
Computer Simulations of Crystal Growth Using a Hard-Sphere Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Crystalline Phase in a Hard-Sphere System
1.2. Colloidal Crystals and Effective Hard-Sphere Model
2. Simulation Methods
2.1. MD Method for Hard Spheres in General
2.2. MC Method Employed for Hard Spheres under Gravity
3. Hard-Sphere Crystal/Fluid Interface
3.1. Crystal–Fluid Coexistence in Equilibrium
3.2. Interface Properties
4. Hard-Sphere System under Gravity
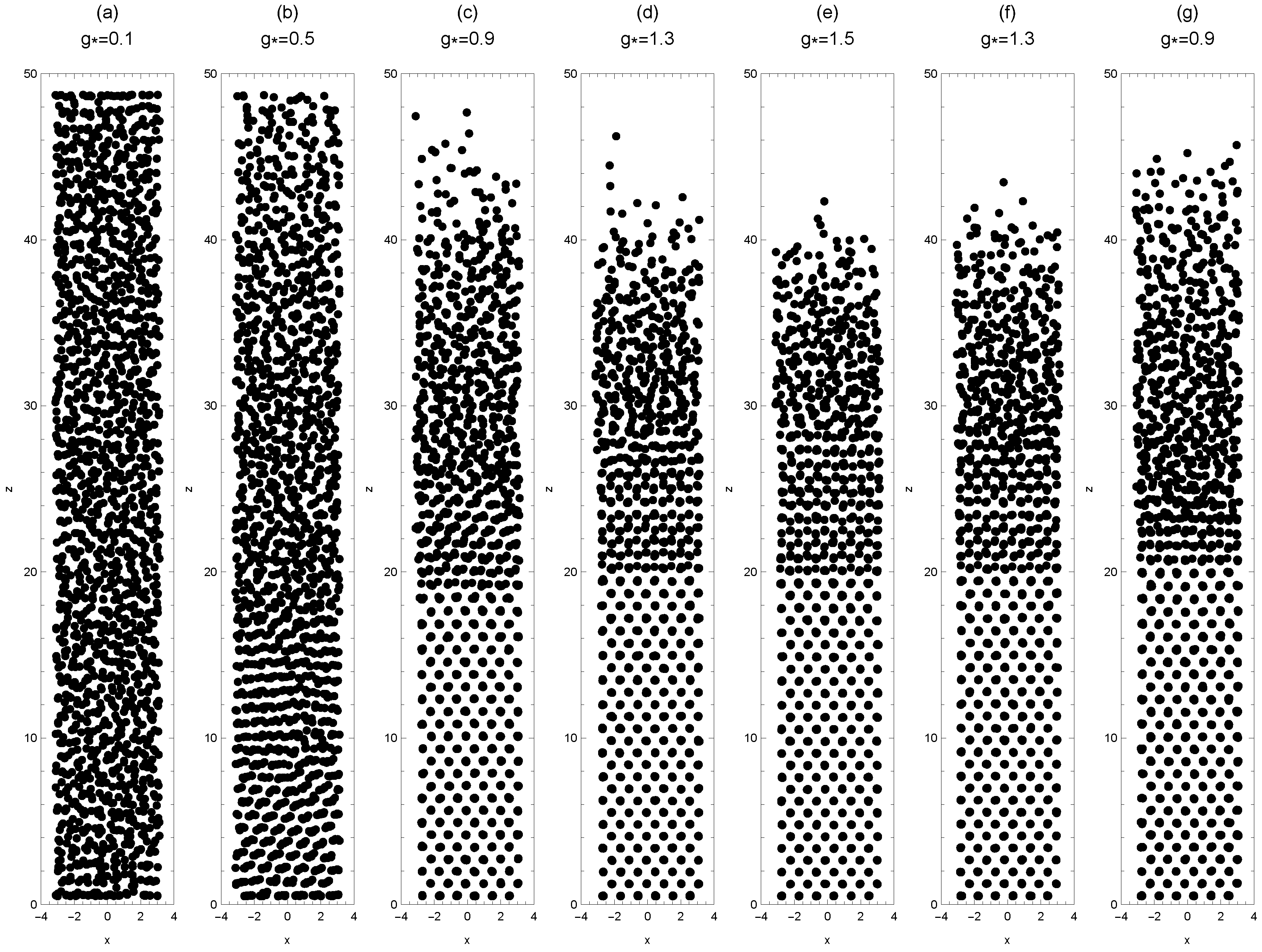
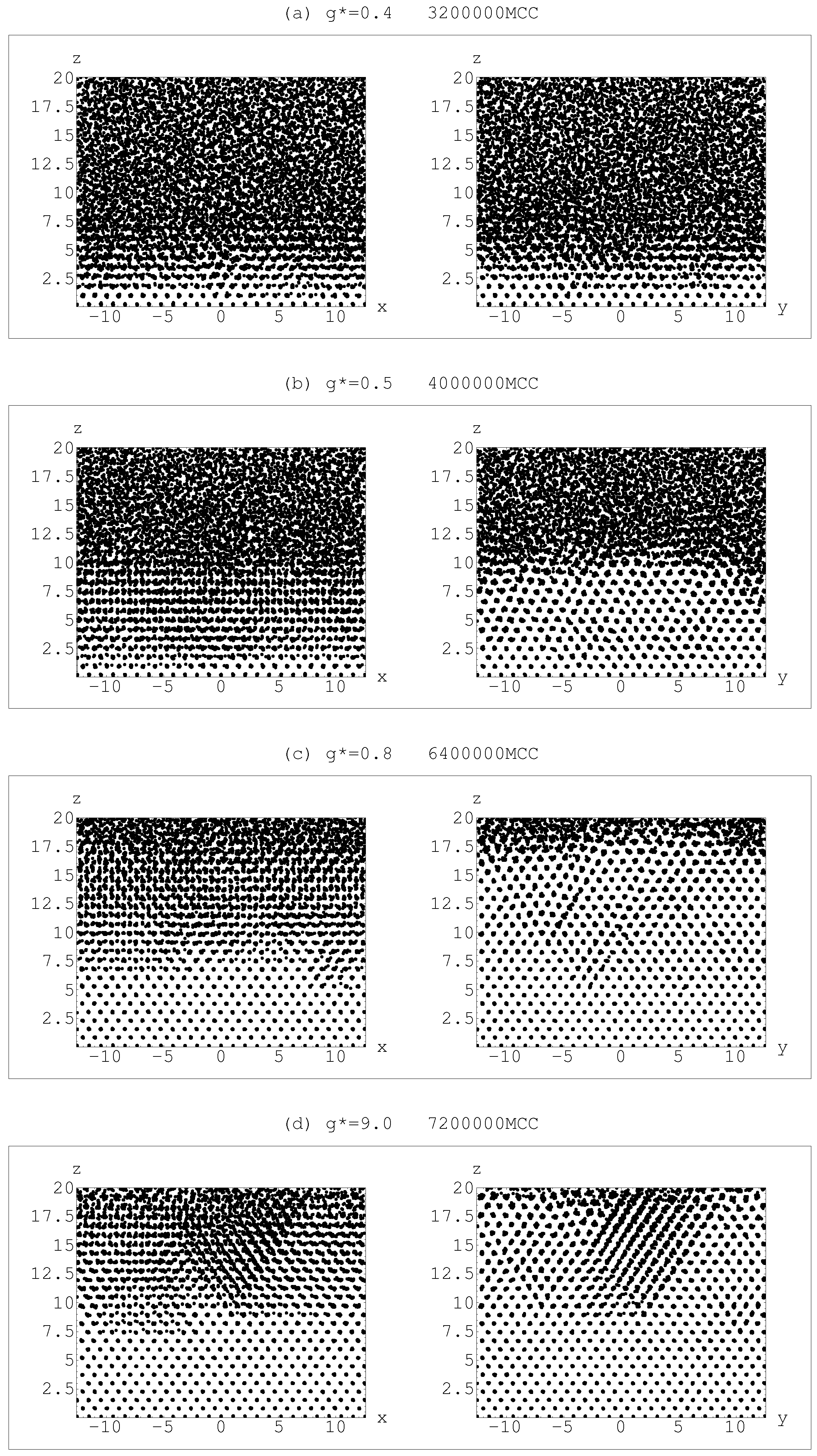
4.1. Sedimentation
4.2. Colloidal Epitaxy
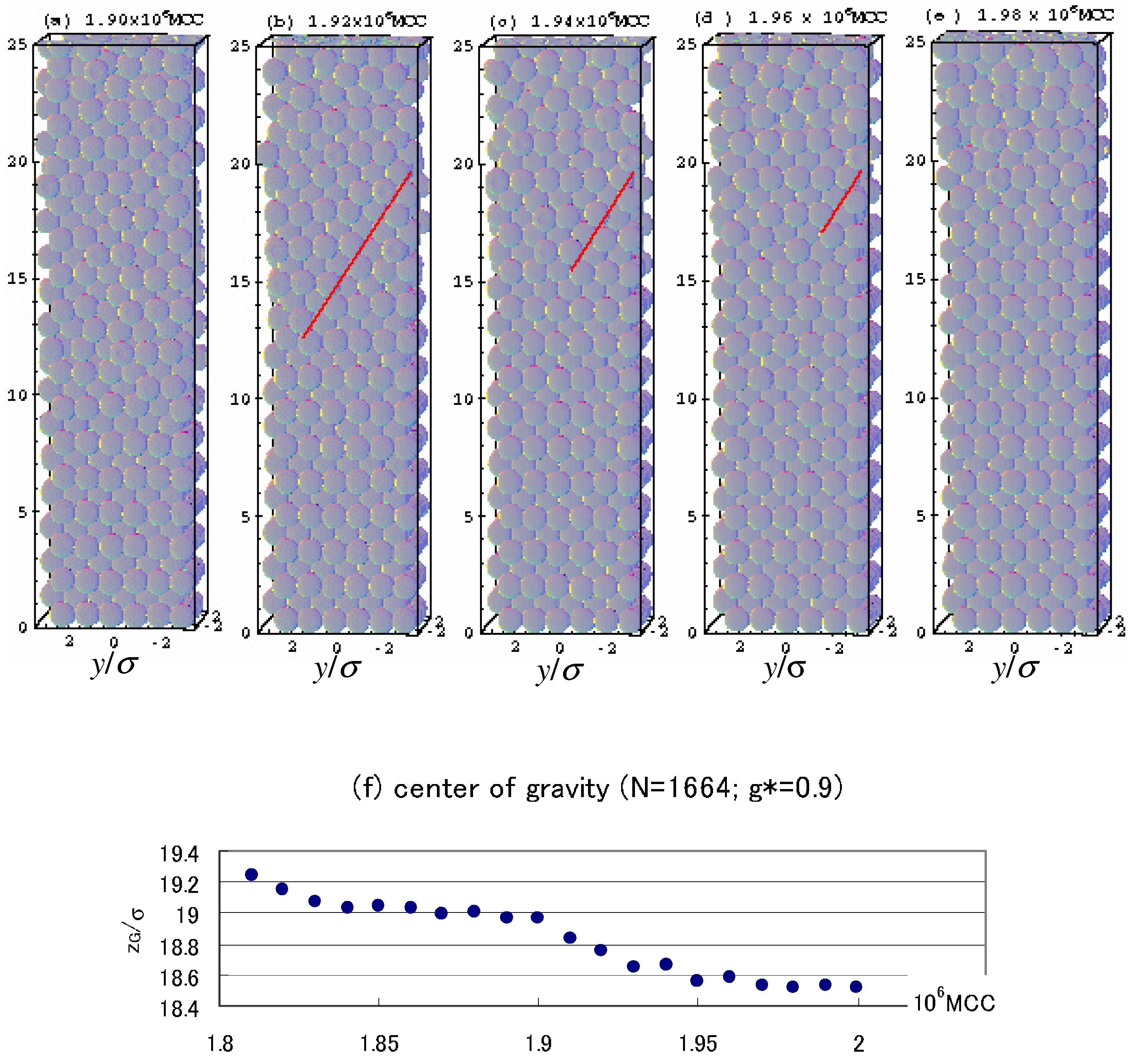
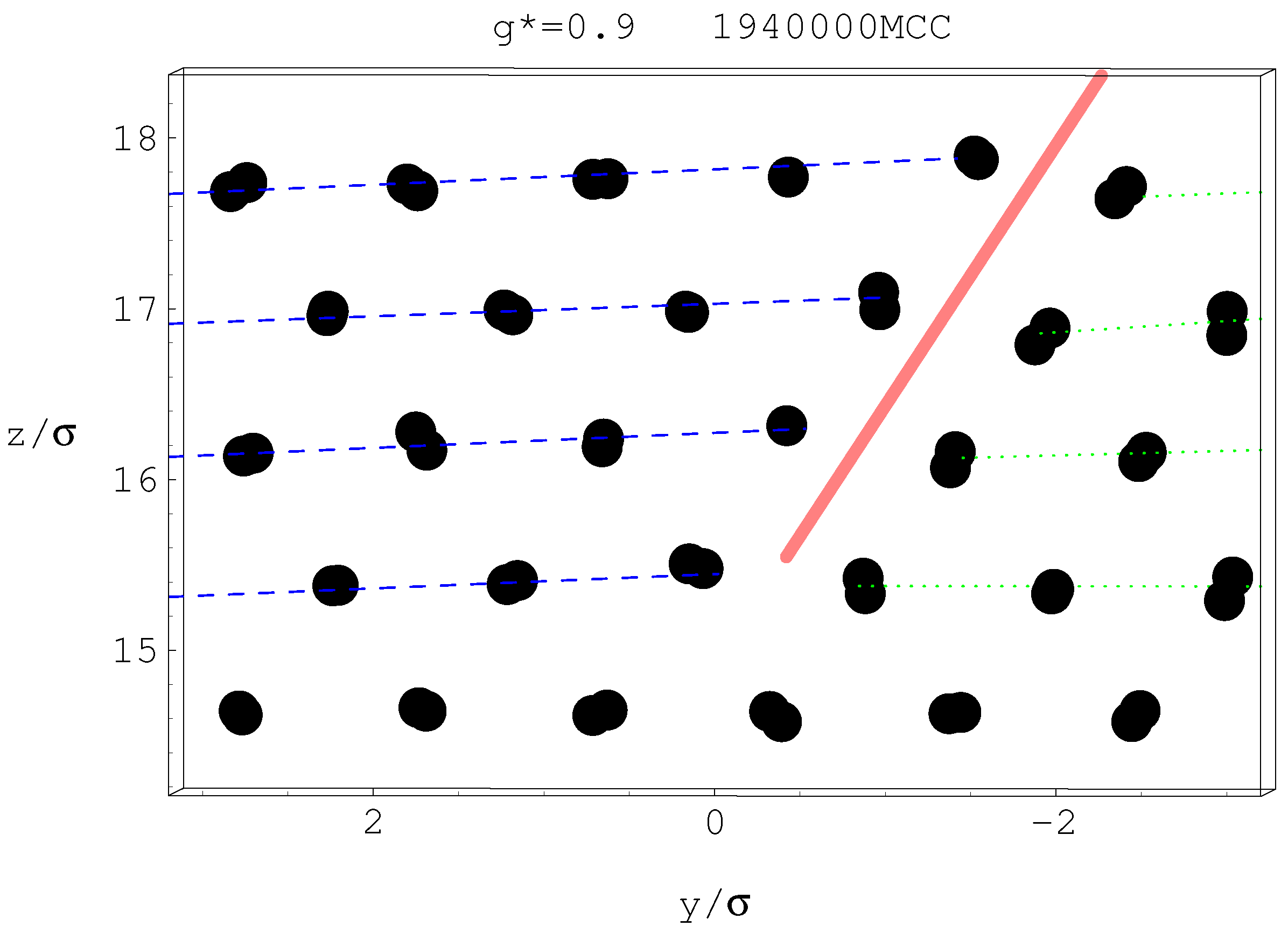
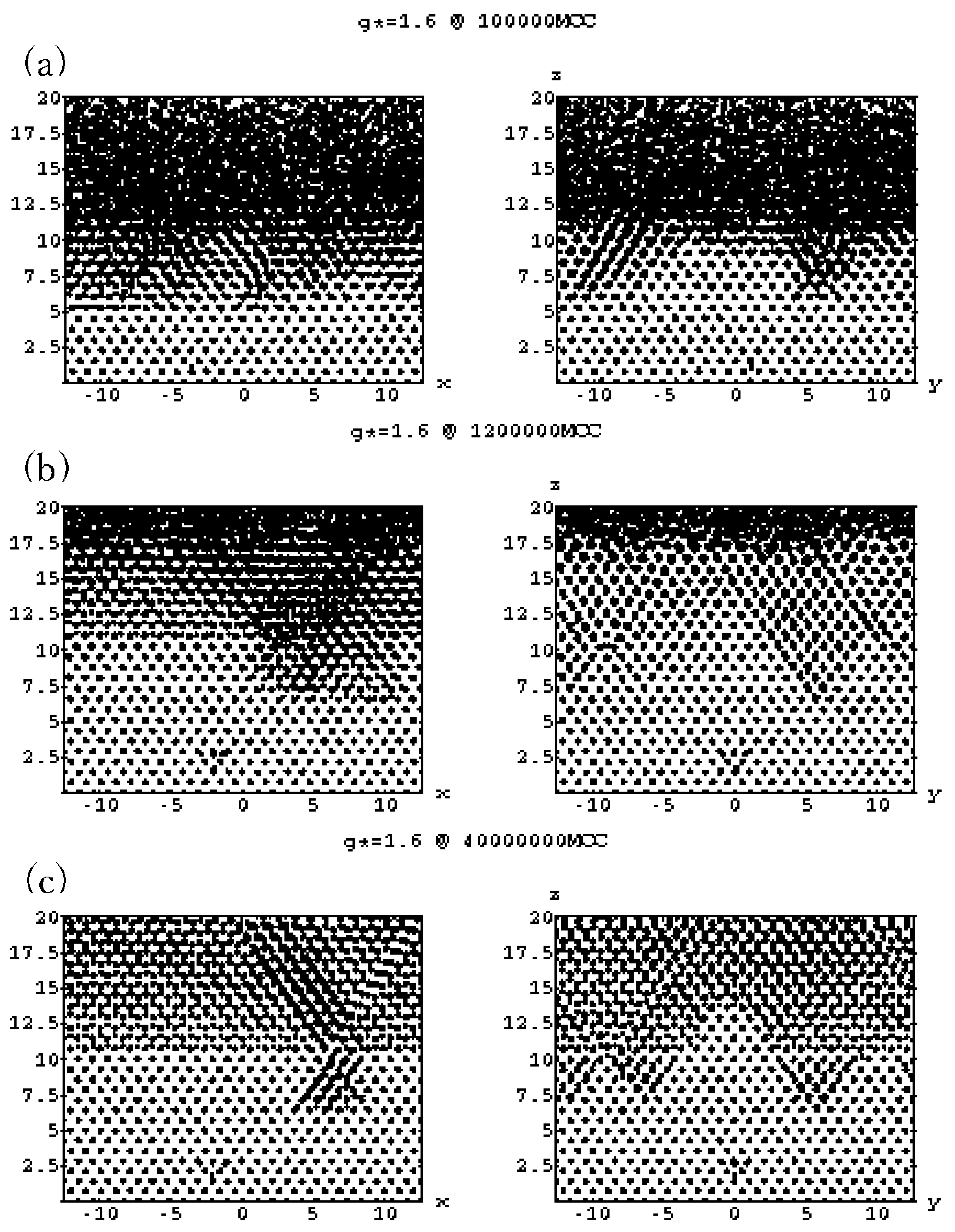
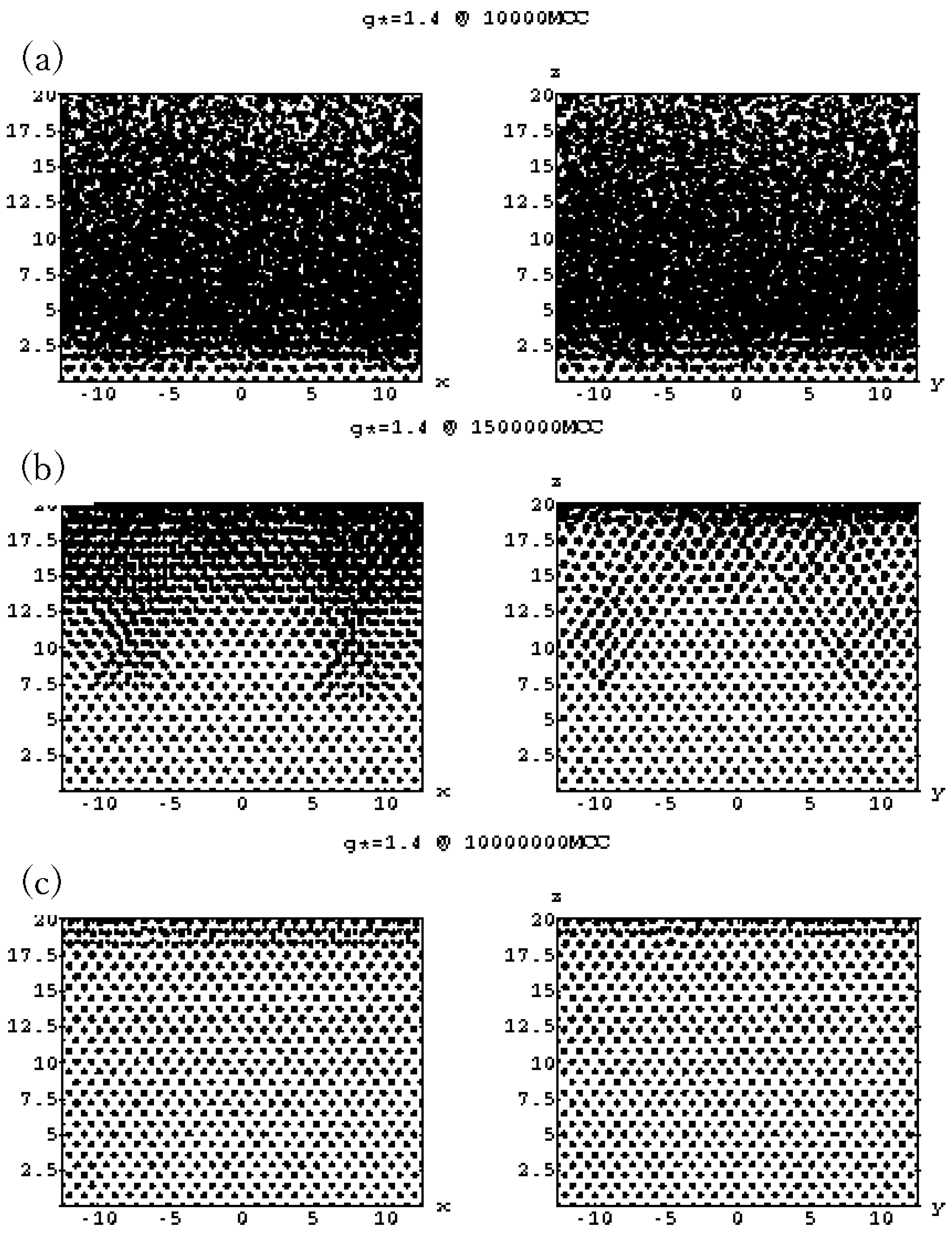
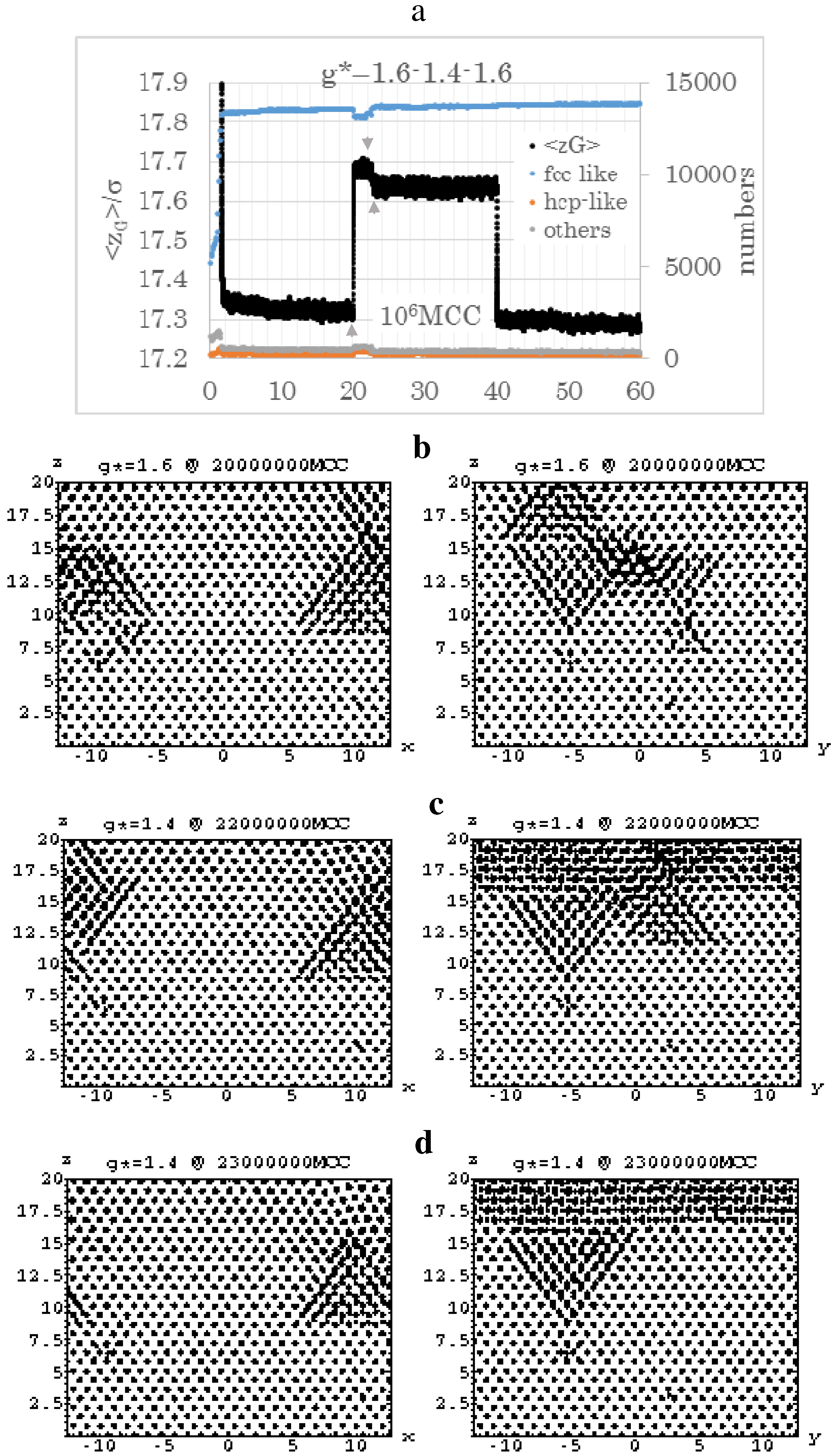
4.3. Gravitational Annealing/Tempering
5. Discussion
5.1. Hard-Sphere Crystallization without External Field
5.2. Processing of Colloidal Crystals Using Centrifugation
6. Summary
Acknowledgments
Conflicts of Interest
References
- Wood, W.W.; Jacobson, J.D. Preliminary Results from a Recalculation of the Monte Carlo Equation of State of Hard Spheres. J. Chem. Phys. 1957, 27, 1207–1208. [Google Scholar] [CrossRef]
- Alder, B.J.; Wainwright, T.E. Studies in Molecular Dynamics. II. Behavior of s Small Number of Elastic Spheres. J. Chem. Phys. 1957, 27, 1208–1209. [Google Scholar] [CrossRef]
- Alder, B.J.; Wainwright, T.E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1960, 33, 1439–1451. [Google Scholar] [CrossRef]
- Percus, J.K. (Ed.) The Many Body Problem; Interscience: New York, NY, USA, 1968; Chaps. XXII and XXVIII. [Google Scholar]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Onsager, L. The Effect of Shape on the Interaction of Colloidal Particles. Ann. NY. Acad. Sci. 1949, 51, 627–659. [Google Scholar] [CrossRef]
- Kimura, H. Statistical Theory of Liquid Crystalline Orderings in Hard Rod Fluids—Nematic, Choresteric, Smectic and Columnar Phases. In Ordering in Macromolecular Systems; Springer-Verlag: Berlin, Germany, 1994; pp. 125–138. [Google Scholar]
- Hoover, W.G.; Ree, F.H. Melting Transition and Communal Entropy for Hard Spheres. J. Chem. Phys. 1968, 49, 3609–3617. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Laird, B.B. Simulation of the Hard-sphere Crystal-melt Interface. J. Chem. Phys. 1998, 108, 9452–9462. [Google Scholar] [CrossRef]
- Zykova-Timan, Z.; Horbach, J.; Binder, K. Monte Carlo Simulations of the Solid-liquid Transition in Hard Spheres and Colloid-polymer Mixtures. J. Chem. Phys. 2010, 133, 014705. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.A.; Martín-Mayor, V.; Seoane, B.; Verrocchio, P. Equilibrium Fluid-Solid Coexistence of Hard Spheres. Phys. Rev. Lett. 2012, 1–8, 165701. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.A. Mechanism of Growth. In Liquid Metals and Solidification; American Society of Metals: Cleveland, OH, USA, 1958; pp. 174–186. [Google Scholar]
