
Chemical Crystallography at the Advanced Light Source


Abstract
:
1. Introduction
2. Benefits of Synchrotron Radiation for Chemical Crystallography
3. Beamline Descriptions
3.1. Beamline 11.3.1
3.2. Beamlines 12.2.1 and 12.2.2
4. In Situ Experiments
4.1. High Pressure
4.2. Photocrystallography
4.3. Environmental Gas/Vacuum
5. The Future of Chemical Crystallography at the ALS
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Bragg, W.L. The structure of some crystals as indicated by their diffraction of X-rays. Proc. R. Soc. Lond. Ser. A 1913, 89, 248–277. [Google Scholar] [CrossRef]
- Bragg, W.H.; Bragg, W.L. The structure of the diamond. R. Soc. Lond. Ser. A 1913, 89, 277–291. [Google Scholar] [CrossRef]
- Becker, K.; Rose, H. Röntgenspektroskopie an organischen verbindungen. Z. Phys. 1923, 14, 369–373. [Google Scholar] [CrossRef]
- Robertson, J.M. 255. An X-ray study of the phthalocyanines. Part II. Quantitative structure determination of the metal-free compound. J. Chem. Soc. (Resumed) 1936, 1, 1195–1209. [Google Scholar] [CrossRef]
- Lipson, H.; Beevers, C.A. An improved numerical method of two-dimensional fourier synthesis for crystals. Proc. Phys. Soc. 1936, 48, 772. [Google Scholar] [CrossRef]
- Tate, M.W.; Eikenberry, E.F.; Barna, S.L.; Wall, M.E.; Lowrance, J.L.; Gruner, S.M. A large-format high-resolution area X-ray-detector based on a fiberoptically bonded charge-coupled-device (CCD). J. Appl. Cryst. 1995, 28, 196–205. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, M.M. Direct methods in crystallography. Rep. Prog. Phys. 1971, 34, 369. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.M. Recording diffraction data for structure determination for very small crystals. J. Synchrotron Radiat. 1996, 3, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.B.; Rosenbaum, G. Synchrotron X-ray sources: A new tool in biological structural and kinetic analysis. Annu. Rev. Biophys. Bioeng. 1976, 5, 239–270. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, A.; Leberman, R.; Schulz, G.E. Comparison of protein crystal diffraction patterns and absolute intensities from synchrotron and conventional X-ray sources. J. Mol. Biol. 1976, 104, 311–314. [Google Scholar] [CrossRef]
- Einfeld, D.; Klotz, W.D.; Muelhaupt, G.; Mueller, T.; Richter, R. Bessy, an 800 MeV storage ring dedicated to synchrotron radiation. IEEE Trans. Nucl. Sci. 1979, 26, 3801–3802. [Google Scholar] [CrossRef]
- Duke, E.M.H.; Johnson, L.N. Macromolecular crystallography at synchrotron radiation sources: Current status and future developments. Proc. R. Soc. A Math. Phys. Eng. Sci. 2010, 466, 3421–3452. [Google Scholar] [CrossRef]
