
Stochastic and Deterministic Crystal Structure Solution Methods in GSAS-II: Monte Carlo/Simulated Annealing Versus Charge Flipping

Abstract
:1. Introduction
2. Methods
2.1. Monte Carlo/Simulated Annealing
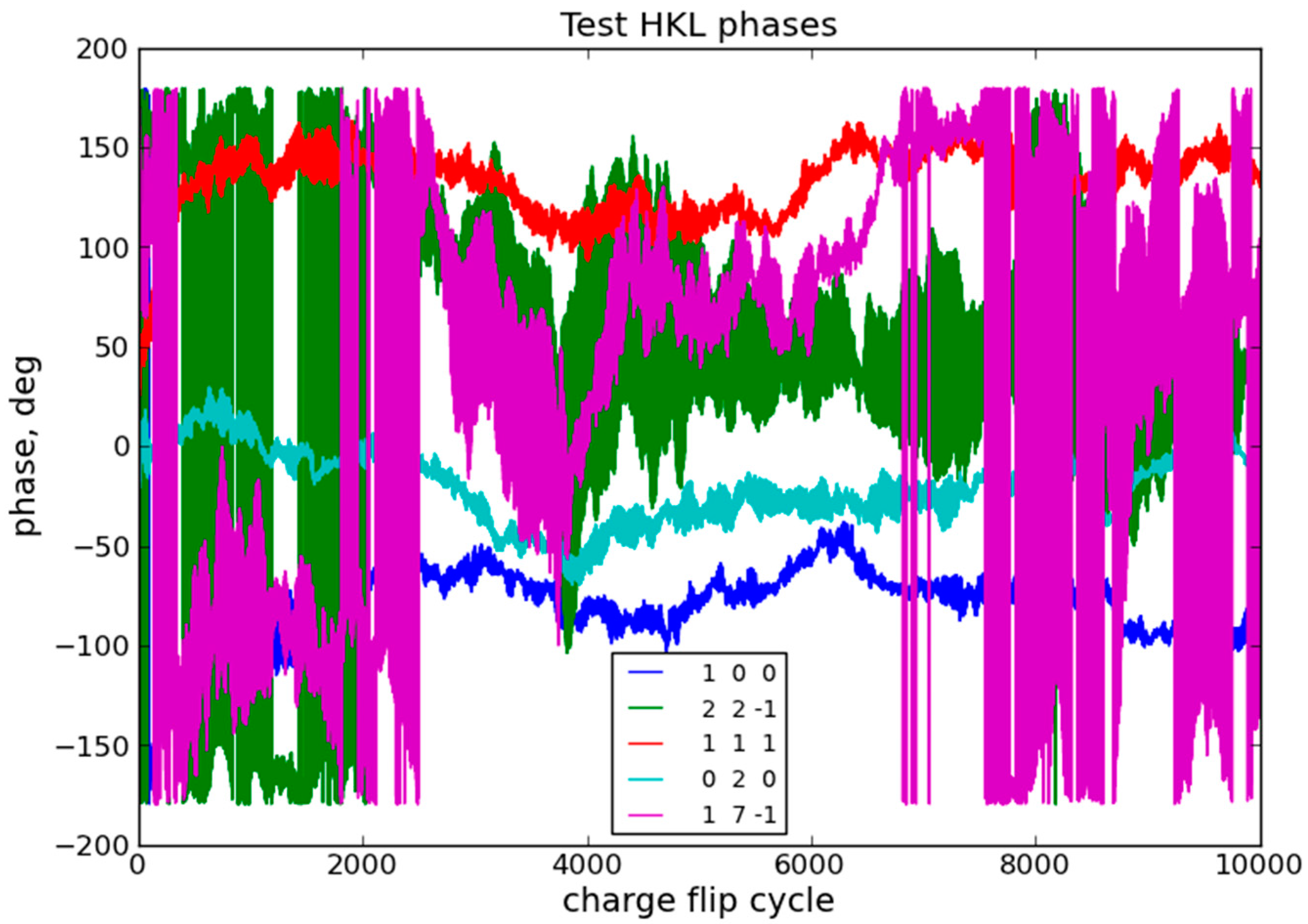
2.2. Charge Flipping
3. Discussion
3.1. Monte Carlo/Simulated Annealing
3.2. Charge Flipping
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Toby, B.H.; Von Dreele, R.B. GSAS-II: the genesis of a modern open-source all-purpose crystallography software package. J. Appl. Crystallogr. 2013, 46, 544–549. GSAS-II can be obtained from the site: https://subversion.xray.aps.anl.gov/trac/pyGSAS. [CrossRef]
- Toby, B.H.; Von Dreele, R.B. What’s new in GSAS-II. Powder Diffr. 2014, 29, 1–5. [Google Scholar] [CrossRef]