- Woodruff, D.P. The Solid-Liquid Interface; Cambridge University Press: London, UK, 1973. [Google Scholar]
- Alfrey, T., Jr.; Braford, E.B.; Vanderhoff, J.W. Optical Properties of Uniform Particle-Size Latexes. J. Opt. Soc. Am. 1954, 44, 603–609. [Google Scholar] [CrossRef]
- Lux, V.W.; Klier, M.; Wesslau, H. Über Bragg-Reflexe mit Sichtbarem Licht an Monodispersen Kunststofflatices. I. Ber. Bunsenges. Phys. Chem. 1963, 67, 75–83. [Google Scholar]
- Derjaguin, B.; Landau, L. Theory of the Stability of Strongly Charged Lyophobic Sols and of the Adhesion of Strongly Charged Particles in Solutions of Electrolytes. Acta Phys. Chim. 1941, 14, 633–662. [Google Scholar]
- Verway, E.J.W.; Overbeek, J.T.G. Theory of the Stability of Lyophobic Colloids; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Watachi, M.; Toda, M. An Evidence for the Existence of Kirkood-Alder Transition. J. Phys. Soc. Jpn. 1972, 32, 1147. [Google Scholar]
- Anti, L.; Goodwin, J.W.; Hill, R.D.; Ottewill, R.H.; Owens, S.M.; Papworth, S. The Preparation of Poly(methyl methacrylate) Lattices in Non-aqueous Media. Coll. Surf. 1986, 17, 67–78. [Google Scholar]
- Pusey, P.N.; van Megen, W. Phase Behavior of Concentrated Suspensions of Nearly Hard Colloidal Spheres. Nature 1986, 320, 340–342. [Google Scholar] [CrossRef]
- Underwood, S.M.; Taylor, J.R.; van Megen, W. Sterically Stabilized Colloidal Particles as Model Hard Spheres. Langmuir 1994, 10, 3550–3554. [Google Scholar] [CrossRef]
- van Blaaderen, A.; Ruel, R.; Wiltzius, P. Template-directed Colloidal Crystallization. Nature 1997, 385, 321–324. [Google Scholar] [CrossRef]
- Zhu, J.; Li., M.; Rogers, R.; Mayer, W.; Ottewill, R.T.; STS-73 Space Shuttle Crew; Russel, W.B.; Chaikin, P.M. Crystallization of Hard-sphere Colloids in Microgravity. Nature 1997, 387, 883–885. [Google Scholar]
- Kegel, W.K.; Dhont, K.G. “Aging” of the Structure of Hard Colloidal Spheres. J. Chem. Phys. 2000, 112, 3431–3436. [Google Scholar] [CrossRef]
- Dolbnya, I.P.; Petukhov, A.V.; Aarts, D.G.A.L.; Vroege, G.J.; Lekkerkerker, H.N.W. Coexistence of RHCP and FCC Phases in Hard-Sphere Colloidal Crystals. Europhys. Lett. 2005, 72, 962–968. [Google Scholar] [CrossRef]
- Hilhorst, J.; de Winter, D.A.M.; Wolters, J.R.; Post, J.A.; Petukov, A.V. Defect Engineering in Sedimentary Colloidal Photonic Crystals. Langmuir 2013, 29, 10011–10018. [Google Scholar] [CrossRef] [PubMed]
- Ohtaka, K. Energy Band of Photons and Low-energy Photon Diffraction. Phys. Rev. B 1979, 19, 5057–5067. [Google Scholar] [CrossRef]
