1. Introduction
Spiropyran and spirooxazine are photochromically active compounds. Their photochromic character stems from a ring-opening isomerization by breaking the C
spiro-O bond upon UV-light absorption [
1]. Spiropyrans are usually found non-photochemically active in solid state at ambient conditions. The limited solid-state response of the compounds is expected to be due to the large structural changes accompanying the ring opening mechanism. In principle, this issue can be tackled using the so-called chemical modification method, in which the spiro compound is given functional groups large enough to open up the crystal structure, allowing the compound to become photochromically active at the solid state. This approach, however, turned out to be challenging [
2]. Another approach consists in modifying the crystal structures by a crystal engineering approach. In this approach, one does not chemically change the form of the original compound, but one targets a modification of the crystal structure. Former investigations of related photo- and thermochromic compounds (i.e., anils) revealed the importance of the present structural environment causing a significant influence on the physical properties [
3]. In that case, the polymorphs of the anils under study differ not only in color but also in their photochromic behavior. Results of Harada et al. showed the opportunity of accessing the photochromic properties of spiropyran and spirooxazine compounds at low temperature in the solid state by suppressing the thermal back reaction [
4]. Combining these two insights, we consider the importance of a basic knowledge about the polymorphic situation of spiropyran and spirooxazines not only as inevitable but also necessary to find a suitable way to enter the photochromism in the solid state.
Thus, our first investigation on this matter set priorities on the intermolecular interactions possibly leading to different arrangements of the analyzed compounds in the crystal lattice. Hereby, mostly non-covalent interactions like van der Waals interactions and hydrogen bonds were decisive and were our main focus [
5]. For the commercially available photochromic compounds that are the target of this work, little to no structural information was available. A search in the Cambridge Structural Database (CSD, Version 5.37, May 2016) [
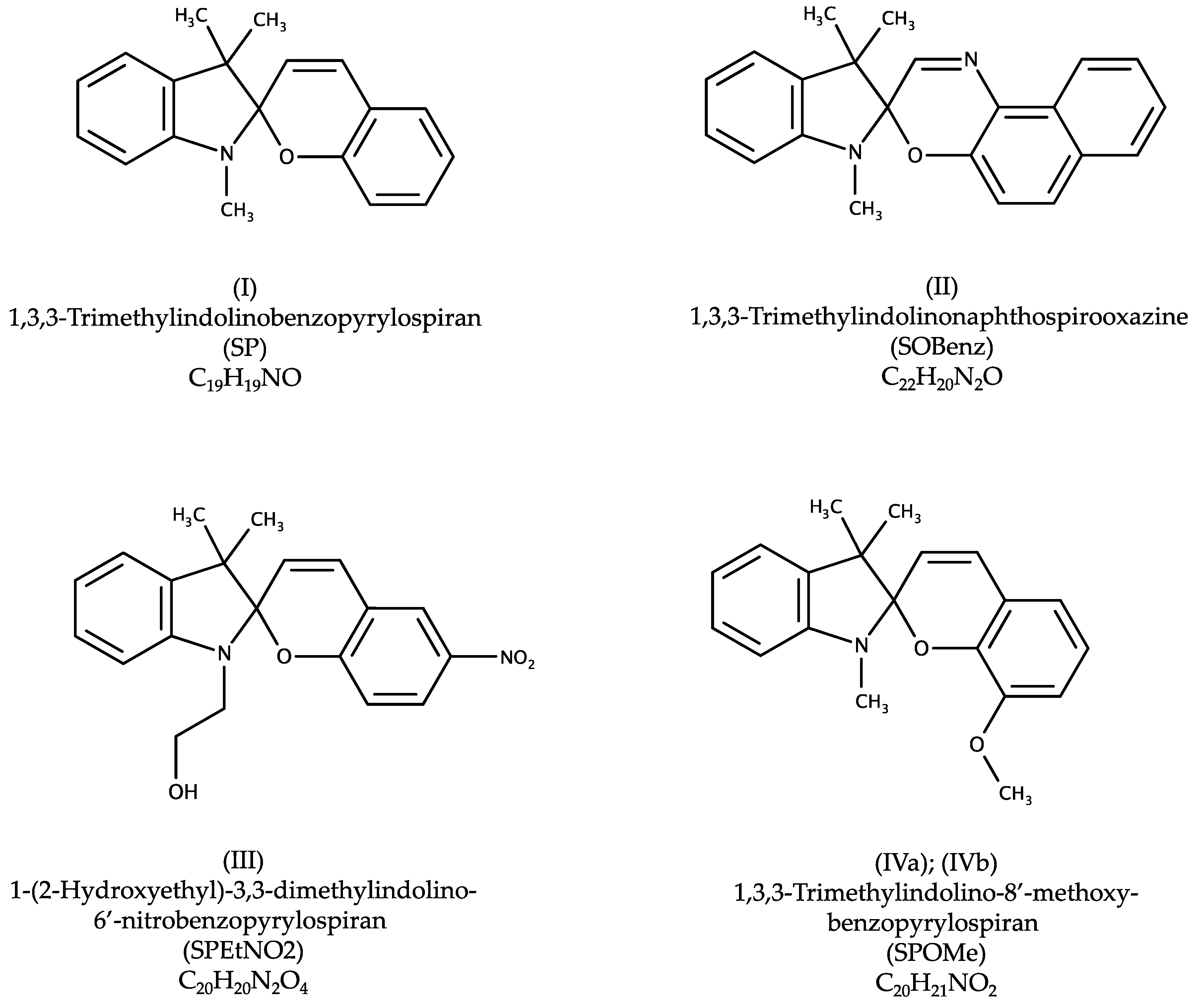
6], restricted to organic and not polymeric structures yielded no match for 1,3,3-Trimethylindolinobenzopyrylospiran (SP) nor 1,3,3-Trimethylindolino-8′-methoxybenzopyrylospiran (SPOMe). Only one hit was found for both 1,3,3-Trimethylindolinonaphtospirooxazine (SOBenz; CSD Refcode JIZJOR/01/02 [
4,
7], polymorph I; JIZJOR03 [
8], polymorph II) and 1-(2-Hydroxyethyl)-3,3-dimethylindolino-6′-nitrobenzopyrylospiran (SPEtNO2; CSD Refcode IHOFOA) [
9]. In order to gain further information on the preferred interaction within crystalline spiro materials, we performed a structural study on the series of commercially available spiro compounds (
Scheme 1) with the goal to gain an overall structural understanding of their interaction patterns. Within this context, we identified five novel crystal structures.