- Pagola, S.; Stephens, P. PSSP, a computer program for the crystal structure solution of molecular materials from X-ray powder diffraction data. J. Appl. Crystallogr. 2010, 43, 370–376. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Cerny, R. FOX, ‘free objects for crystallography’: A modular approach to ab initio structure determination from powder diffraction. J. Appl. Crystallogr. 2002, 35, 734–743. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van deStreek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Cryst. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Scipy.optimize.anneal. Available online: https://docs.scipy.org/doc/scipy-0.13.0/reference/generated/scipy.optimize.anneal.html (accessed on 25 June 2017).
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Crystallogr. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- LeBail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mat. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Scipy.optimize.minimize (method=’L-BFGS-B’). Available online: https://docs.scipy.org/doc/scipy-0.19.0/reference/generated/scipy.optimize.anneal.html (accessed on 25 June 2017).
- Oszlányi, G.; Sütő, A. Ab initio structure solution by charge flipping. Acta Crystallogr. 2004, A60, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Oszlányi, G.; Sütő, A. The charge flipping algorithm. Acta Crystallogr. 2008, A64, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Solving an Organic Structure (Cimetidine—C 10 H 16 N6 S) from Powder Diffraction Data. Available online: http://www.ccp14.ac.uk/solution/powder_structure_solution_pathways/cimetidine.html (accessed on 27 June 2017).
- Wales, D.J.; Doye, J.P.K. Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. J. Phys. Chem. A 1997, 101, 5111–5116. [Google Scholar] [CrossRef]
- Scipy.optimize.basinhopping. Available online: https://docs.scipy.org/doc/scipy-0.19.0/reference/generated/scipy.optimize.basinhopping.html (accessed on 25 June 2017).











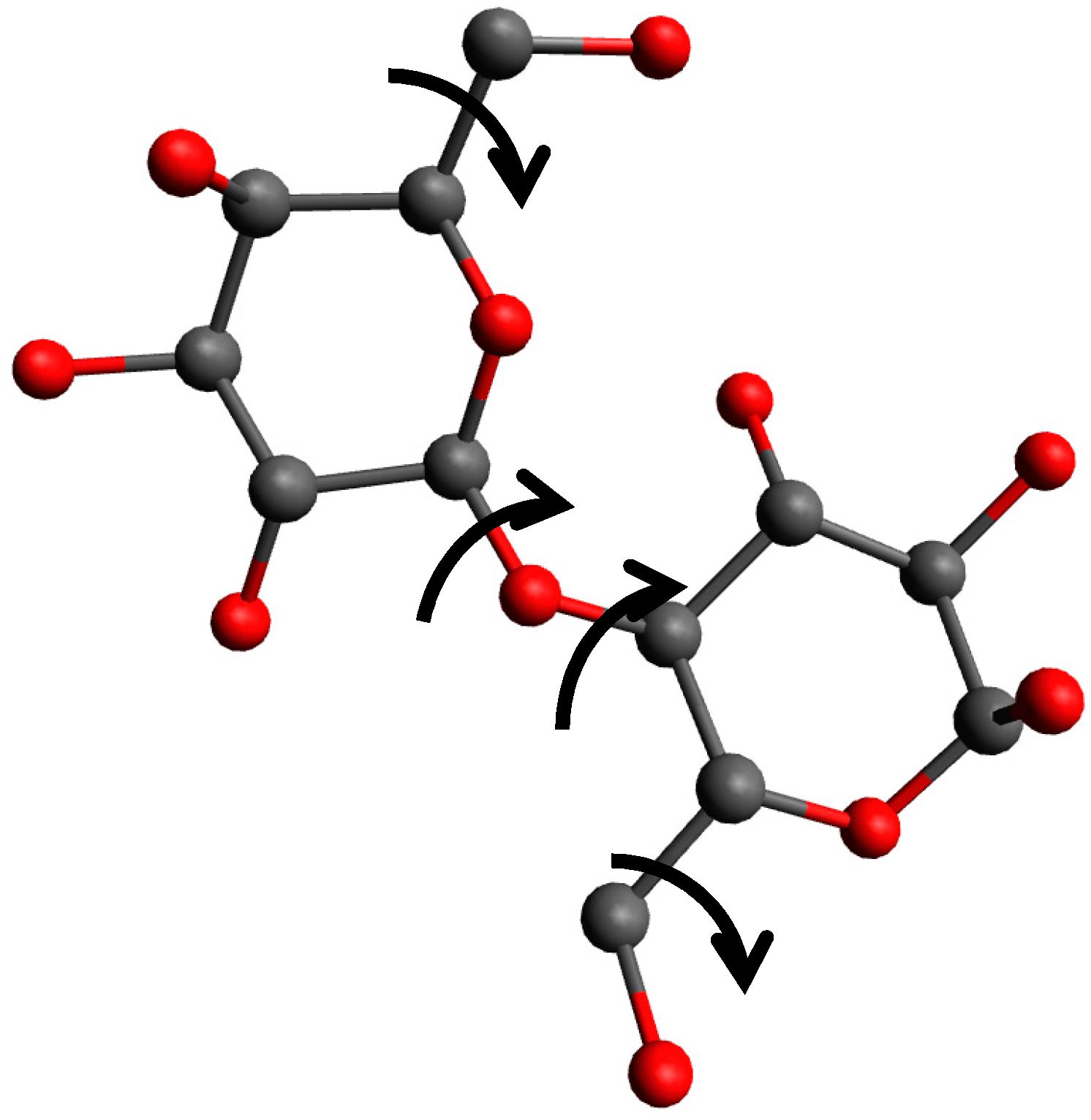{kind=link}
{kind=link}
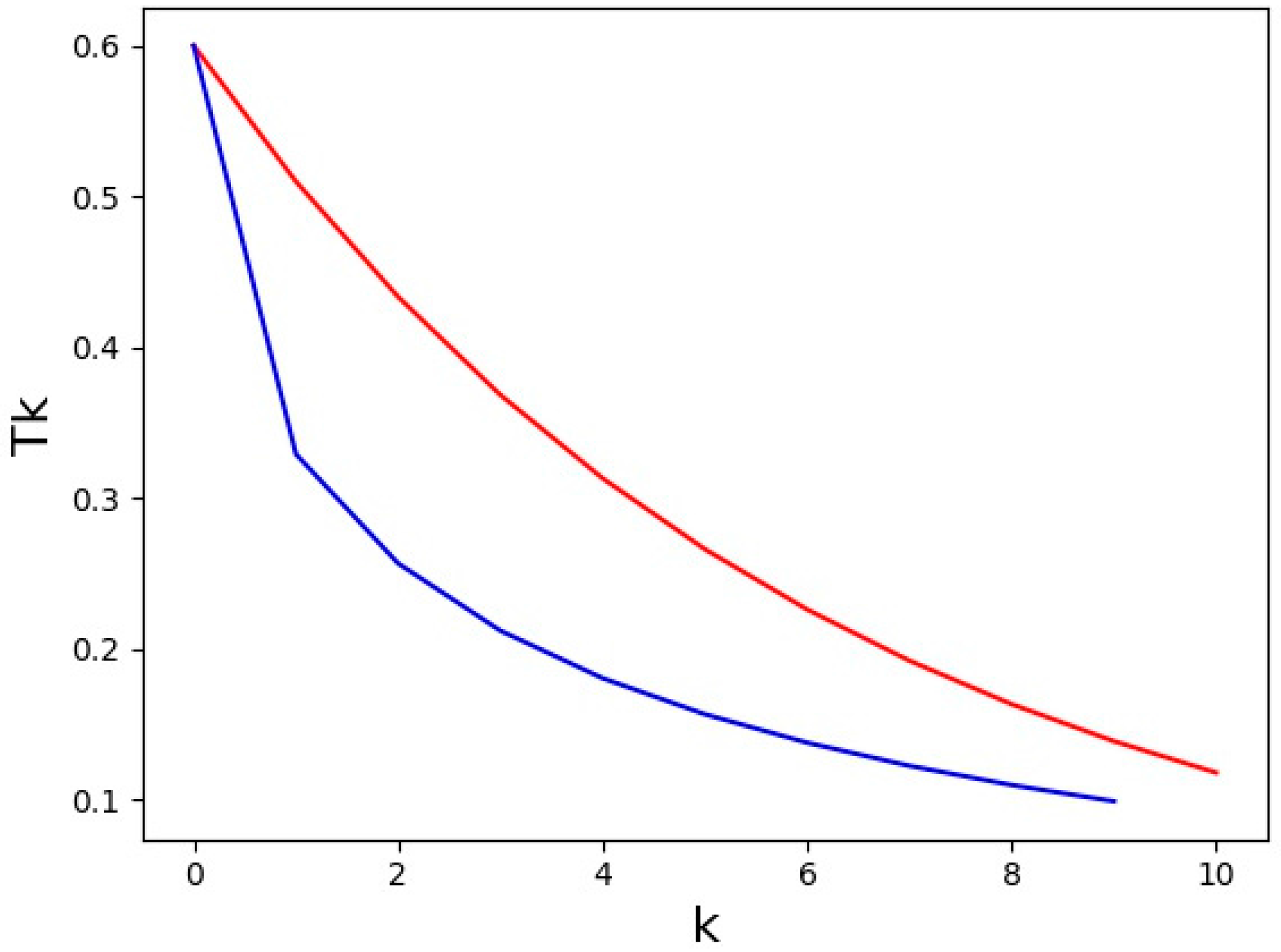{kind=link}
{kind=link}
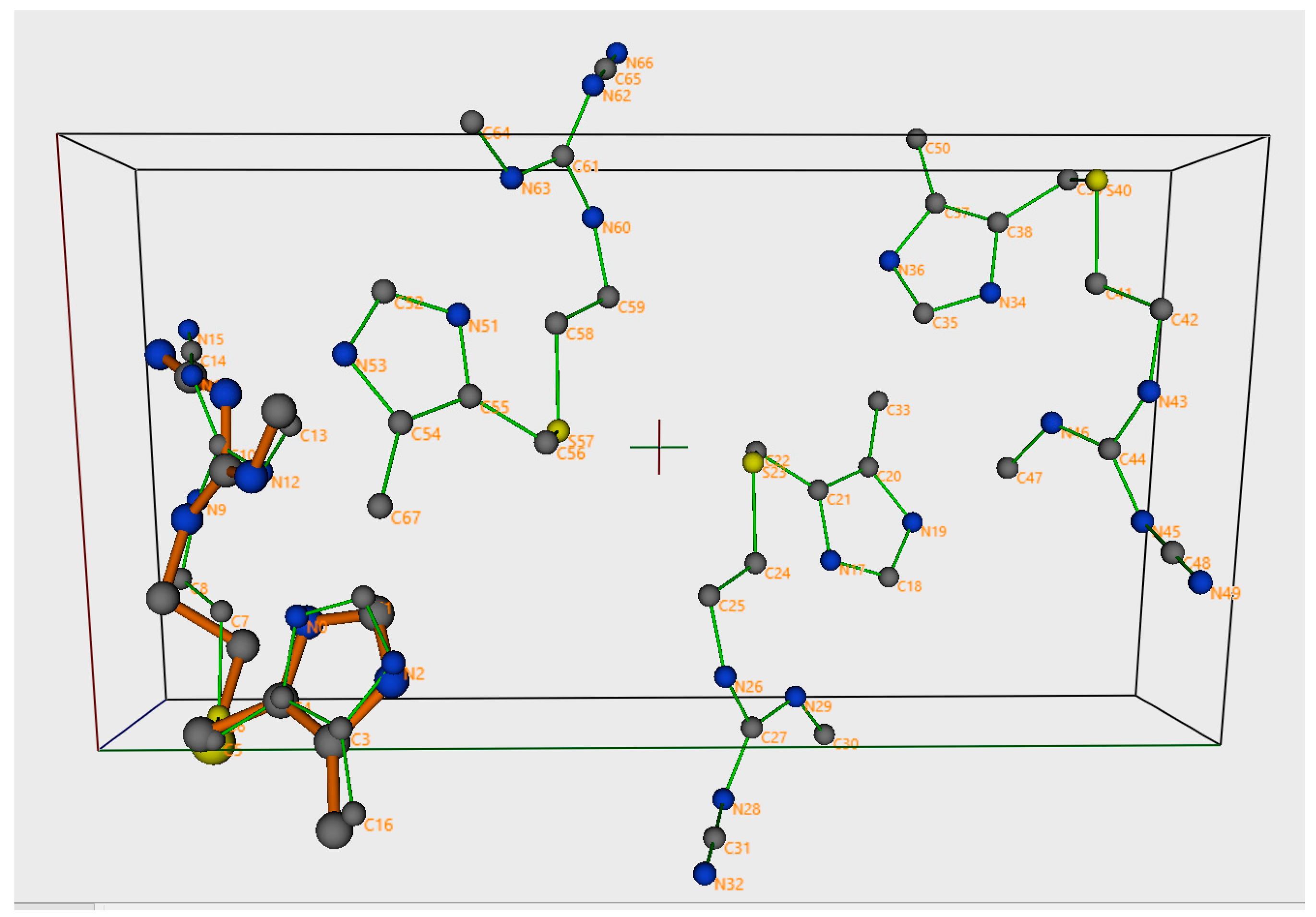{kind=link}
{kind=link}
{kind=link}
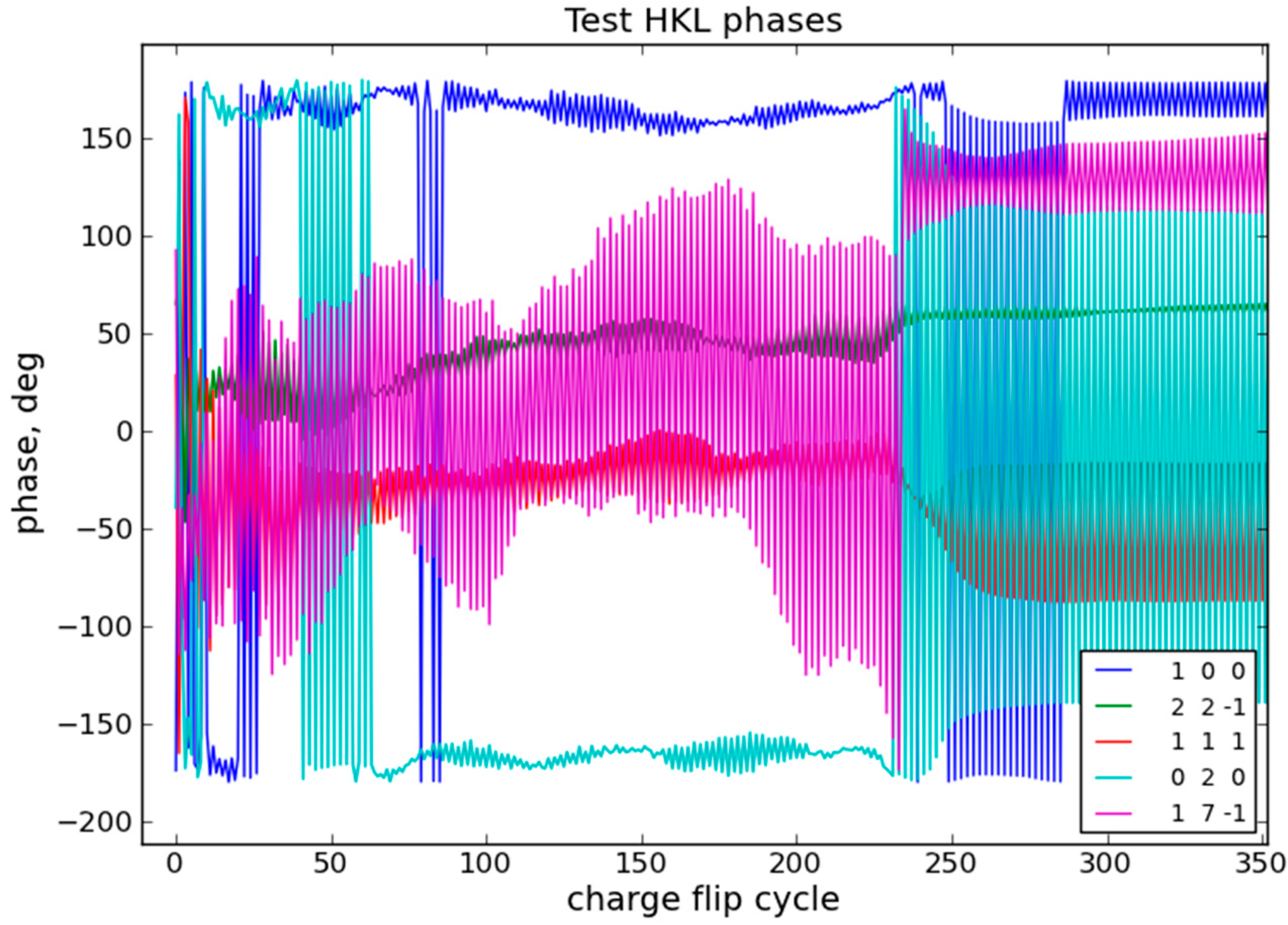{kind=link}
{kind=link}
{kind=link}