- Yablonovitch, E. Inhibited Spontaneous Emission in Solid-State Physics and Electronics. Phys. Rev. Lett. 1987, 58, 2059–2062. [Google Scholar] [CrossRef] [PubMed]
- John, S. Strong Localization of Photons in Certain Disordered Dielectric Superlattices. Phys. Rev. Lett. 1987, 58, 2486–2489. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Ladd, A.J.C. New Monte Carlo Method to Compute the Free Energy of Arbitrary Solids. Application to the fcc and hcp Phases of Hard Spheres. J. Chem. Phys. 1984, 81, 3188–3198. [Google Scholar] [CrossRef]
- Woodcock, L.V. Entropy Difference Between the Face-centered Cubic and Hexagonal Close-packed Crystal Structures. Nature 1997, 385, 141–143. [Google Scholar] [CrossRef]
- Bruce, A.D.; Wilding, N.B.; Ackerson, G.A. Free Energy of Crystalline Solids: A Lattice-Switch Monte Carlo Method. Phys. Rev. Lett. 1997, 79, 3002–3005. [Google Scholar] [CrossRef]
- Pronk, S.; Frenkel, D. Can Stacking Faults in Hard-sphere Crystals Anneal out Spontaneously? J. Chem. Phys. 1999, 110, 4589–4592. [Google Scholar] [CrossRef]
- Mau, S.-C.; Huse, D.A. Stacking Entropy of Hard-sphere Crystals. Phys. Rev. E 1999, 59, 4396–4401. [Google Scholar] [CrossRef]
- Elser, V. Phonon Contribution to the Entropy of Hard-sphere Crystals. Phys. Rev. E 2014, 89, 052404. [Google Scholar] [CrossRef] [PubMed]
- Alder, B.J.; Wainwright, T.E. Studies in Molecular Dynamics. I. General Method. J. Chem. Phys. 1959, 31, 459–466. [Google Scholar] [CrossRef]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation, 2nd ed.; Cambridge University Press: Cambridge, UK, 1997. [Google Scholar]
- Quentrec, B.; Brot, C. New Method for Searching for Neighbors in Molecular Dynamics Calculations. J. Comp. Phys. 1975, 13, 430–432. [Google Scholar] [CrossRef]
- Rapaport, D.C. The Event Scheduling Problem in Molecular Dynamic Simulation. J. Comp. Phys. 1980, 34, 184–201. [Google Scholar] [CrossRef]
- Isobe, M. Simple and Efficient Algorithm for Large Scale Molecular Dynamics Simulation in Hard Disk System. Int. J. Mod. Phys. C 1999, 10, 1281–2183. [Google Scholar] [CrossRef]
- Bannerman, M.N.; Sargant, R.; Lue, L. DynamO: A FreeO(N) General Event-Driven Molecular Dynamics Simulator. J. Comp. Chem. 2011, 32, 3329. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Manabe, R.; Nishioka, K. Construction and Investigation of a Hard-sphere Crystal-melt Interface by a Molecular Dynamics Simulation. Phys. Rev. E 1995, 51, R3831–R3833. [Google Scholar] [CrossRef]
- Kyrlidis, A.; Brown, R. Density-functional Theory and Atomistic Simulation of the Hard-sphere Melt-solid Interface. Phys. Rev. E 1995, 51, 5832–5845. [Google Scholar] [CrossRef]