2. Results
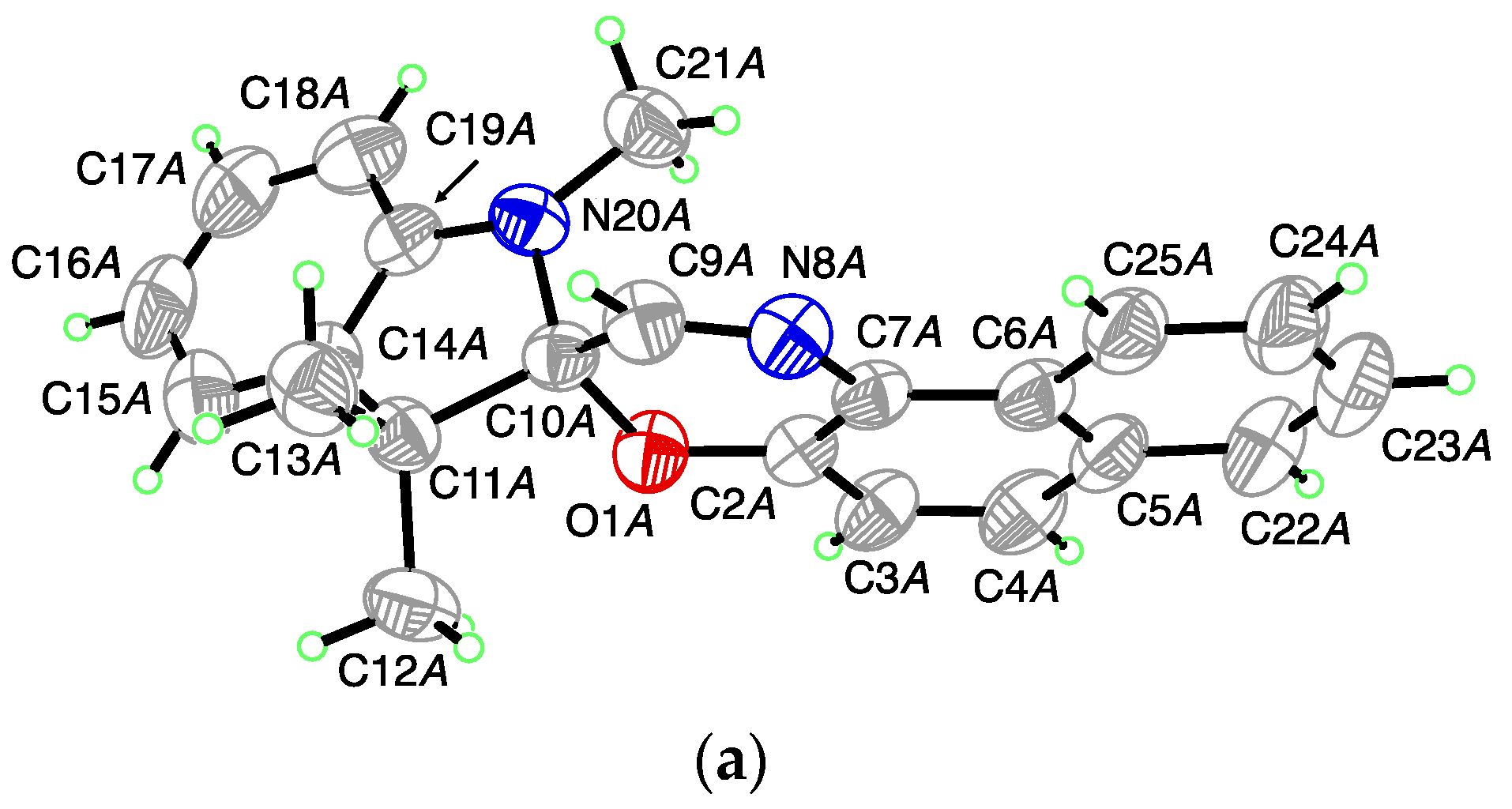
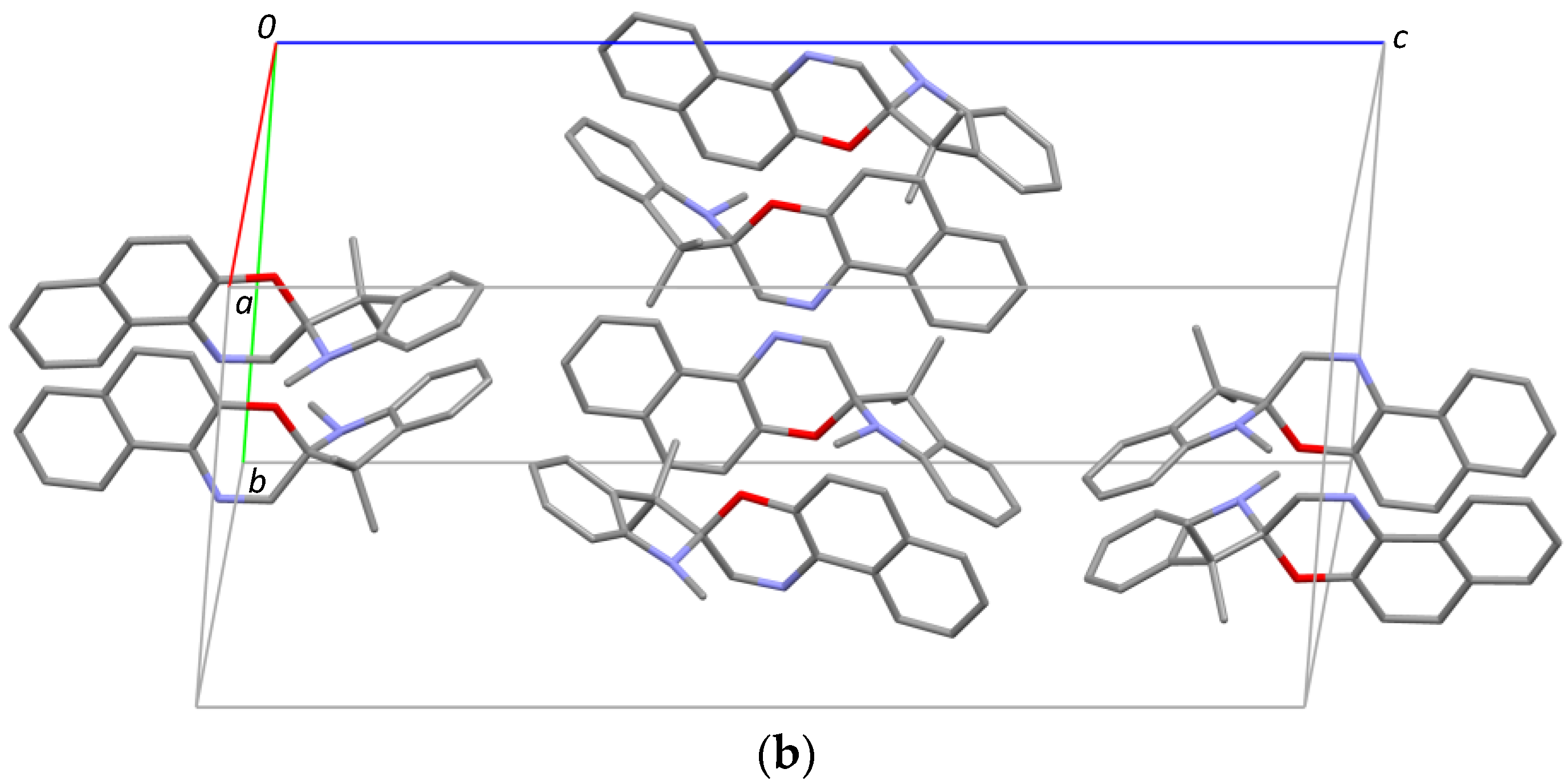
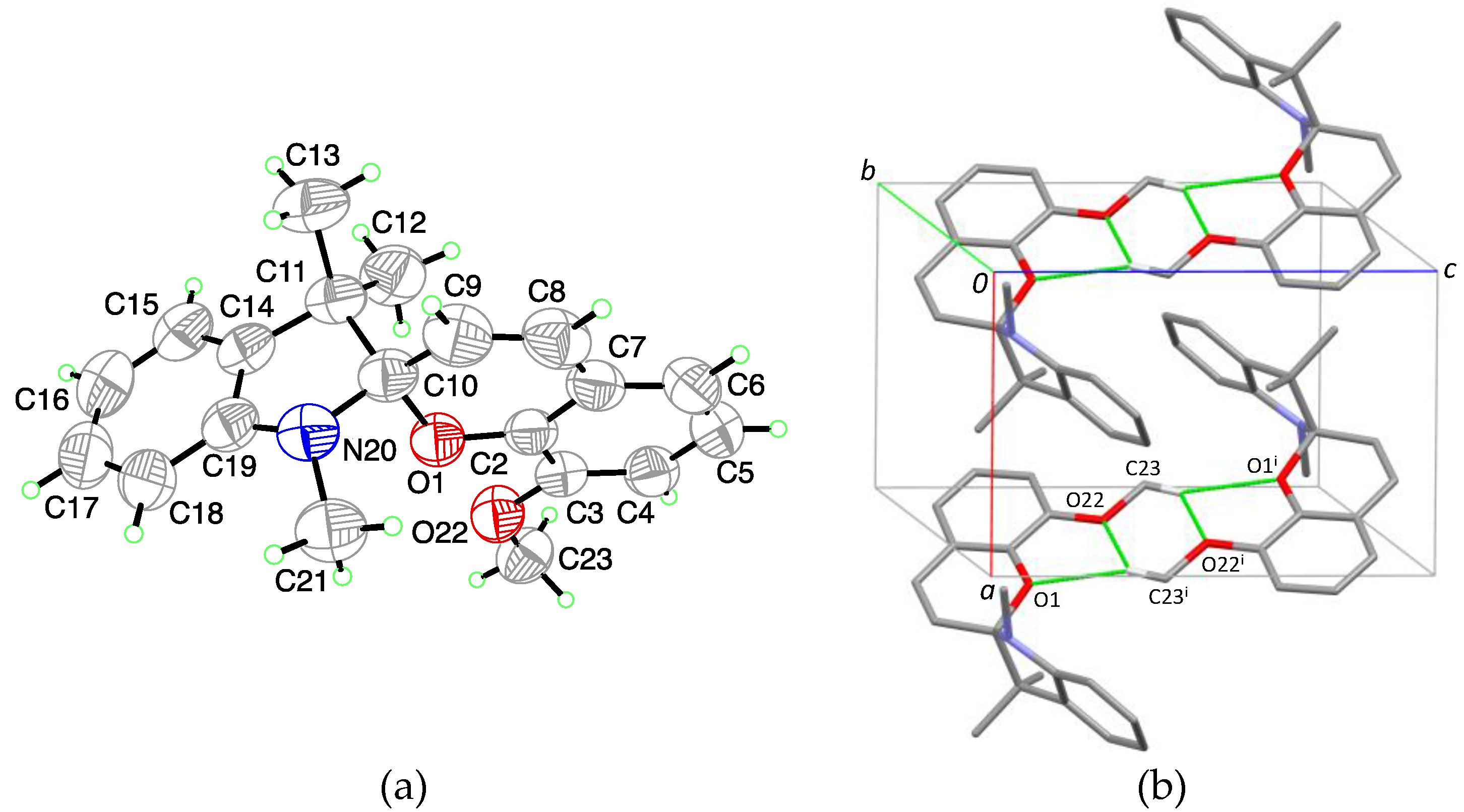
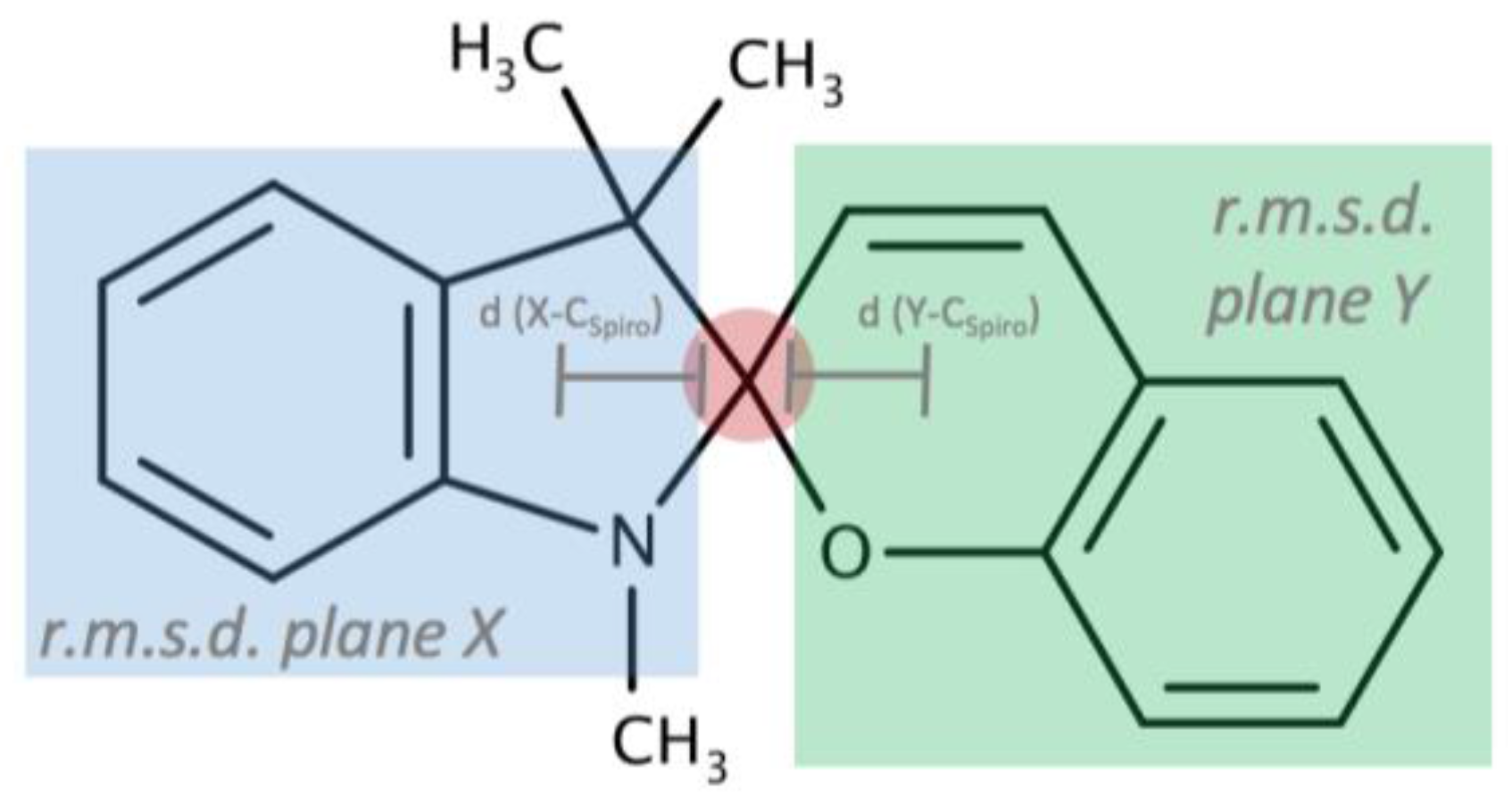
For 1,3,3-Trimethylindolinobenzopyrylospiran (SP), no crystal structure was identified in the literature. (I) crystallizes in the monoclinic space group
P2
1/
c with one molecule in the asymmetric unit (
Figure 1a). Bound together via a spiro junction (C10), the indoline plane (r.m.s. deviation for all non-H-atoms excluding C10 = 0.014 Å) and the chromene plane (r.m.s. deviation for all non-H-atoms excluding C10 = 0.023 Å) shows a dihedral angle of 80.03 (3)° with a distance for the indoline plane of 0.4528 (14) Å and 0.3081 (13) Å for the chromene plane to the chiral spiro center (C10). The C
Spiro-O bond length is 1.4714 (13) Å and the C
Spiro-N bond length is 1.4456 (15) Å. The crystal packing shows isolated units without specific intermolecular interactions (
Figure 1b).
