The Effects of Nanostructure on the Hydrogen Sorption Properties of Magnesium-Based Metallic Compounds: A Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hydrogen in Nanostructured Alloys and Compounds
2.1. Nanoscale Effects on Thermodynamics
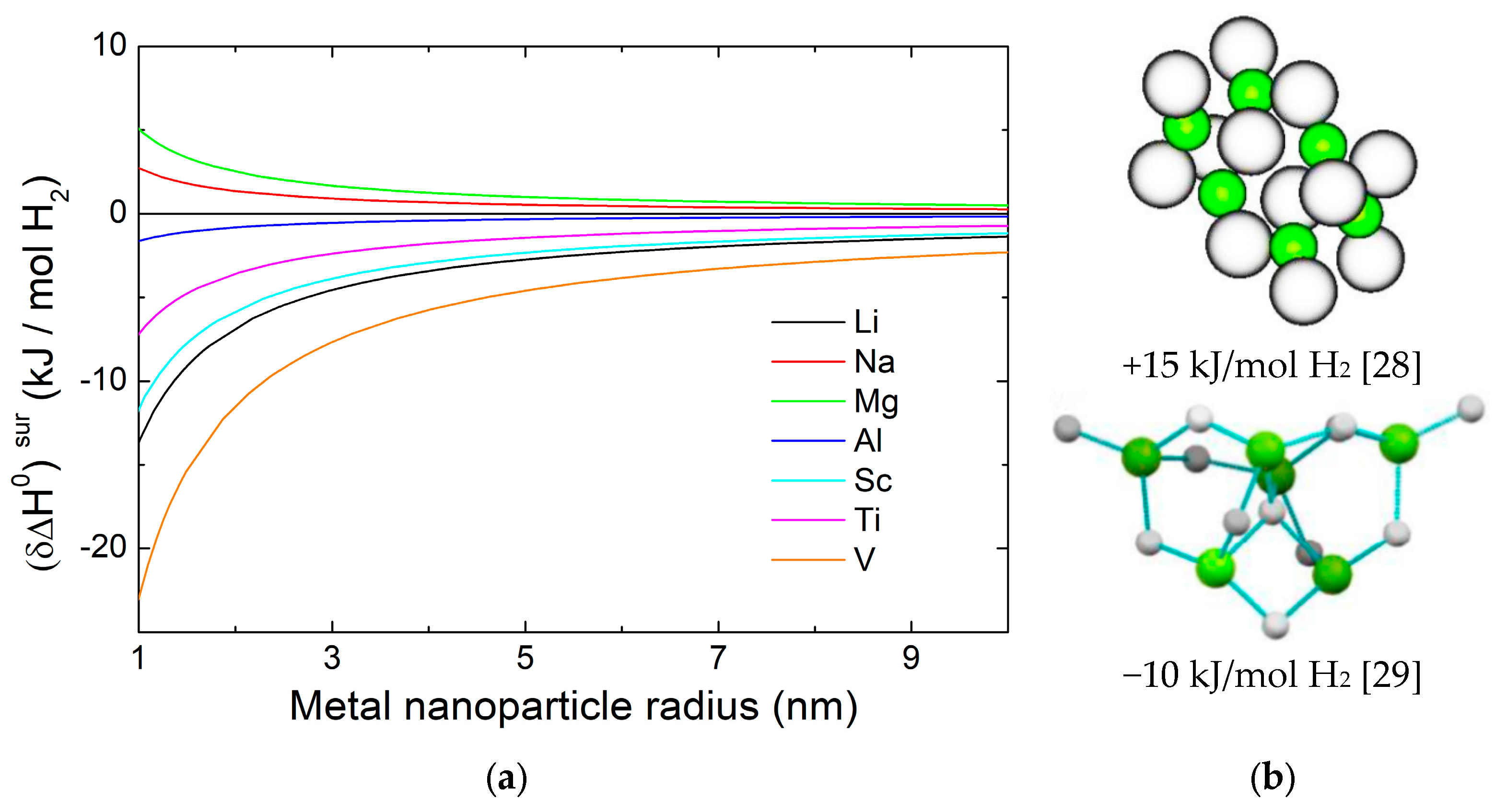
2.1.1. Surface Free Energy