- Vega, C.; Sanz, E.; Abascal, J.L.F.; Noya, E.G. Determination of Phase Diagrams via Computer Simulation: Methodology and Applications to Water, Electrolytes and Proteins. J. Phys. Condens. Matter 2008, 20, 153101. [Google Scholar] [CrossRef]
- Cahn, J.W. Theory of Crystal Growth and Interface Motion in Crystalline Materials. Acta Metal. 1960, 8, 554–562. [Google Scholar] [CrossRef]
- Cape, J.N.; Woodcock, L.V. Molecular Dynamics Calculation of Phase Coexistence Properties: The Soft-Sphere Melting Transition. Chem. Phys. Lett. 1978, 59, 271–274. [Google Scholar] [CrossRef]
- Ladd, A.J.C.; Woodcock, L.V. Structure of the Lennard-Jones (100) Crystal-liquid Interface. J. Phys. C Solid State Phys. 1978, 11, 3565–3576. [Google Scholar] [CrossRef]
- Hiwatari, Y.; Stoll, E.; Schneider, T. Molecular-dynamics Investigation of Solid-liquid Coexistence. J. Chem. Phys. 1978, 68, 3401–3404. [Google Scholar] [CrossRef]
- Broughton, J.Q.; Abraham, F.F. A Comparison of the fcc (111) and (100) Crystal-Melt Interface by Molecular Dynamics Simulation. Chem. Phys. Lett. 1980, 71, 456–459. [Google Scholar] [CrossRef]
- Cape, J.N.; Woodcock, L.V. Soft-sphere Model for the Crystal-liquid Interface: A Molecular Dynamics Simulation of the Surface Stress. J. Chem. Phys. 1980, 73, 2420–2429. [Google Scholar] [CrossRef]
- Ueda, A.; Takada, J.; Hiwatari, Y. Molecular Dynamics Studies on Solid-Liquid Interface of Soft-Core Model. J. Phys. Soc. Jpn. 1981, 59, 307–314. [Google Scholar] [CrossRef]
- Broughton, J.Q.; Bonissent, A.; Abraham, F.F. The fcc (111) and (100) Crystal-Melt Interfaces: A Comparison by Molecular Dynamics Simulation. J. Chem. Phys. 1981, 74, 4029–4039. [Google Scholar] [CrossRef]
- Broughton, J.Q.; Gilmer, G.H. Molecular Dynamics of the Crystal-fluid Interface. V. Structure and Dynamics of Crystal-melt Systems. J. Chem. Phys. 1986, 84, 5749–5758. [Google Scholar] [CrossRef]
- Temkin, D.E. Molecular Roughness of the Crystal-Melt Boundary. In Crystallization Process; Consultant Bureau: New York, NY, USA, 1996; pp. 15–23. [Google Scholar]
- Chernov, A.A. Modern Crystallography III; Springer-Verlag: Berlin, Germany, 1984. [Google Scholar]
- Vere, A.W. Crystal Growth; Plenum: New York, NY, USA, 1987. [Google Scholar]
- Markov, I.V. Crystal Growth for Beginners; World Scientific: Singapore, 1995. [Google Scholar]
- Homma, S.; Yoshida, U.; Nakano, H. Theory of Solid-Liquid Interface. J. Phys. Soc. Jpn. 1981, 50, 2175–2179. [Google Scholar] [CrossRef]
- Ishibashi, Y. On the Activation Energy of Solid-Melt Interface in the Temkin Model. J. Phys. Soc. Jpn. 1986, 55, 2099–2101. [Google Scholar] [CrossRef]
- Mori, A.; Maksmov, I.L. On the Temkin Model of Solid-Liquid Interface. J. Cryst. Growth 1999, 200, 297–304. [Google Scholar] [CrossRef]