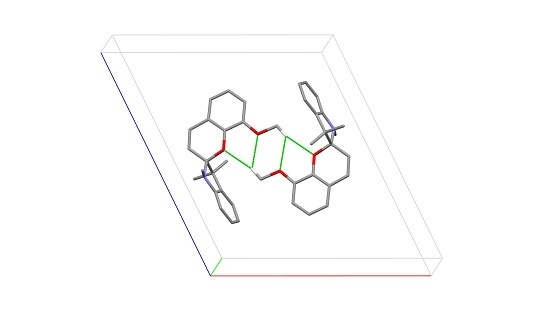
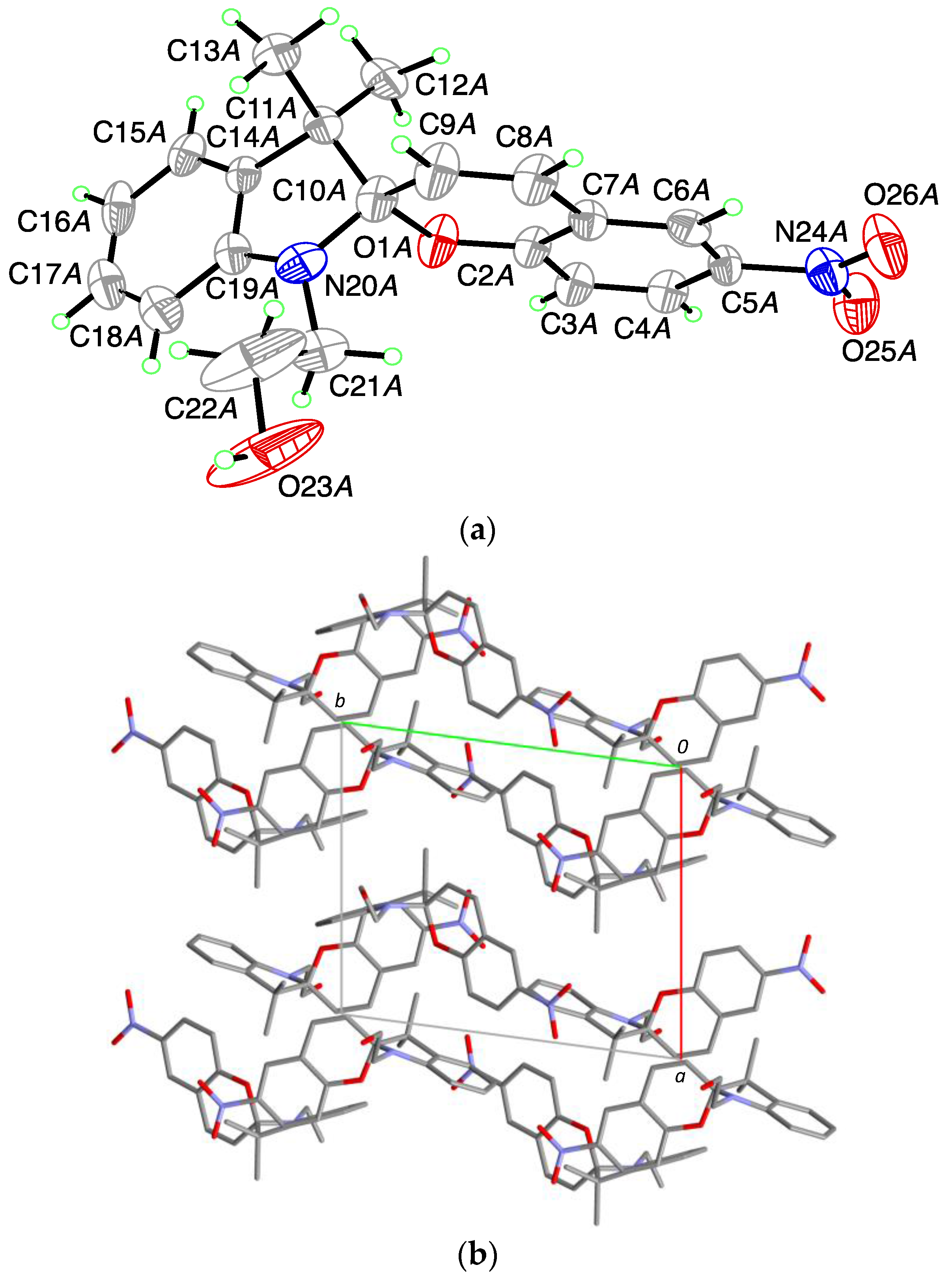
1,3,3-Trimethylindolinonaphthospirooxazine, (SOBenz), has two known polymorphic crystal structure in the CSD crystallizing in the orthorhombic space group
Pbca and
P2
1 (CSD Refcode JIZJOR/01/02 and JIZJOR03, respectively). Here, we identified another structure (II) for this compound, which crystallizes in the monoclinic space group
Pc with four molecules in the asymmetric unit (
A,
B,
C and
D;
Figure 2a). Each of the molecules show a different dihedral angle between the indoline plane (r.m.s. deviation for all non H-atoms excluding C10 = 0.020 Å for
A; 0.020 Å for
B; 0.026 Å for
C and 0.021 Å for
D) and the benzochromene plane (r.m.s. deviation for all non H-atoms excluding C10 = 0.046 Å for
A; 0.043 Å for
B; 0.013 Å for
C and 0.022 Å for
D), of respectively:
A: 79.74 (8)°;
B: 81.99 (10)°;
C: 86.46 (9)°;
D: 83.75 (8)°. This leads to a distance of the chiral spiro center (C10) from the above defined planes for the indoline part of respectively 0.360 (4) Å for
A; 0.365 (4) Å for
B, 0.371 (4) Å for
C and 0.375 (4) Å for
D and for the chromene part of respectively 0.399 (4) Å for
A, 0.342 (4) Å for
B, 0.168 (4) Å for
C and 0.249 (4) Å for
D. Within the asymmetric unit, two S- and two R-enantiomers are observed. The C
Spiro-O-bond length is on average 1.459 (4) Å (1.458 (4) Å for
A and
D; 1.461 (4) Å for
B and 1.460 (4) Å for
C). The C
Spiro-N-bond length is on average 1.442 (5) Å (1.434 (4) Å for
A, 1.443 (5) Å for
B, 1.446 (5) Å for
C and 1.445 (5) Å for
D). In the crystal packing (
Figure 2b) no strong intermolecular interactions are observed showing
A and
C as well as
B and
D related through a pseudo twofold axis along
a.