- Wu, G.; Rodrigues, B.L.; Coppens, P. The correction of reflection intensities for incomplete absorption of high-energy X-rays in the CCD phosphor. J. Appl. Cryst. 2002, 35, 356–359. [Google Scholar] [CrossRef]
- There is a discrepency in how instruments were referred to at the SRS versus most modern light source facilites. At the SRS, beamlines were referred to as “Stations,” probably due to the fact that multiple instrumental stations were arranged about a common beamline. At most modern facilites, beamlines are single purpose, and branchlines are indicated in the facilites’ naming convention.
- Cernik, R.J.; Clegg, W.; Catlow, C.R.A.; Bushnell-Wye, G.; Flaherty, J.V.; Greaves, G.N.; Burrows, I.; Taylor, D.J.; Teat, S.J.; Hamichi, M. A new high-flux chemical and materials crystallography station at the SRS Daresbury. 1. Design, construction and test results. J. Synchrotron Radiat. 1997, 4, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Bom, A.; Bradley, M.; Cameron, K.; Clark, J.K.; Van Egmond, J.; Feilden, H.; MacLean, E.J.; Muir, A.W.; Palin, R.; Rees, D.C.; et al. A novel concept of reversing neuromuscular block: Chemical encapsulation of rocuronium bromide by a cyclodextrin-based synthetic host. Angew. Chem. Int. Ed. 2002, 41, 265–270. [Google Scholar] [CrossRef]
- Rajaraman, G.; Murugesu, M.; Sañudo, E.C.; Soler, M.; Wernsdorfer, W.; Helliwell, M.; Muryn, C.; Raftery, J.; Teat, S.J.; Christou, G.; Brechin, E.K. A family of manganese rods: Syntheses, structures, and magnetic properties. J. Am. Chem. Soc. 2004, 126, 15445–15457. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.; Allan, D.R.; Belmonte, S.A.; Clark, S.J.; David, W.I.F.; McGregor, P.A.; Parsons, S.; Pulham, C.R.; Sawyer, L. Effect of high pressure on the crystal structures of polymorphs of glycine. Cryst. Growth Des. 2005, 5, 1415–1427. [Google Scholar] [CrossRef]
- Warren, M.R.; Brayshaw, S.K.; Johnson, A.L.; Schiffers, S.; Raithby, P.R.; Easun, T.L.; George, M.W.; Warren, J.E.; Teat, S.J. Reversible 100% linkage isomerization in a single-crystal to single-crystal transformation: Photocrystallographic identification of the metastable [Ni(dppe)(η1-ONO)Cl] isomer. Angew. Chem. Int. Ed. 2009, 48, 5711–5714. [Google Scholar] [CrossRef] [PubMed]