- Mori, A.; Suzuki, Y. Grand Potential Formalism of Interfacial Thermodynamics for Critical Nucleus. Natl. Sci. 2013, 5, 631–639. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Laird, B.B. Direct Calculation of the Hard-Sphere Crystal/Melt Interfacial Free Energy. Phys. Rev. Lett. 2000, 85, 4751–4754. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.Q.; Gilmer, G.H. Molecular Dynamics of the Crystal-fluid Interface. VI. Excess Surface Free Energies of Crystal-melt Systems. J. Chem. Phys. 1986, 84, 5759–5768. [Google Scholar] [CrossRef]
- Laird, B.B.; Davidchack, R.L. Direct Calculation of the Crystal-Melt Interfacial Free Energy via Molecular Dynamics Computer Simulation. J. Phys. Chem. B 2005, 109, 17802–17812. [Google Scholar] [CrossRef] [PubMed]
- Davidchack, R.L. Hard Sphere Revisited: Accurate Calculation of the Solid-liquid Interfacial Free Energy. J. Chem. Phys. 2010, 133, 23470. [Google Scholar] [CrossRef] [PubMed]
- Härtel, A.; Oettel, M.; Rozas, R.E.; Egelhaaf, S.U.; Horbach, J.; Löwen, H. Tension and Stiffness of the Hard Sphere Crystal-Fluid Interface. Phys. Rev. Lett. 2012, 108, 226101. [Google Scholar]
- Davis, K.E.; Russel, W.B.; Glantschnig, W.J. Disorder-to-Order Transition in Settling Suspensions of Colloidal Silica: X-Ray Measurements. Science 1989, 245, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Pusey, P.N.; van Megen, W.; Bartlett, P.; Ackerson, B.J.; Rarity, J.G.; Underwood, S.M. Structure of Crystals of Hard Colloidal Spheres. Phys. Rev. Lett. 1989, 63, 2753–2756. [Google Scholar] [CrossRef] [PubMed]
- Martelozzo, V.C.; Schofield, A.B.; Poon, W.C.K.; Pusey, P.N. Structural Aging of Crystals of Hard-sphere Colloids. Phys. Rev. E 2002, 66, 021408. [Google Scholar] [CrossRef] [PubMed]
- Biben, T.; Ohnesorge, R.; Löwen, H. Crystallization in Sedimentation Profiles of Hard Spheres. Europhys. Lett. 1994, 28, 665–670. [Google Scholar] [CrossRef]
- Volkov, I.; Cieplak, M.; Koplik, K.; Banavar, J.R. Molecular Dynamics Simulations of Crystallization of Hard Spheres. Phys. Rev. E 2002, 66, 061404. [Google Scholar] [CrossRef] [PubMed]
- Yanagiya, S.-I.; Mori, A.; Suzuki, Y.; Miyoshi, Y.; Kasuga, M.; Sawada, T.; Ito, K.; Inoue, T. Enhancement of Crystallization of Hard Spheres by Gravity: Monte Carlo Simulation. Jpn. J. Appl. Phys. 2005, 44, 5113–5116. [Google Scholar] [CrossRef]
- Mori, A.; Yanagiya, S.-I.; Suzuki, Y.; Sawada, T.; Ito, K. Monte Carlo Simulation of Crystal-fluid Coexistence States in the Hard-Sphere System under Gravity with Stepwise Control. J. Chem. Phys. 2006, 124, 174507. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Suzuki, Y.; Yanagiya, S.-I.; Sawada, T.; Ito, K. Shrinking Stacking Fault through Glide of the Shockley Partial Dislocation in Hard-sphere Crystal under Gravity. Mol. Phys. 2007, 105, 1377–1383. [Google Scholar] [CrossRef]