For 1-(2-Hydroxyethyl)-3,3-dimethylindolino-6′-nitrobenzopyrylospiran, (SPEtNO2), a single entry was found in the CCD, with the compound crystallizing in the monoclinic space group
I2/
a (IHOFOA). We obtained a polymorphic form for this compound, (III), which crystallizes in the triclinic space group
P with two disordered molecules in the asymmetric unit capturing the S- (
A) and the R-Conformer (
B), (
Figure 3a). The indoline plane (r.m.s. deviation for all non H-atoms excluding C10 = 0.017 Å for
A and 0.016 for
B) and the chromene plane (r.m.s. deviation for all non H-atoms excluding C10 = 0.018 Å for
A and 0.014 Å for
B) present a dihedral angle of 88.61 (12)° for
A and 89.45 (11)° for
B with the chiral center displaced by 0.402 (5) Å for
A and 0.405 (5) Å for
B from the indole plane and 0.073 (8) Å for
A and 0.046 (7) Å for
B from the chromene moiety. The C
Spiro-O-bond length is 1.485 (5) Å for
A and 1.488 (5) Å for
B. The C
Spiro-N-bond length is 1.427 (6) Å for
A and 1.426 (6) Å for
B. In the crystal packing, molecules of
A and
B are isolated, showing no specific intermolecular interactions (
Figure 3b).
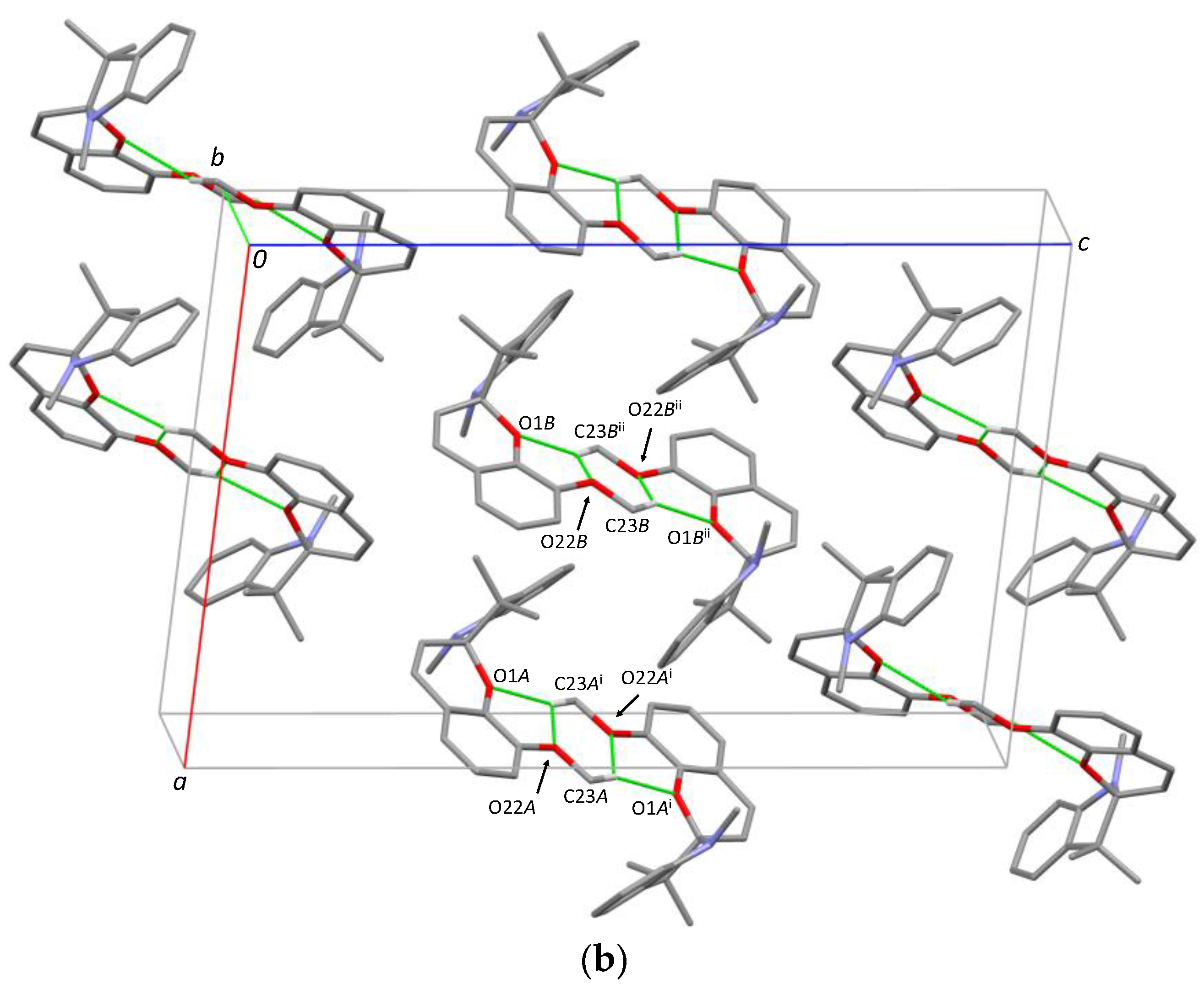
For 1,3,3-Trimethylindolino-8′-methoxybenzopyrylospiran, (SPOMe), we were able to crystallize two polymorphs, (IVa) and (IVb). (IVa) belongs to the monoclinic space group
P2
1/
c, with two molecules in the asymmetric unit (
A and
B;
Figure 4a). The dihedral angle of the indoline plane (r.m.s. deviation for all non H-atoms excluding C10 = 0.014 Å for
A and 0.017 Å for
B) and chromene plane (r.m.s deviation for all non H-atoms excluding C10 = 0.049 Å for
A and 0.028 Å for
B) encloses 77.06 (3)° for
A and 79.11 (4)° for
B. For molecule
A, the distance to the chiral center C10 is 0.382 (2) Å for the indoline plane and 0.326 (2) Å for the chromene plane. The respective distances in
B are 0.394 (2) Å and 0.311 (2) Å. The arrangement of the methoxy substituent is in both cases antiperiplanar to C2 and slightly rotated out of the planar indoline part (2.29 (2)° for
A and 6.13 (2)° for
B). The C
Spiro-O bond shows a length of 1.4635 (19) Å for
A and 1.4672 (18) Å for
B and the C
Spiro-N bond a length of 1.445 (2) Å for both
A and
B. In the crystal packing (
Figure 4b), additional intermolecular C-H
…O van der Waals interactions are formed for
A (C23
A-H
…O1
Ai; C23
A-H
…O22
Ai) as well for
B (C23
B-H
…O1
BiI; C23
B-H
…O22
BiI) around the inversion center. [Symmetry code: (i) 2 − x, 1 − y, 1 − z; (ii) 1 − x, 1 − y, 1 − z].