- Cerenius, Y.; Stahl, K.; Svensson, L.A.; Ursby, T.; Oskarsson, A.; Albertsson, J.; Liljas, A. The crystallography beamline I711 at MAX II. J. Synchrotron Radiat. 2000, 7, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Nowell, H.; Barnett, S.A.; Christensen, K.E.; Teat, S.J.; Allan, D.R. I19, the small-molecule single-crystal diffraction beamline at Diamond Light Source. J. Synchrotron Radiat. 2012, 19, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, R.; Kohler, H.; Schulz, H.; Weber, H.P. Structure investigation of a 6μm CaF2 crystal with synchrotron radiation. Acta Cryst. Sect. A Found. Cryst. 1985, 41, 35–40. [Google Scholar] [CrossRef]
- Caciuffo, R.; Melone, S.; Rustichelli, F.; Boeuf, A. Monochromators for X-ray synchrotron radiation. Phys. Rep. 1987, 152, 1–71. [Google Scholar] [CrossRef]
- Beaumont, J.H.; Hart, M. Multiple Bragg reflection monochromators for synchrotron X radiation. J. Phys. E Sci. Instrum. 1974, 7, 823. [Google Scholar] [CrossRef]
- Clegg, W. Synchrotron chemical crystallography. J. Chem. Soc. Dalton Trans. 2000, 19, 3223–3232. [Google Scholar] [CrossRef]
- Helliwell, M. Anomalous scattering for small-molecule crystallography. J. Synchrotron Radiat. 2000, 7, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Merrill, L.; Bassett, W.A. Miniature diamond anvil pressure cell for single crystal X-ray diffraction studies. Rev. Sci. Instrum. 1974, 45, 290–294. [Google Scholar] [CrossRef]
- Thompson, A.C.; Padmore, H.A.; Oliver, A.G.; Teat, S.J.; Celestre, R.S.; Clark, S.M.; Domning, E.E.; Franck, K.D.; Morrison, G.Y. A simple high performance beamline for small molecule chemical crystallography. AIP Conf. Proc. 2004, 708, 482–493. [Google Scholar]
- Padmore, H.A.; Thompson, A.C.; Jun, F. A double crystal monochromator using tangentially bent crystals in combination with toroidal mirror focusing. Nucl. Instrum. Methods Phys. Res. A 2001, 467, 650–653. [Google Scholar] [CrossRef]
- Flechsig, U.; Jaggi, A.; Spielmann, S.; Padmore, H.A.; MacDowell, A.A. The optics beamline at the Swiss Light Source. Nucl. Instrum. Methods Phys. Res. A 2009, 609, 281–285. [Google Scholar] [CrossRef]
- There is often confusion when crystallographers first become acquainted with a beamline, due to the synchrotron tendency to describe beamlines and instruments by energy and not wavelength. Attempts have been made here to only refer to wavelengths, but a simple aproximation can be done if energy units are needed: E(keV) = 12/(lambda(Å)).
- Robin, D.; Benjegerdes, R.; Biocca, A.; Bish, P.; Brown, W.; Byrne, W.; Calais, D.; Chin, M.; Corradi, C.; Coulomb, D.; et al. Successful completion of the ALS superbend project. Presented at the European Particle Accelerator Conference, Paris, France, 3–7 June 2002; pp. 215–217. [Google Scholar]