- Mori, A.; Suzuki, Y.; Matsuo, S. Disappearance of a Stacking Fault in Hard-Sphere Crystals under Gravity. Prog. Theor. Phys. Suppl. 2009, 178, 33–40. [Google Scholar] [CrossRef]
- Mori, A.; Suzuki, Y. Interplay between Elastic Fields due to Gravity and a Partial Dislocation for a Hard-sphere Crystal Coherently Grown under Gravity: Driving Force for Defect Disappearance. Mol. Phys. 2010, 108, 1731–1738. [Google Scholar] [CrossRef]
- Mori, A.; Yanagiya, S.-I.; Suzuki, Y.; Sawada, T.; Ito, K. Crystal Structure of Hard Spheres under Gravity by Monte Carlo Simulation. Sci. Technol. Adv. Mater. 2006, 7, 296–302. [Google Scholar] [CrossRef]
- Marechal, M.; Dijkstra, M. Crystallization of Colloidal Hard Spheres under Gravity. Phys. Rev. E 2007, 75, 061404. [Google Scholar] [CrossRef] [PubMed]
- Ramsteiner, I.B.; Jensen, K.E.; Weitz, D.A.; Spaepen, F. Experimental Observation of the Crystallization of Hard-sphere Colloidal Particles by Sedimentation onto Flat and Patterned Surfaces. Phys. Rev. E 2009, 79, 011403. [Google Scholar] [CrossRef] [PubMed]
- Marechal, M.; Hermes, M.; Dijkstra, M. Stacking in Sediments of Colloidal Hard Spheres. J. Chem. Phys. 2011, 135, 034510. [Google Scholar] [CrossRef] [PubMed]
- Heni, M.; Löwen, H. Interfacial Free Energy of Hard-Sphere Fluids and Solids near a Hard Wall. Phys. Rev. E 1999, 93, 7057–7065. [Google Scholar] [CrossRef]
- Laird, B.B.; Davidchack, R.L. Wall-Induced Prefreezing in Hard Spheres: A Thermodynamic Perspective. J. Phys. Chem. C 2007, 111, 15952–15956. [Google Scholar] [CrossRef]
- Laird, B.B.; Davidchack, R.L. Calculation of the Interfacial Free Energy of a Fluid at a Static wall by Gibbs–Cahn Integration. J. Chem. Phys. 2010, 132, 204101. [Google Scholar] [CrossRef] [PubMed]
- Courtemanche, D.J.; van Swol, F. Wetting State of a Crystal-Fluid System of Hard Spheres. Phys. Rev. Lett. 1992, 69, 2078–2081. [Google Scholar] [CrossRef] [PubMed]
- Auer, S.; Frenkel, D. Line Tension Controls Wall-Induced Crystal Nucleation in Hard-Sphere Colloids. Phys. Rev. Lett. 2003, 91, 015703-1. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-H.; Crocker, J.C.; Prasad, V.; Schofield, A.; Weitz, D.A.; Lubensky, T.C.; Yodh, A.G. Entropically Driven Colloidal Crystallization on Patterned Surfaces. Phys. Rev. Lett. 2000, 85, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Seo, E.-M.; Kim, D.-Y. Surface-modulation-Controlled Three-dimensional Colloidal Crystals. Appl. Phys. Lett. 2002, 80, 225–227. [Google Scholar] [CrossRef]