The polymorphic structure (IVb) crystallizes in the triclinic space group
P. The asymmetric unit contains one molecule for which the indoline plane (r.m.s. deviation for all non H-atoms excluding C10 = 0.022 Å) and chromene plane (r.m.s. deviation for all non H-atoms excluding C10 = 0.032 Å) enclose a dihedral angle of 78.07° (3). The distance to the chiral center C10 is 0.3849 (17) Å for the indoline plane and 0.2836 (18) Å for the chromene plane. The methoxy substituent is slightly rotated outwards by 1.81° (2) with respect to the indoline plane and takes an antiperiplanar arrangement to C2. The C
Spiro-O and C
Spiro-N bonds show respectively bond lengths of 1.4672 (16) Å and 1.444 (2) Å. In the crystal packing weak C-H
…O van-der Waals interactions (C23-H
…O1
i; C23-H
…O22
i) are obtained similar to structure (IVa) (
Figure 5b). [Symmetry code: (i) − x, 1 − y, 1 − z].
3. Discussion
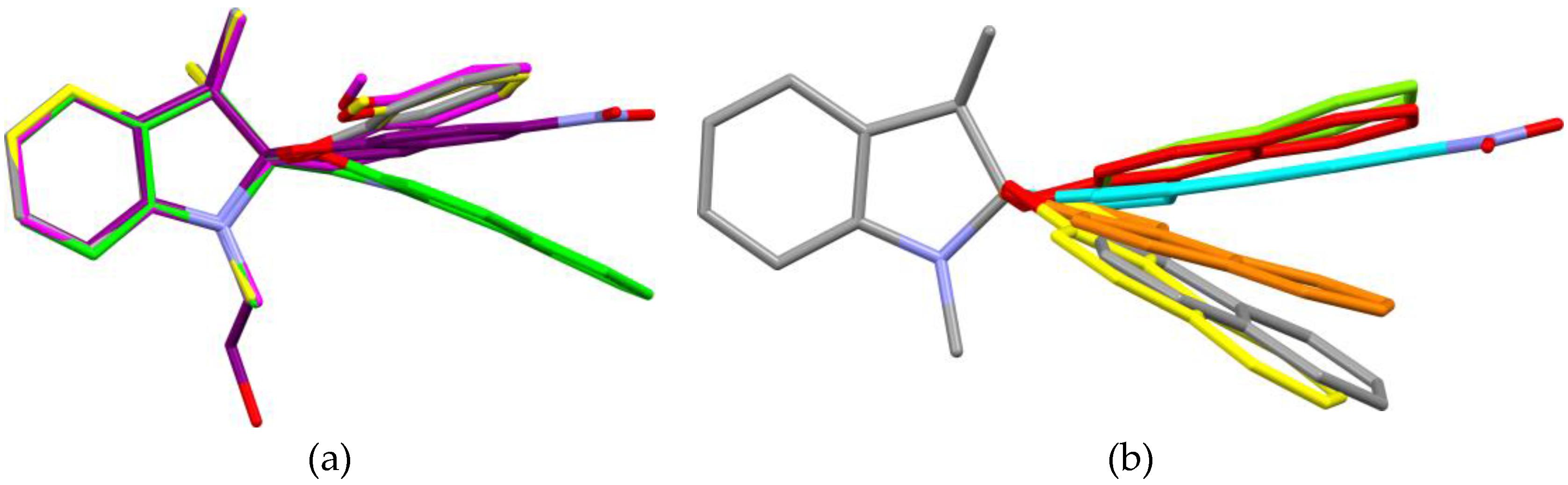
A closer look at the structural features shows a high similarity between the different compounds. All compounds crystallize out as racemic compounds, containing both R and S enantiomers (
Appendix Table A1). Furthermore, in all cases, the indoline heterocycle plane (excluding the central chiral center C10) shows the arrangement of a flattened envelope with a distance of approximately 0.4 Å to the chiral center C10. The chromene plane (excluding the C10 atom) makes an angle with respect to the first plane. All angles can be found in a 13° range from one another. A strict overlay of the indoline heterocycles in all five crystal structures (
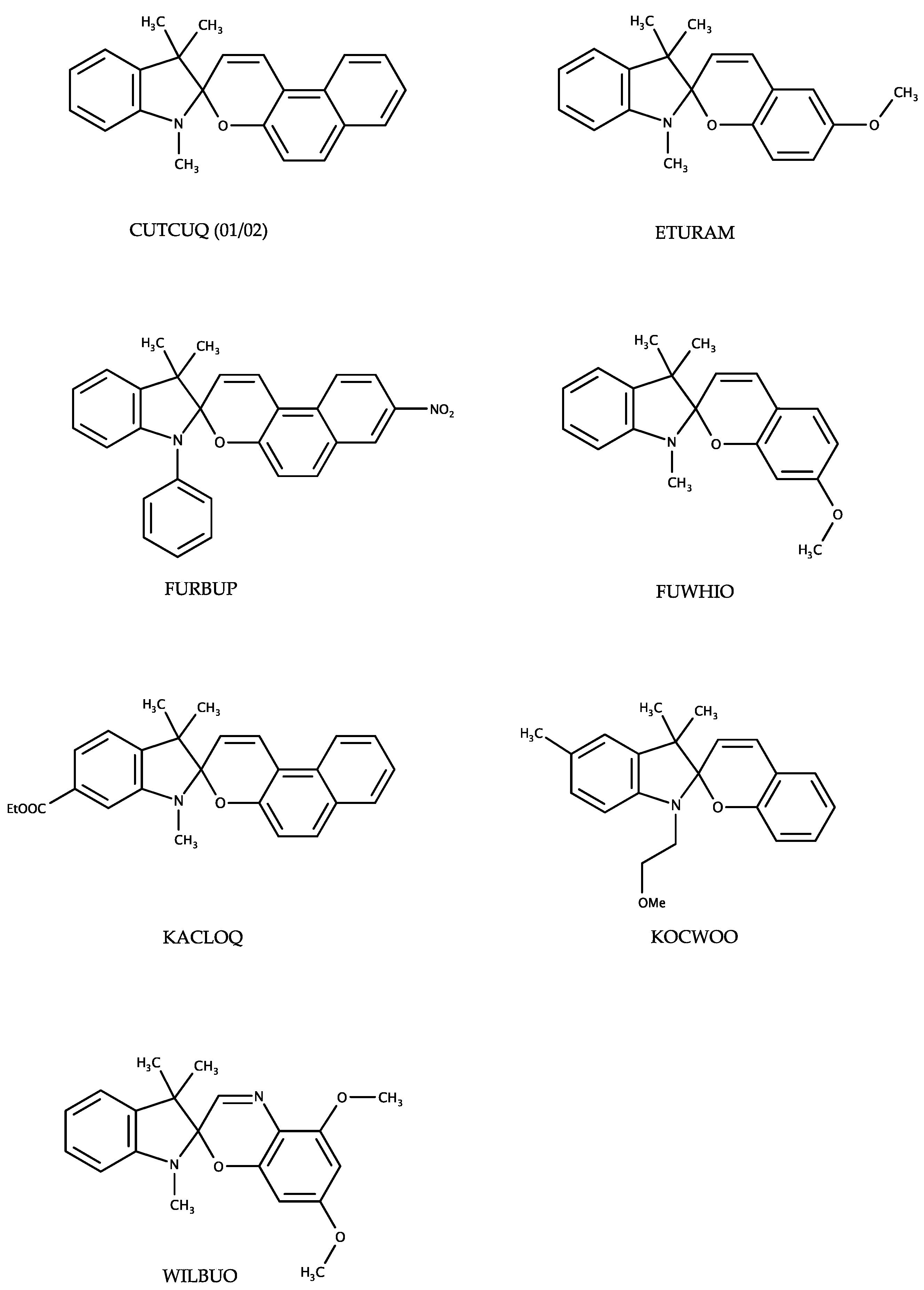
Figure 6a) shows the structural variety of the chromene plane. The distance of the latter plane to the chiral center C10 varies from 0.4 Å to 0.0 Å. The chromene plane can bend either towards the geminal methyl groups or away from these. Focusing on chromene heterocyles of structural related entries in the CSD (CUTQUQ01 [
4], CUTCUQ 02 [
10], FURBUP [
11], KACLOQ [
12], KOCWOO [
13]) the same flexibility of the angle is observed (
Figure 6b).
However, in all structures the typical weakening of the CSpiro-O bond to 1.47 Å is observed, which correspond to the bond length indicated for spiropyrans and spirooxazines in all known crystal structures of the CSD. The bond lengths extensions amount 0.06 Å compared to normal C-O bonds (1.41 Å) in six-membered heterocylces extracted from overall crystal structures in the CSD database. Simultaneously, the strengthening of the CSpiro-N bond agrees with the gained results. It is shortening in average about 0.03 Å from 1.47 Å for C-N bonds in five-membered heterocycles to 1.44 Å in spiropyrans and spirooxazines.