2.1.2. Interface Free Energy
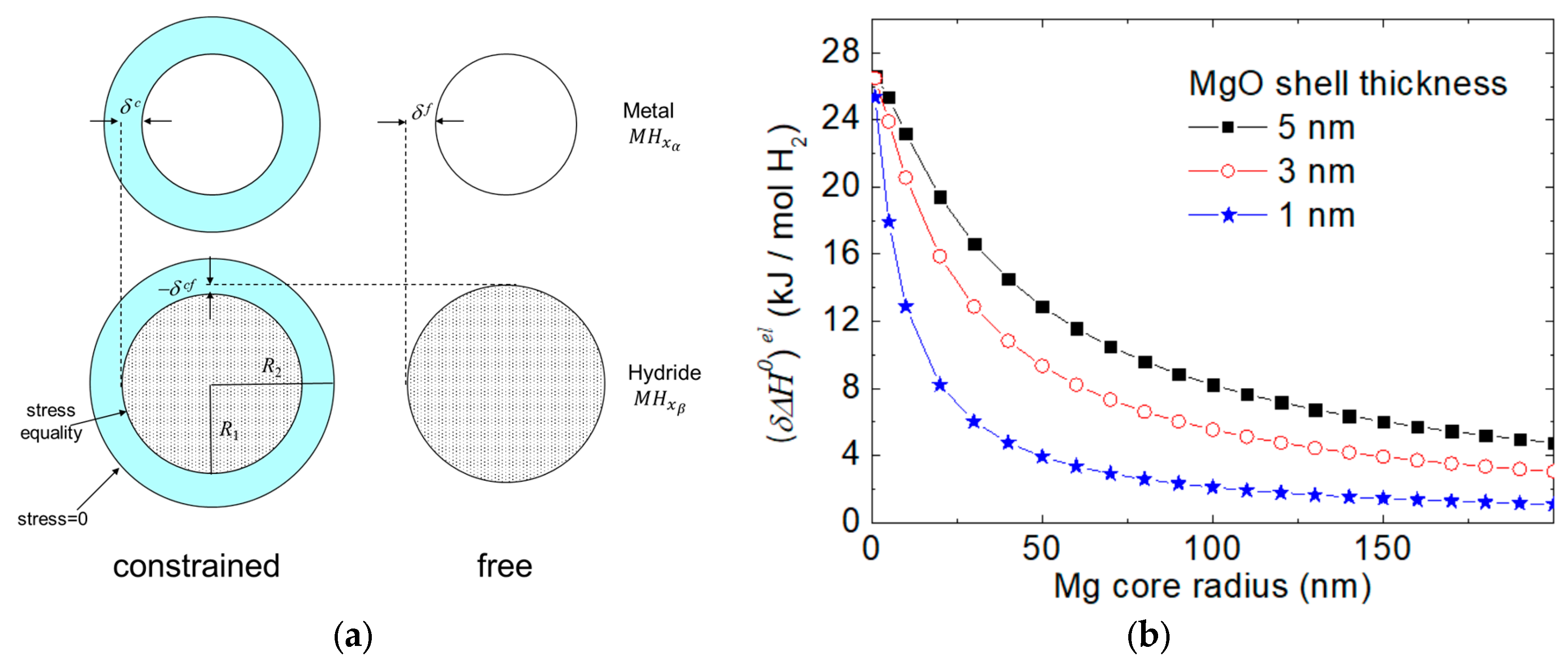
2.1.3. Elastic Constraints
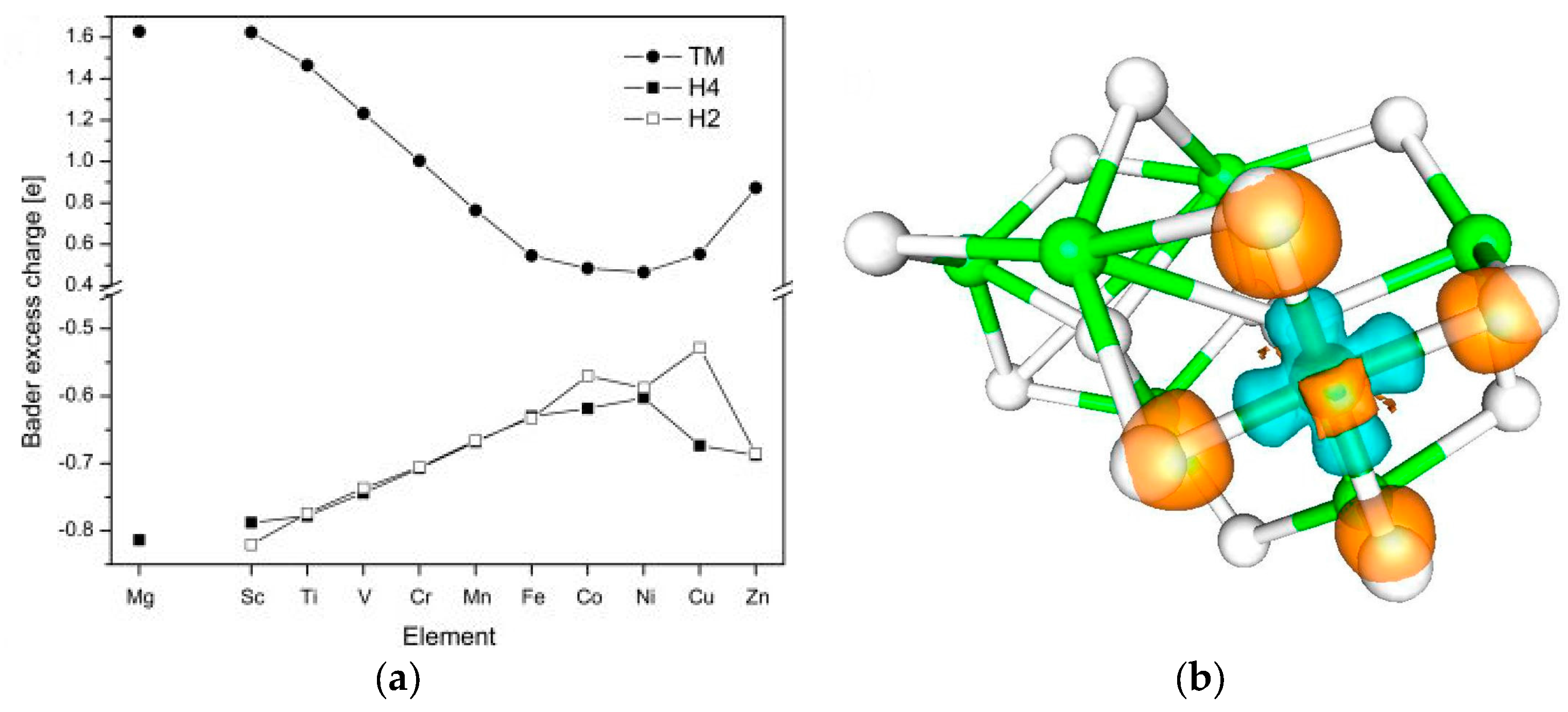
2.1.4. Alloying Effects
- Nanoscale dispersion of phases is required to keep the solid-state diffusion lengths short, thus making it possible kinetic reversibility of reactions (13) and (15).