- MacDowell, A.A.; Celestre, R.S.; Howells, M.; McKinney, W.; Krupnick, J.; Cambie, D.; Domning, E.E.; Duarte, R.M.; Kelez, N.; Plate, D.W.; et al. Suite of three protein crystallography beamlines with single superconducting bend magnet as the source. J. Synchrotron Radiat. 2004, 11, 447–455. [Google Scholar] [CrossRef] [PubMed]
- MacDowell, A.; Parkinson, D.; Haboub, A.; Schaible, E.; Nasiatka, J.; Yee, C.; Jameson, J.; Ajo-Franklin, J.; Brodersen, C.R.; McElrone, A.J. X-ray micro-tomography at the Advanced Light Source. Proc. SPIE 2012. [Google Scholar] [CrossRef]
- Kunz, M.; MacDowell, A.A.; Caldwell, W.A.; Cambie, D.; Celestre, R.S.; Domning, E.E.; Duarte, R.M.; Gleason, A.E.; Glossinger, J.M.; Kelez, N.; et al. A beamline for high-pressure studies at the Advanced Light Source with a superconducting bending magnet as the source. J. Synchrotron Radiat. 2005, 12, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Kunz, M.; Chen, K.; Celestre, R.S.; MacDowell, A.A.; Warwick, T. A superbend X-ray microdiffraction beamline at the advanced light source. Mater. Sci. Eng. A 2009, 524, 28–32. [Google Scholar] [CrossRef]
- Katrusiak, A. High-pressure crystallography. Acta Cryst. Sect. A 2008, 64, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Boldyreva, E.V. High-pressure diffraction studies of molecular organic solids. A personal view. Acta Cryst. Sect. A 2008, 64, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Ballaran, T.B.; Kurnosov, A.; Trots, D. Single-crystal X-ray diffraction at extreme conditions: A review. High Press. Res. 2013, 33, 453–465. [Google Scholar] [CrossRef]
- Moggach, S.A.; Allan, D.R.; Parsons, S.; Warren, J.E. Incorporation of a new design of backing seat and anvil in a Merrill-Bassett diamond anvil cell. J. Appl. Cryst. 2008, 41, 249–251. [Google Scholar] [CrossRef]
- Kantor, I.; Prakapenka, V.; Kantor, A.; Dera, P.; Kurnosov, A.; Sinogeikin, S.; Dubrovinskaia, N.; Dubrovinsky, L. BX90: A new diamond anvil cell design for X-ray diffraction and optical measurements. Rev. Sci. Instrum. 2012, 83, 125102. [Google Scholar] [CrossRef] [PubMed]
- Woodall, C.H.; Brayshaw, S.K.; Schiffers, S.; Allan, D.R.; Parsons, S.; Valiente, R.; Raithby, P.R. High-pressure crystallographic and spectroscopic studies on two molecular dithienylethene switches. CrystEngComm 2014, 16, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, K.J.; Beavers, C.M.; Clearfield, A. MOFs under pressure: The reversible compression of a single crystal. J. Am. Chem. Soc. 2013, 135, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.U.; Boutin, A.; Gagnon, K.J.; Clearfield, A.; Coudert, F.X. Remarkable pressure responses of metal-organic frameworks: Proton transfer and linker coiling in zinc alkyl gates. J. Am. Chem. Soc. 2014, 136, 11540–11545. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, E.A.; Lee, S.; Alshammari, A.S.; Yaghi, O.M. Molecular retrofitting adapts a metal-organic framework to extreme pressure. ACS Cent. Sci. 2017, 3, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Woodall, C.H.; Fuertes, S.; Beavers, C.M.; Hatcher, L.E.; Parlett, A.; Shepherd, H.J.; Christensen, J.; Teat, S.J.; Intissar, M.; Rodrigue-Witchel, A.; et al. Tunable trimers: Using temperature and pressure to control luminescent emission in Gold(I) pyrazolate-based trimers. Chem. Eur. J. 2014, 20, 16933–16942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, H.; Tonge, G.; Hatcher, L.; Bryant, M.; Knichal, J.; Raithby, P.; Halcrow, M.; Kulmaczewski, R.; Gagnon, K.; Teat, S. A high pressure investigation of the order-disorder phase transition and accompanying spin crossover in [FeL12](ClO4)2 (L1 = 2,6-bis{3-methylpyrazol-1-yl}-pyrazine). Magnetochemistry 2016, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Velamazán, J.A.; Fabelo, O.; Beavers, C.M.; Natividad, E.; Evangelisti, M.; Roubeau, O. A Multifunctional Magnetic Material under Pressure. Chem. Eur. J. 2014, 20, 7956–7961. [Google Scholar] [CrossRef] [PubMed]
- Woodall, C.H.; Christensen, J.; Skelton, J.M.; Hatcher, L.E.; Parlett, A.; Raithby, P.R.; Walsh, A.; Parker, S.C.; Beavers, C.M.; Teat, S.J.; et al. Observation of a re-entrant phase transition in the molecular complex tris([mu]2-3,5-diisopropyl-1,2,4-triazolato-[kappa]2N1:N2)trigold(I) under high pressure. IUCrJ 2016, 3, 367–376. [Google Scholar] [CrossRef] [PubMed]
- O’Bannon, E.; Beavers, C.M.; Kunz, M.; Williams, Q. The high-pressure phase of lawsonite: A single crystal study of a key mantle hydrous phase. J. Geophys. Res. Solid Earth 2017, 122, 6294–6305. [Google Scholar] [CrossRef]
- Jaffe, A.; Lin, Y.; Beavers, C.M.; Voss, J.; Mao, W.L.; Karunadasa, H.I. High-Pressure single-crystal structures of 3D lead-halide hybrid perovskites and pressure effects on their electronic and optical properties. ACS Cent. Sci. 2016, 2, 201–209. [Google Scholar] [CrossRef] [PubMed]
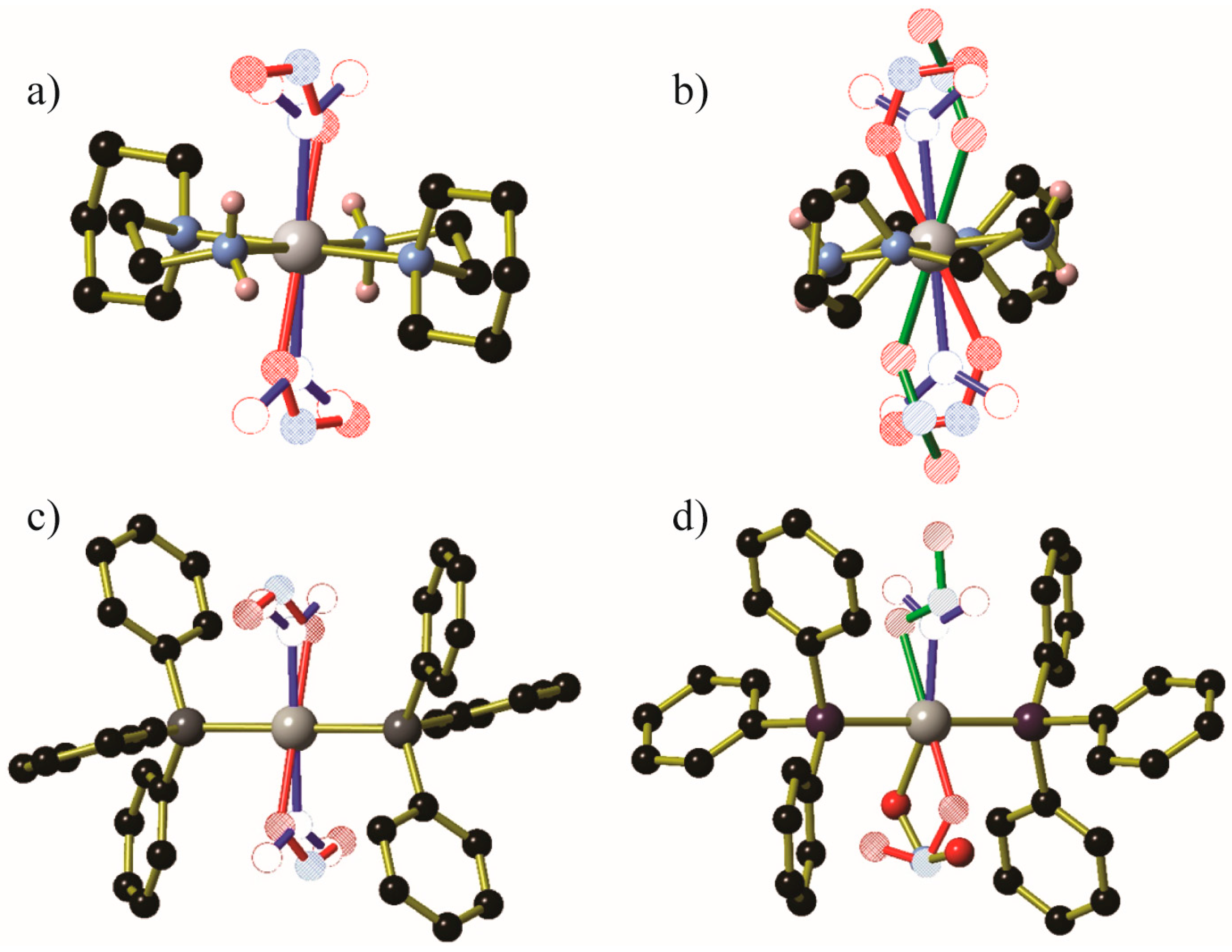
- Brayshaw, S.K.; Knight, J.W.; Raithby, P.R.; Savarese, T.L.; Schiffers, S.; Teat, S.J.; Warren, J.E.; Warren, M.R. Photocrystallography — Design and methodology for the use of a light-emitting diode device. J. Appl. Cryst. 2010, 43, 337–340. [Google Scholar] [CrossRef]