Comparing the structures (I)–(IV), no strong interactions in terms of inter- or intramolecular hydrogen bonding are observed. Even introduction of additional functional groups, such as -NO2 and -OMe, only lead to weak C-H…O interactions as shown in structures (IVa) and (IVb) by two facing methoxy groups in a motif. In all cases, the methoxy group shows an approximate coplanar arrangement compared to the chromene plane, with atoms C4 and C23 adopting a synperiplanar conformation.
A CSD search of spiropyran and spirooxazine compounds with one or more methoxy substituents connected to the chromene moiety (restricted to organic and not polymeric structures; selection of one polymorph per data set) yielded three hits (ETURAM [
14], FUWHIO [
11] and WILBUO [
15];
Appendix Scheme A1). In ETURAM, a similar interaction is given by the methoxy group compared to (IVa) and (IVb). This is also displayed by one of the methoxy substituents in WILBUO. Another
van der Waals interaction can be formed in FUWHIO. In every presented crystal structure, an almost coplanar arrangement of the methoxy substituent with respect to the chromene heterocycle is found. A nitro substituent attached to a Spiropyran molecule, for example in (III) and IHOFOA shows less directionality resulting in weak N-O
…H van der Waals contacts in direct comparison to a methoxy group, yet the coplanar arrangement regarded to the chromene moiety is maintained.
A direct comparison of the structural polymorphic pairs (III) and IHOFOA, (II) and JIZJOR03 as well as (IVa) and (IVb) show high similarity in the packing features. Whereas the first-mentioned pair can be assumed to be temperature polymorphs, the remaining pairs show an almost perfect overlay, despite different space groups. Close examination reveals that JIZJOR03 structure was determined using inappropriate selection of unit cell settings, most probably due to weak diffraction of the crystal on Mo radiation. The author refined the resulting structure using twinning and disorder in order to describe the structure using their model. Transformation matrix from our unit cell setting to their setting is (0 1 0, −1 0 0, 0.5 −0.5 1). This situation became possible due to an unfortunate combination of circumstances: Z′ = 4 and packing features of the structure.
The gained insights into the five new presented crystal structures are helpful in order to improve the basic understanding of the intermolecular interactions occurring in the solid state necessary to selectively modify the crystal structure for any future work. With the aid of the additional gained knowledge of the polymorphism we intend to engineer the crystal structure selectively in our upcoming work.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


















