–79. [Google Scholar] [CrossRef]
- Marechal, Y. The Hydrogen Bond and the Water Molecule; Elsevier: London, UK, 2007. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A. 02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, Release 16a, August 2013, Editor: Russell D. Johnson III. Available online: http://cccbdb.nist.gov/ (accessed on 11 November 2015).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 2a | 3a |
|---|---|---|
| Chemical formula | C12H7ClN2O2 | C12H7ClN2O2 |
| Mr | 246.65 | |
| Crystal system, space group | Monoclinic, Pc | Monoclinic, P21/n |
| Temperature (K) | 100 | |
| a, b, c (Å) | 4.0250 (12), 6.5937 (6), 19.3214 (13) | 11.1550 (3), 7.9114 (2), 12.0047 (3) |
| β (°) | 90.673 (12) | 97.554 (3) |
| V (Å3) | 512.75 (16) | 1050.24 (5) |
| Z | 2 | 4 |
| Radiation type | Mo Kα | |
| µ (mm−1) | 0.36 | 0.35 |
| Crystal size (mm) | 0.38 × 0.05 × 0.04 | 0.56 × 0.22 × 0.03 |
| Diffractometer | Oxford Diffraction diffractometer with Sapphire3 detector | |
| Absorption correction | Multi-scan CrysAlis RED, Oxford Diffraction Ltd., Version 1.171.32.29 Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. | |
| Tmin, Tmax | 0.875, 0.984 | 0.911, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 3745, 1261, 1144 | 7654, 1995, 1657 |
| Rint | 0.036 | 0.026 |
| (sin θ/λ)max (Å−1) | 0.609 | 0.610 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.032, 0.078, 1.00 | 0.027, 0.072, 1.03 |
| No. of reflections | 1261 | 1995 |
| No. of parameters | 164 | 160 |
| No. of restraints | 2 | - |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | |
| (Δ)max, (Δ)min (e Å−3) | 0.61, −0.25 | 0.30, −0.20 |
| Absolute structure | Refined as an inversion twin. | - |
| Absolute structure parameter | 0.95 (14) | - |
| D–H···A | D–H | H···A | D···A | <D–H···A |
|---|---|---|---|---|
| 6-chloro-7-propargylamine-5,8-quinolinedione 2a | ||||
| C4–H4···O2 | 0.95 (1) | 2.373 (4) | 3.219 (4) | 148.1 |
| N2–H2N···O1 | 0.83 (1) | 2.403 (4) | 3.015 (4) | 131.2 |
| C9–H9A···O1 | 0.83 (2) | 2.629 (2) | 3.207 (2) | 127.9 |
| C11–H11···N1 | 0.95 (1) | 2.300 (2) | 3.158 (3) | 149.9 |
| 7-chloro-6-propargylamine-5,8-quinolinedione 3a | ||||
| N2–H2N···N1 | 0.85 (1) | 2.181 (1) | 2.957 (1) | 152.5 |
| C11–H11···O2 | 0.95 (1) | 2.338 (2) | 3.282 (1) | 172.2 |
| Experimental | Calculated | Assignment |
|---|---|---|
| 6-chloro-7-propargylamine-5,8-quinolinedione 2a | ||
| 581 | 579 | C–C aliphatic bend |
| 652 | 650 | C≡C–H bend |
| 748–696 | 723–689 | C–C ring stretch, C–H ring stretch |
| 826–818 | 803 | C–Cl bend |
| 1153–1139 | 1175–1123 | C–C ring bend, C–H ring bend |
| 1120 | 1220 | HN–C ring bend, C–H ring bend |
| 1283–1250 | 1272 | C–C ring stretch |
| 1332–1310 | 1299 | C–C ring bend |
| 1353 | 1332 | C–H aliphatic bend |
| 1427 | 1423 | C–H aliphatic stretch |
| 1506 | 1504 | N–H bend |
| 1599–1565 | 1534–1550 | C–H ring bend, C–H aliphatic stretch |
| 1643 | 1652 | C=O sym stretch |
| 1700 1680 | 1695 | C=O asym stretch, N–H bend |