- Zhang, J.; Alsayed, A.; Lin, K.H.; Sanyal, S.; Zhang, F.; Pao, W.-J.; Balagurusamy, V.S.K.; Heiney, P.A.; Yodh, A.G. Template-directed Convective Assembly of Three-dimensional Face-centered Cubic Colloidal Crystals. Appl. Phys. Lett. 2002, 81, 3176–3178. [Google Scholar] [CrossRef]
- Stenger, N.; Rehspringer, J.-L.; Hirlimann, C. Template-directed Self-organized Silica Beads on Square and Penrose-like Patterns. J. Lumin. 2006, 121, 278–281. [Google Scholar] [CrossRef]
- Heni, M.; Löwen, H. Surface Freezing on Patterned Substrates. Phys. Rev. Lett. 2000, 85, 3668–3671. [Google Scholar] [CrossRef] [PubMed]
- Heni, M.; Löwen, H. Precrystallization of Fluids Induced by Patterned Substrates. J. Phys. Condens. Matter 2001, 13, 4675–4696. [Google Scholar] [CrossRef]
- Cacciuto, A.; Frenkel, D. Simulation of Colloidal Crystallization on Finite Structured Templates. Phys. Rev. E 2005, 72, 041604. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-S.; Sun, Z.-Y.; An, L.-J. Heterogeneous Crystallization of Hard Spheres on Patterned Substrates. J. Chem. Phys. 2010, 132, 144506. [Google Scholar] [CrossRef] [PubMed]
- Mori, A. Disappearance of Stacking Fault in Colloidal Crystals under Gravity. World J. Eng. 2011, 8, 117–122. [Google Scholar] [CrossRef]
- Mori, A. Monte Carlo Simulation of Growth of Hard-sphere Crystals on a Square Pattern. J. Cryst. Growth 2011, 318, 66–71. [Google Scholar] [CrossRef]
- Mori, A.; Suzuki, Y.; Matsuo, S. Possibility of Gravitational Tempering in Colloidal Epitaxy to Obtain a Perfect Crystal. Chem. Lett. 2012, 41, 1069–1071. [Google Scholar] [CrossRef]
- Mori, A.; Suzuki, Y. Identification of Triangular Shaped Defect Often Appeared in Hard-sphere Crystals Grown on a Square Pattern under Gravity by Monte Carlo Simulations. Physica B 2014, 452, 58–65. [Google Scholar] [CrossRef]
- Jensen, K.E.; Pennachio, D.; Recht, D.; Weitz, D.A.; Spaepen, F. Rapid Growth of Large, Defect-free Colloidal Crystals. Soft Matter 2013, 9, 320–328. [Google Scholar] [CrossRef]
- Russo, J.; Maggs, A.C.; Bonn, D.; Tanaka, H. The Interplay of Sedimentation and Crystallization in Hard-sphere Suspensions. Soft Matter 2013, 9, 3769–3783. [Google Scholar] [CrossRef]
- Nakamura, S.; Pearton, S.; Fasol, G. The Blue Laser Diode: The Complete Story; Springer: Berlin, Germany, 2000. [Google Scholar]
- Mori, A.; Suzuki, Y.; Matsuo, S. Monte Carlo Simulation of Defects in Hard-sphere Crystal Grown on a Square Pattern. World J. Eng. 2012, 9, 37–44. [Google Scholar] [CrossRef]
- Mori, A.; Suzuki, Y.; Sato, M. Gravitational Tempering in Colloidal Epitaxy To Reduce Defects Further. Cryst. Growth Des. 2014, 14, 2086–2088. [Google Scholar] [CrossRef]