- Brayshaw, S.K.; Easun, T.L.; George, M.W.; Griffin, A.M.E.; Johnson, A.L.; Raithby, P.R.; Savarese, T.L.; Schiffers, S.; Warren, J.E.; Warren, M.R.; et al. Photocrystallographic identification of metastable nitrito linkage isomers in a series of nickel(ii) complexes. Dalton Trans. 2012, 41, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.R.; Brayshaw, S.K.; Hatcher, L.E.; Johnson, A.L.; Schiffers, S.; Warren, A.J.; Teat, S.J.; Warren, J.E.; Woodall, C.H.; Raithby, P.R. Photoactivated linkage isomerism in single crystals of nickel, palladium and platinum di-nitro complexes — A photocrystallographic investigation. Dalton Trans. 2012, 41, 13173–13179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
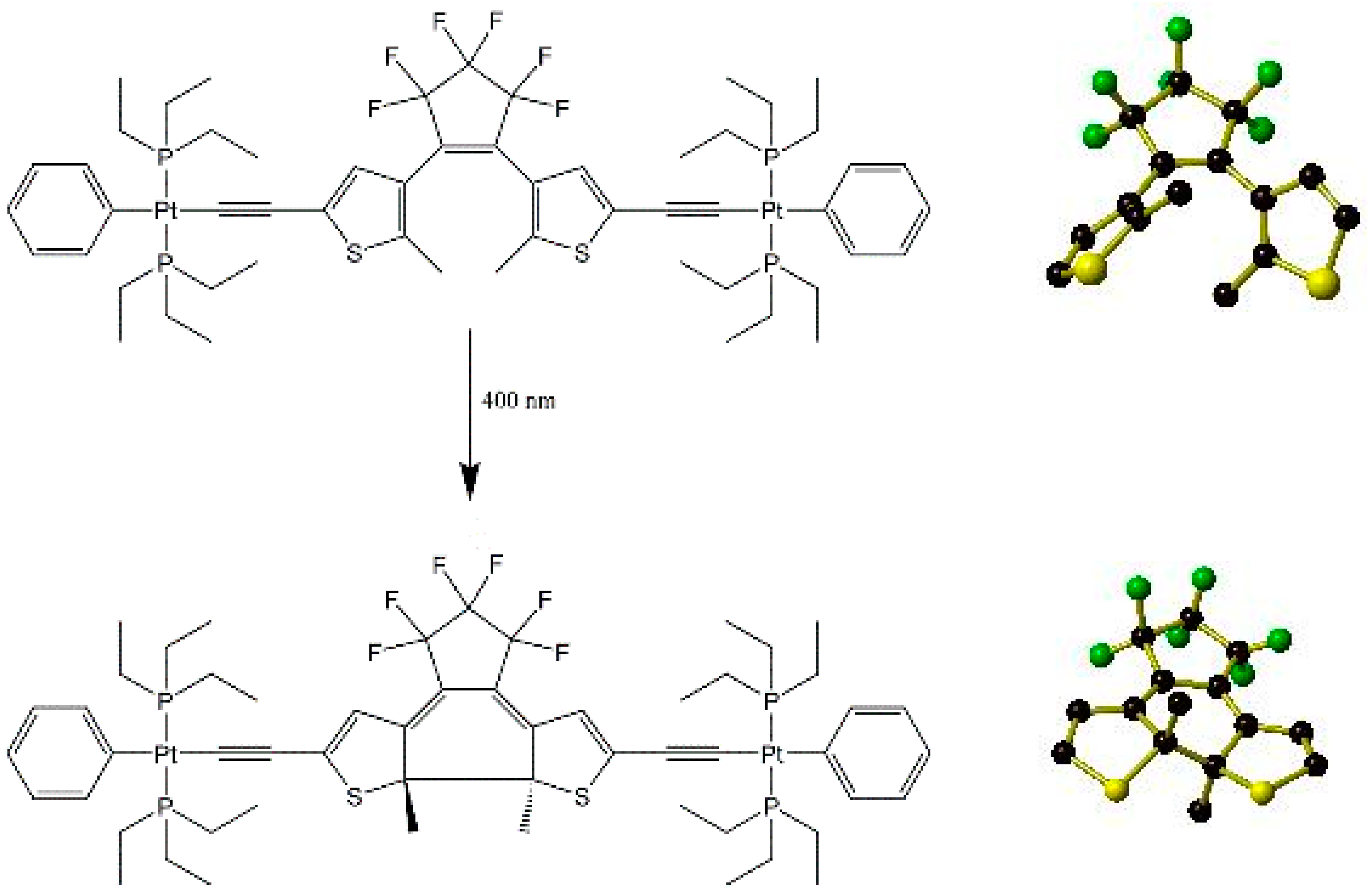
- Brayshaw, S.K.; Schiffers, S.; Stevenson, A.J.; Teat, S.J.; Warren, M.R.; Bennett, R.D.; Sazanovich, I.V.; Buckley, A.R.; Weinstein, J.A.; Raithby, P.R. Highly efficient visible-light driven photochromism: Developments towards a solid-state molecular switch operating through a triplet-sensitised pathway. Chem. Eur. J. 2011, 17, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.E.; Pritchard, R.G.; Abram, D.; Davies, H.M.; Savarese, T.L.; Cash, R.J.; Raithby, P.R.; Morris, R.; Jones, R.H.; Teat, S.J. A prototype environmental gas cell for in situ small-molecule X-ray diffraction. J. Appl. Cryst. 2009, 42, 457–460. [Google Scholar] [CrossRef]
- Cox, J.M.; Walton, I.M.; Benson, C.A.; Chen, Y.S.; Benedict, J.B. A versatile environmental control cell for in situ guest exchange single-crystal diffraction. J. Appl. Cryst. 2015, 48, 578–581. [Google Scholar] [CrossRef]
- Allan, P.K. A Study of Metal-Organic Frameworks for the Storage and Release of Medical Gases. Ph.D. Thesis, University of St Andrews, St Andrews, UK, 2012. [Google Scholar]
- Renouf, C.L. Coordinatively Unsaturated Metal-Organic Frameworks for Olefin Separations. Ph.D. Thesis, University of St Andrews, St Andrews, UK, 2013. [Google Scholar]
- Gonzalez, M.I.; Mason, J.A.; Bloch, E.D.; Teat, S.J.; Gagnon, K.J.; Morrison, G.Y.; Queen, W.L.; Long, J.R. Structural characterization of framework-gas interactions in the metal-organic framework Co2(dobdc) by in situ single-crystal X-ray diffraction. Chem. Sci. 2017, 8, 4387–4398. [Google Scholar] [CrossRef] [PubMed]
- Mercado, R.; Vlaisavljevich, B.; Lin, L.C.; Lee, K.; Lee, Y.; Mason, J.A.; Xiao, D.J.; Gonzalez, M.I.; Kapelewski, M.T.; Neaton, J.B.; et al. Force field development from periodic density functional theory calculations for gas separation applications using metal-organic frameworks. J. Phys. Chem. C 2016, 120, 12590–12604. [Google Scholar] [CrossRef]