| 2113 | 2145 | C≡C stretch |
| 2957 | 2964 | C–H aliphatic stretch |
| 3085–3038 | 3109–3064 | C–H ring stretch |
| 3190 | 3354 | C≡CH stretch |
| 7-chloro-6-propargylamine-5,8-quinolinedione 3a | ||
| 595 | 611–580 | C–C aliphatic bend |
| 657 | 657 | C≡C–H bend |
| 749–681 | 725–687 | C–C ring stretch, C–H ring stretch |
| 832–807 | 806 | C–Cl bend |
| 1147–1076 | 1182–1091 | C–C ring bend, C–H ring bend |
| 1207 | 1224 | HN–C ring bend, C–H ring bend |
| 1269 | 1261 | C–C ring stretch |
| 1324–1310 | 1293 | C–C ring bend |
| 1346 | 1331 | C–H aliphatic bend |
| 1419 | 1407 | C–H aliphatic stretch |
| 1461 | 1438 | C–H ring bend, C–C ring bend |
| 1517 | 1503 | N–H bend |
| 1597–1569 | 1584–1551 | C–H ring bend, C–H aliphatic stretch |
| 1682 | 1664 | C=O sym stretch, N–H bend |
| 1692 | 1679 | C=O asym stretch |
| 2119 | 2143 | C≡C stretch |
| 2996 | 2965 | C–H aliphatic stretch |
| 3168–3058 | 3108–3030 | C–H ring stretch |
| 3250 | 3354 | C≡CH stretch |
| 3271 | 3399 | N–H stretch |
| Compound | Cytotoxic Activity IC50 (µg/mL) | ||
|---|---|---|---|
| C-32 | SNB-19 | T47D | |
| 1 | 42.48 ± 2.02 | 2.77 ± 0.07 | 8.26 ± 0.32 |
| 2a | 0.61 ± 0.02 | 0.26 ± 0.02 | 8.50 ± 0.54 |
| 2b | 0.75 ± 0.05 | 0.97 ± 0.01 | 9.22 ± 0.77 |
| 2c | 0.67 ± 0.01 | 0.98 ± 0.01 | 9.14 ± 0.77 |
| 2d | 0.63 ± 0.02 | 0.88 ± 0.05 | 9.03 ± 0.10 |
| 2e | 0.64 ± 0.03 | 0.50 ± 0.04 | 8.54 ± 0.41 |
| 3a | 0.58 ± 0.03 | 0.09 ± 0.01 | 1.01 ± 0.05 |
| 3b | 0.67 ± 0.05 | 0.44 ± 0.01 | 8.48 ± 0.55 |
| 3c | 0.65 ± 0.05 | 0.92 ± 0.07 | 4.57 ± 0.62 |
| 3d | 0.61 ± 0.03 | 0.79 ± 0.03 | 7.07 ± 0.28 |
| 3e | 0.64 ± 0.01 | 0.28 ± 0.05 | 3.05 ± 5.65 |
| cisplatin | 1.51 ± 0.49 | 0.79 ± 0.07 | 62.65 ± 2.70 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadela-Tomanek, M.; Jastrzębska, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Kusz, J.; Tarnawska, D.; Boryczka, S. New Acetylenic Amine Derivatives of 5,8-Quinolinediones: Synthesis, Crystal Structure and Antiproliferative Activity. Crystals 2017, 7, 15. https://doi.org/10.3390/cryst7010015
Kadela-Tomanek M, Jastrzębska M, Bębenek E, Chrobak E, Latocha M, Kusz J, Tarnawska D, Boryczka S. New Acetylenic Amine Derivatives of 5,8-Quinolinediones: Synthesis, Crystal Structure and Antiproliferative Activity. Crystals. 2017; 7(1):15. https://doi.org/10.3390/cryst7010015
Chicago/Turabian StyleKadela-Tomanek, Monika, Maria Jastrzębska, Ewa Bębenek, Elwira Chrobak, Małgorzata Latocha, Joachim Kusz, Dorota Tarnawska, and Stanisław Boryczka. 2017. "New Acetylenic Amine Derivatives of 5,8-Quinolinediones: Synthesis, Crystal Structure and Antiproliferative Activity" Crystals 7, no. 1: 15. https://doi.org/10.3390/cryst7010015
APA StyleKadela-Tomanek, M., Jastrzębska, M., Bębenek, E., Chrobak, E., Latocha, M., Kusz, J., Tarnawska, D., & Boryczka, S. (2017). New Acetylenic Amine Derivatives of 5,8-Quinolinediones: Synthesis, Crystal Structure and Antiproliferative Activity. Crystals, 7(1), 15. https://doi.org/10.3390/cryst7010015