- Schall, P.; Cohen, I.; Weitz, D.A.; Spaepen, F. Visualization of Dislocation Dynamics in Colloidal Crystals. Science 2004, 305, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Maire, E.; Redston, E.; Gulda, M.P.; Weitz, D.A.; Spaepen, F. Imaging Grain Boundary Grooves in Hard-sphere Colloidal Bicrystals. Phys. Rev. E 2016, 94, 042604. [Google Scholar] [CrossRef] [PubMed]
- Mori, A. Effect of Mass Flow in Melt on the Motion of Crystal-Melt Interface of Hard Spheres: A Molecular Dynamics Study. J. Phys. Soc. Jpn. 1997, 66, 1579–1582. [Google Scholar] [CrossRef]
- Mori, A. Hydrothermodynamic Consideration on the Steady-state Motion of a Solid/liquid Interface. J. Chem. Phys. 1999, 110, 8679–8686. [Google Scholar] [CrossRef]
- Mori, A. Steady-State Interface Motion: Formulation Extended to the Isothermal Case. J. Phys. Soc. Jpn. 1999, 68, 876–880. [Google Scholar] [CrossRef]
- Ackerson, B.J.; Paulin, S.E.; Johnson, B. Crystallization by Settling in Suspensions of Hard Spheres. Phys. Rev. E 1999, 59, 6903–6913. [Google Scholar] [CrossRef]
- Hoogenboom, J.P.; Derks, D.; Vergeer, P.; van Blaaderena, A. Stacking Faults in Colloidal Crystals Grown by Sedimentation. J. Chem. Phys. 2002, 117, 11320–11328. [Google Scholar] [CrossRef]
- Royall, C.P.; Dzubiella, J.; Schmidt, M.; van Blaaderen, A. Nonequilibrium Sedimentation of Colloids on the Particle Scale. Phys. Rev. Lett. 2007, 98, 188304. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, J.P.; Vergeer, P.; van Blaaderena, A. A Real-space Analysis of Colloidal Crystallization in a Gravitational Field at a Flat Bottom Wall. J. Chem. Phys. 2003, 119, 3371–3383. [Google Scholar] [CrossRef]
- Megens, M.; van Kats, C.M.; Bösecke, P.; Vos, W.L. Synchrotron Small-Angle X-ray Scattering of Colloids and Photonic Colloidal Crystals. J. Appl. Crystallogr. 1997, 30, 637–641. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sawada, T.; Tamura, K. Colloidal Crystallization by a Centrifugation Method. J. Cryst. Growth 2011, 318, 780–783. [Google Scholar] [CrossRef]
- Suzuki, Y.; Endoh, J.; Mori, A.; Yabutani, T.; Tamura, K. Gravitational Annealing of Colloidal Crystals. Defect Diffus. Forum 2012, 232–235, 555–558. [Google Scholar] [CrossRef]











© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, A. Computer Simulations of Crystal Growth Using a Hard-Sphere Model. Crystals 2017, 7, 102. https://doi.org/10.3390/cryst7040102
Mori A. Computer Simulations of Crystal Growth Using a Hard-Sphere Model. Crystals. 2017; 7(4):102. https://doi.org/10.3390/cryst7040102
Chicago/Turabian StyleMori, Atsushi. 2017. "Computer Simulations of Crystal Growth Using a Hard-Sphere Model" Crystals 7, no. 4: 102. https://doi.org/10.3390/cryst7040102
APA StyleMori, A. (2017). Computer Simulations of Crystal Growth Using a Hard-Sphere Model. Crystals, 7(4), 102. https://doi.org/10.3390/cryst7040102