- Furukawa, H.; Gándara, F.; Zhang, Y.B.; Jiang, J.; Queen, W.L.; Hudson, M.R.; Yaghi, O.M. Water adsorption in porous metal-organic frameworks and related materials. J. Am. Chem. Soc. 2014, 136, 4369–4381. [Google Scholar] [CrossRef] [PubMed]
- Trickett, C.A.; Gagnon, K.J.; Lee, S.; Gándara, F.; Bürgi, H.B.; Yaghi, O.M. Definitive molecular level characterization of defects in UiO-66 crystals. Angew. Chem. Int. Ed. 2015, 54, 11162–11167. [Google Scholar] [CrossRef] [PubMed]
- Allan, P.K.; Xiao, B.; Teat, S.J.; Knight, J.W.; Morris, R.E. In situ single-crystal diffraction studies of the structural transition of metal-organic framework copper 5-sulfoisophthalate, Cu-SIP-3. J. Am. Chem. Soc. 2010, 132, 3605–3611. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Godfrey, H.G.W.; da Silva, I.; Cheng, Y.; Savage, M.; Tuna, F.; McInnes, E.J.L.; Teat, S.J.; Gagnon, K.J.; Frogley, M.D.; et al. Modulating supramolecular binding of carbon dioxide in a redox-active porous metal-organic framework. Nat. Commun. 2017, 8, 14212. [Google Scholar] [CrossRef] [PubMed]
- Queen, W.L.; Hudson, M.R.; Bloch, E.D.; Mason, J.A.; Gonzalez, M.I.; Lee, J.S.; Gygi, D.; Howe, J.D.; Lee, K.; Darwish, T.A.; et al. Comprehensive study of carbon dioxide adsorption in the metal-organic frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn). Chem. Sci. 2014, 5, 4569–4581. [Google Scholar] [CrossRef]
- Xiao, D.J.; Gonzalez, M.I.; Darago, L.E.; Vogiatzis, K.D.; Haldoupis, E.; Gagliardi, L.; Long, J.R. Selective, tunable O2 binding in Cobalt(II)–triazolate/pyrazolate metal-organic frameworks. J. Am. Chem. Soc. 2016, 138, 7161–7170. [Google Scholar] [CrossRef] [PubMed]
- Siegelman, R.L.; McDonald, T.M.; Gonzalez, M.I.; Martell, J.D.; Milner, P.J.; Mason, J.A.; Berger, A.H.; Bhown, A.S.; Long, J.R. Controlling cooperative CO2 adsorption in diamine-appended Mg2(dobpdc) metal-organic frameworks. J. Am. Chem. Soc. 2017, 139, 10526–10538. [Google Scholar] [CrossRef] [PubMed]
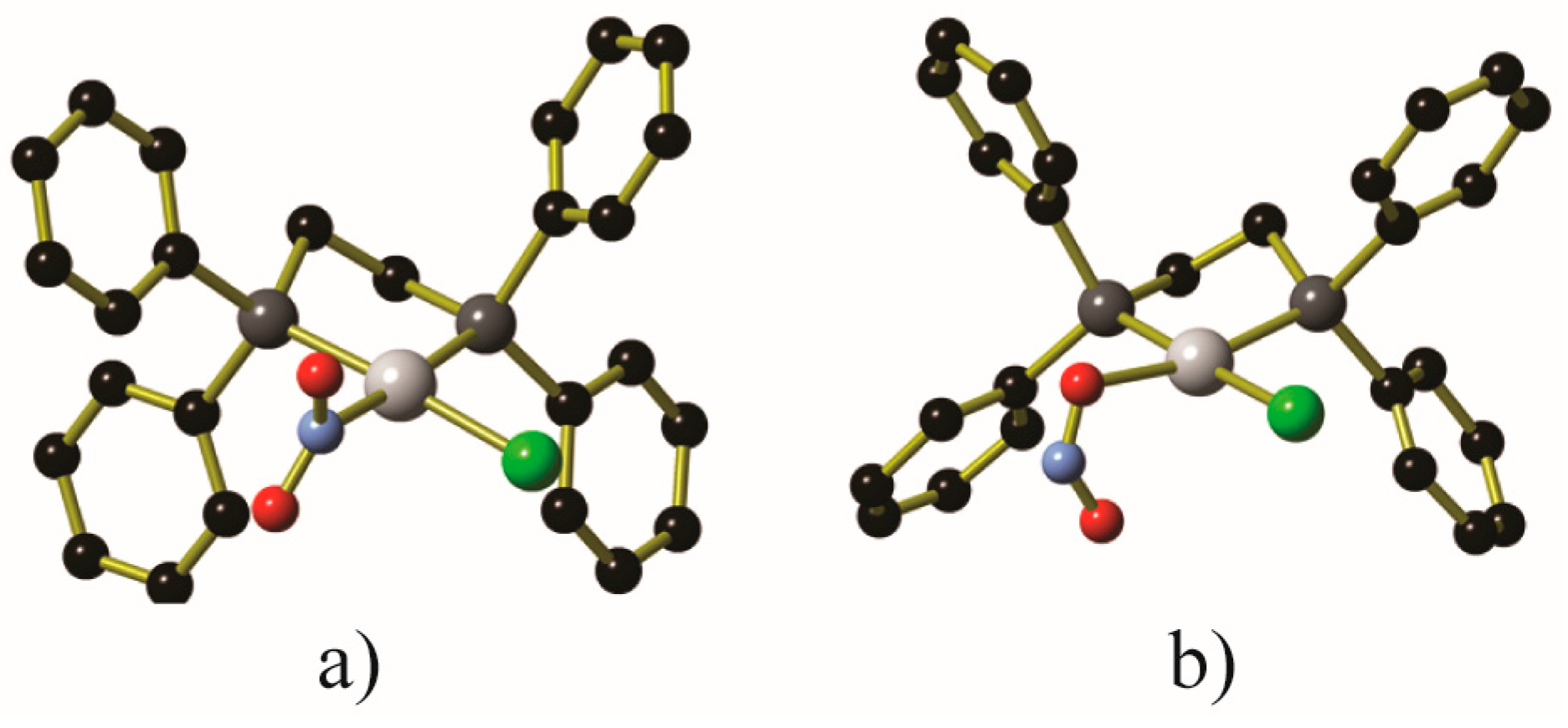
- Martell, J.D.; Porter-Zasada, L.B.; Forse, A.C.; Siegelman, R.L.; Gonzalez, M.I.; Oktawiec, J.; Runčevski, T.; Xu, J.; Srebro-Hooper, M.; Milner, P.J.; et al. Enantioselective recognition of ammonium carbamates in a chiral metal-organic framework. J. Am. Chem. Soc. 2017, 139, 16000–16012. [Google Scholar] [CrossRef] [PubMed]
- Milner, P.J.; Siegelman, R.L.; Forse, A.C.; Gonzalez, M.I.; Runčevski, T.; Martell, J.D.; Reimer, J.A.; Long, J.R. A diaminopropane-appended metal-organic framework enabling efficient CO2 capture from coal flue gas via a mixed adsorption mechanism. J. Am. Chem. Soc. 2017, 139, 13541–13553. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