| CErho = np.real(fft.fftn(fft.fftshift(CEhkl)))*(1. + 0j) |
| CEsig = np.std(CErho) |
| CFrho = np.where(np.real(CErho) >= flipData[‘k-factor’]*CEsig,CErho,-CErho) |
| CFrho = np.where(np.real(CErho) <= flipData[‘k-Max’]*CEsig,CFrho,-CFrho) |
| CFhkl = fft.ifftshift(fft.ifftn(CFrho)) |
| CFhkl = np.where(CFhkl,CFhkl,1.0) |
| phase = CFhkl/np.absolute(CFhkl) |
| CEhkl = np.absolute(CEhkl)*phase |
| Ncyc += 1 |
| sumCF = np.sum(ma.array(np.absolute(CFhkl),mask = Emask)) |
| DEhkl = np.absolute(np.absolute(Ehkl)/sumE-np.absolute(CFhkl)/sumCF) |
| Rcf = min(100.,np.sum(ma.array(DEhkl,mask = Emask)*100.)) |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Von Dreele, R. Stochastic and Deterministic Crystal Structure Solution Methods in GSAS-II: Monte Carlo/Simulated Annealing Versus Charge Flipping. Crystals 2017, 7, 264. https://doi.org/10.3390/cryst7090264
Von Dreele R. Stochastic and Deterministic Crystal Structure Solution Methods in GSAS-II: Monte Carlo/Simulated Annealing Versus Charge Flipping. Crystals. 2017; 7(9):264. https://doi.org/10.3390/cryst7090264
Chicago/Turabian StyleVon Dreele, Robert. 2017. "Stochastic and Deterministic Crystal Structure Solution Methods in GSAS-II: Monte Carlo/Simulated Annealing Versus Charge Flipping" Crystals 7, no. 9: 264. https://doi.org/10.3390/cryst7090264
APA StyleVon Dreele, R. (2017). Stochastic and Deterministic Crystal Structure Solution Methods in GSAS-II: Monte Carlo/Simulated Annealing Versus Charge Flipping. Crystals, 7(9), 264. https://doi.org/10.3390/cryst7090264