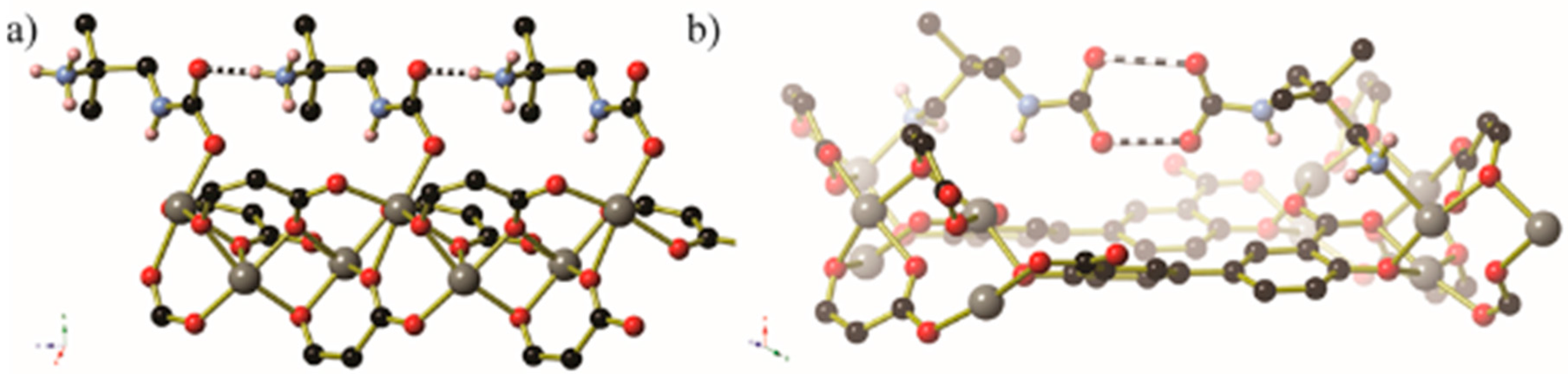{kind=link}
| 11.3.1. | 12.2.1. * | 12.2.2. | ||
|---|---|---|---|---|
| End Station 1 * | End Station 2 | |||
| Wavelength Range (Å) | 2–0.7 | 2–0.5 | 2–0.3 | 2–0.3 |
| Energy Range (keV) | 6–17 | 6–24 | 6–40 | 6–40 |
| Peak Flux photons/s/0.01%BW | 1.7 × 1010 at 1.239 Å | 1.08 × 1012 at 1.068 Å ** | 1.4 × 1011 at 0.6199 Å | |
| Source | 1.27 T Bend | 4.37 T Superbend | 5.29 T Superbend | |
| Spot Size (μm²) | 280 × 90 | 30 × 80 | 30 × 80 | 10 × 10 |
| 30 × 30 | ||||
| Diffractometer | Bruker D8 | Stoe Stadi Vari | Huber | |
| Scannable axes | ϕ/ω | ϕ/ω | ϕ | |
| Detector | PHOTON II CPAD | RDI CMOS | Perkin Elmer a-Si | |
| RDI CMOS * | ||||
| Temperature control | Oxford Cryostream 800+ Cryocool-G2B-LT | Oxford Cryostream 700+ | ||
| 18–500 K | 85–500 K | |||
| DAC capabilities | Merrill-Bassett or smaller | ≤1 kg BX90 | ≤5 kg, e.g., BX90, Mao-Bell | |
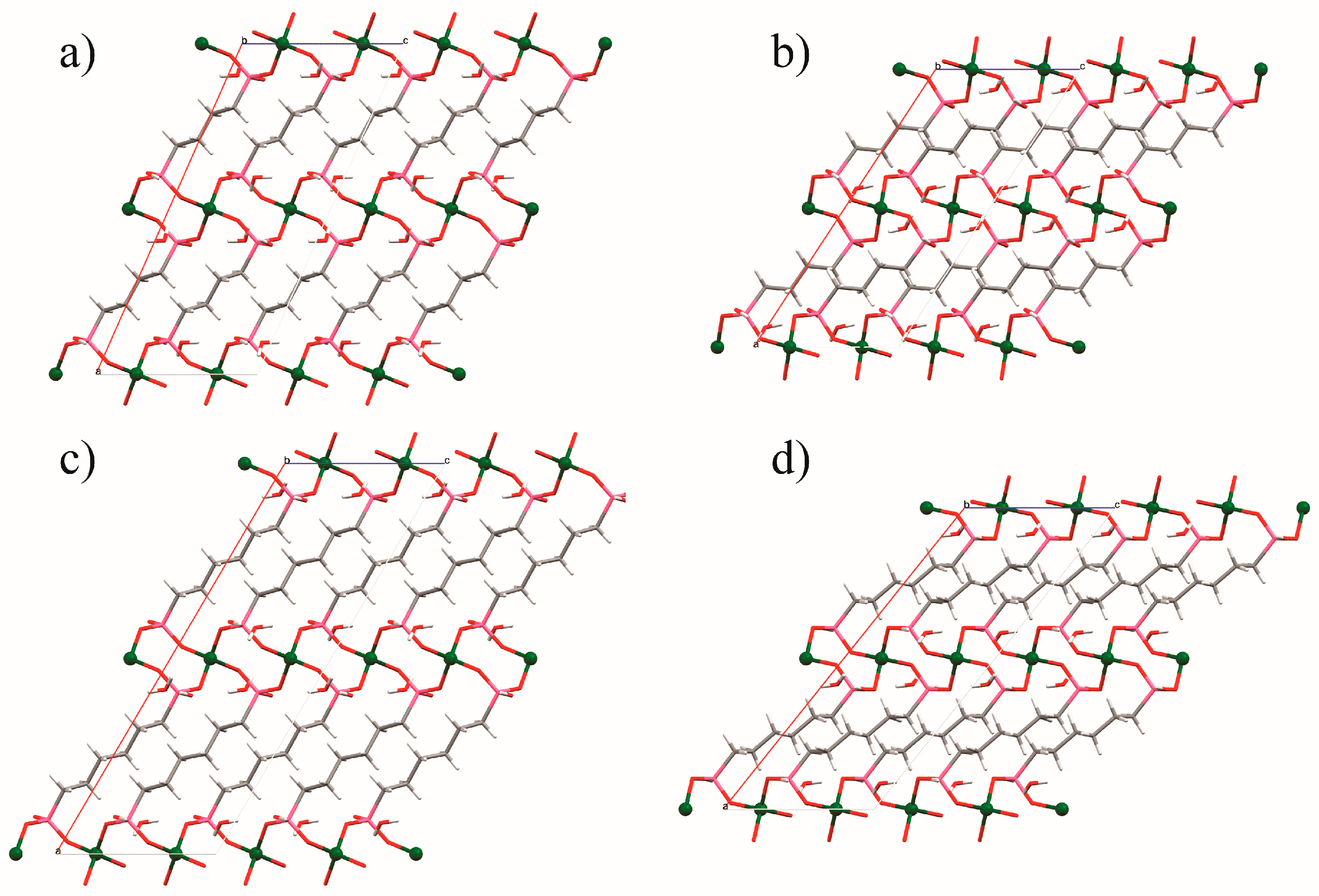
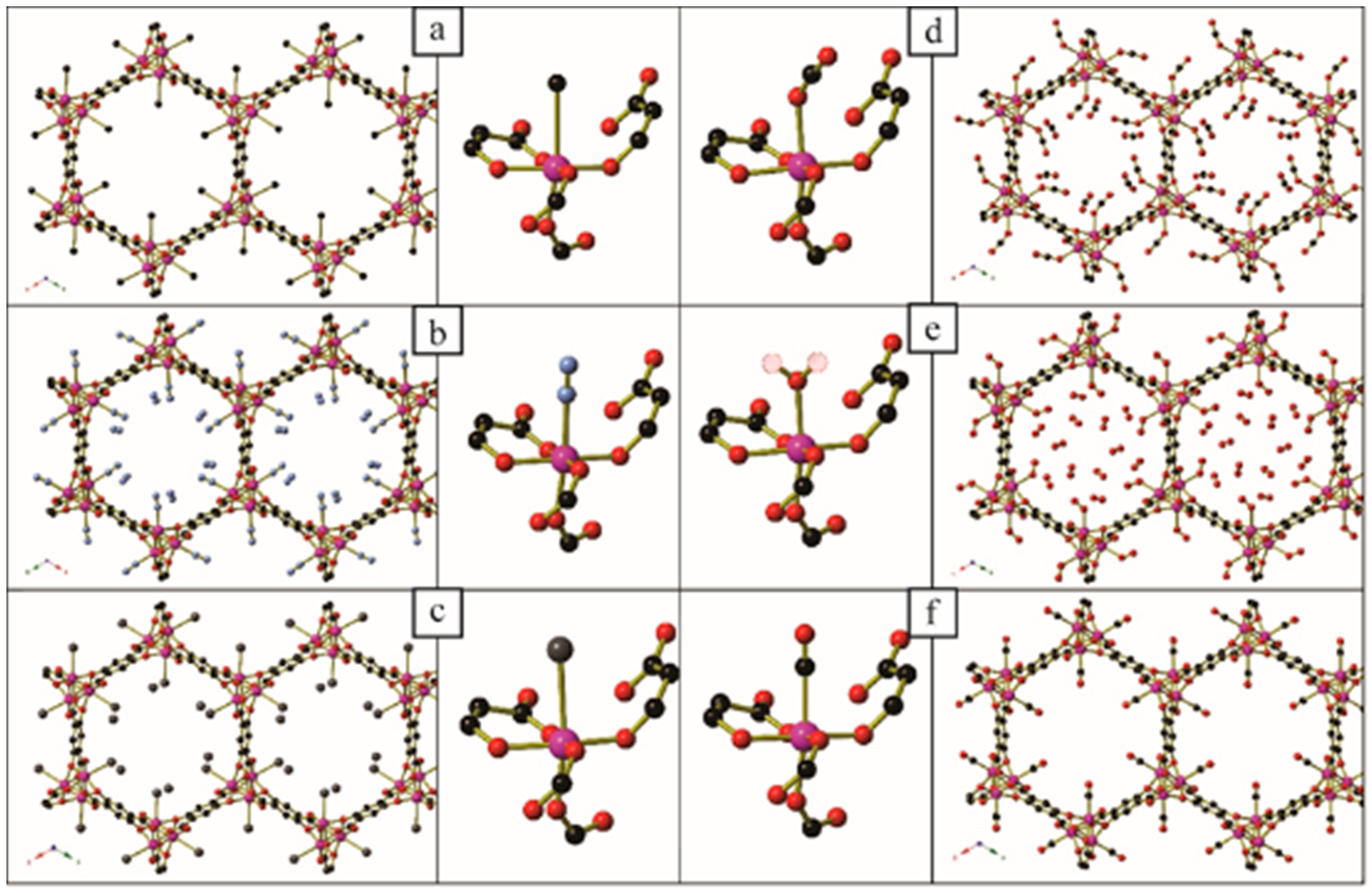
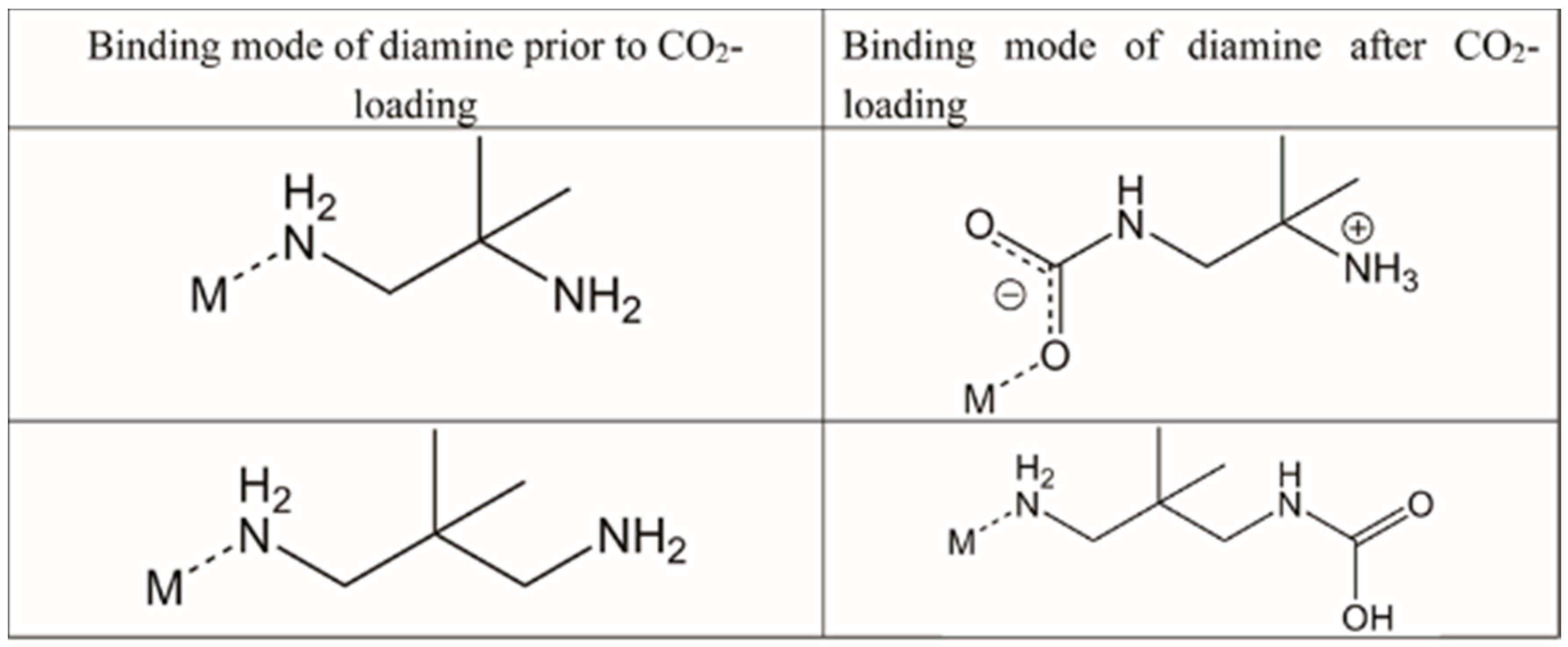
| Diamine Coordinated via 1°, 2° or 3° Amine | Diamine Molecules Adsorbed per Zn2(dobpdc) Formula Unit | Carbamate Oxygen Atom Involved in Intermolecular N-H···O-C Hydrogen Bonds | CO2 Molecules Adsorbed per Zn2(dobpdc) Formula Unit | |
|---|---|---|---|---|
 | 1° | Inconclusive | Bifurcated | 1.62 |
| 2° | Non-coordinated (intramolecular H-bonds to coordinated O) | |||
 | 1° | 1.68 | Coordinated (major) Non-coordinated (minor) | 1.5 |
 | 1° | 1.94 | Coordinated | 1 |
 | 2° | 1.5 | Non-coordinated | 1.5 |
 | 2° | 1 | Non-coordinated | 1 |
 | 1° | 1.83 | * | * |
 | 1° | 1.65 | * | * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCormick, L.J.; Giordano, N.; Teat, S.J.; Beavers, C.M. Chemical Crystallography at the Advanced Light Source. Crystals 2017, 7, 382. https://doi.org/10.3390/cryst7120382
McCormick LJ, Giordano N, Teat SJ, Beavers CM. Chemical Crystallography at the Advanced Light Source. Crystals. 2017; 7(12):382. https://doi.org/10.3390/cryst7120382
Chicago/Turabian StyleMcCormick, Laura J., Nico Giordano, Simon J. Teat, and Christine M. Beavers. 2017. "Chemical Crystallography at the Advanced Light Source" Crystals 7, no. 12: 382. https://doi.org/10.3390/cryst7120382
APA StyleMcCormick, L. J., Giordano, N., Teat, S. J., & Beavers, C. M. (2017). Chemical Crystallography at the Advanced Light Source. Crystals, 7(12), 382. https://doi.org/10.3390/cryst7120382