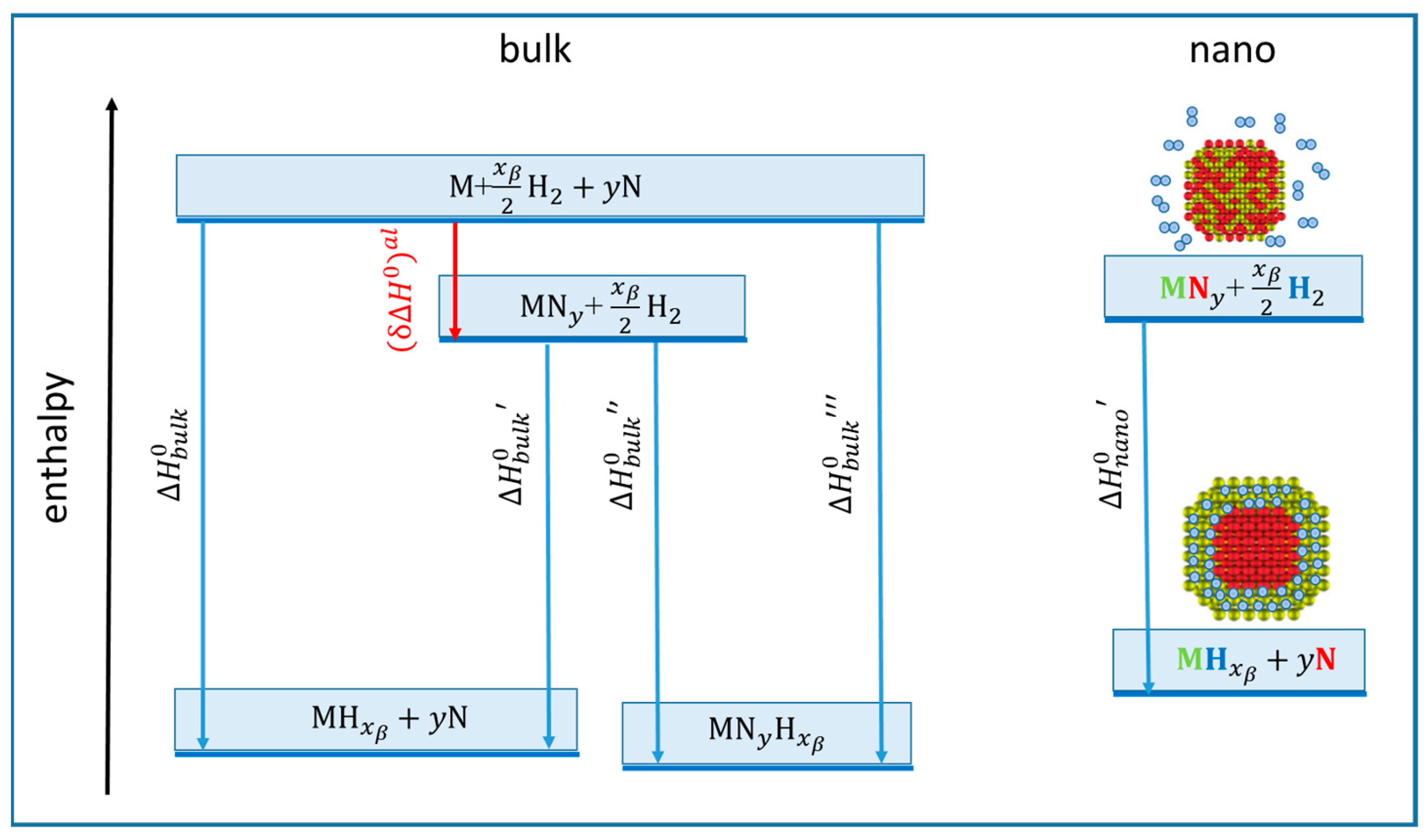
- In nanoalloys, the phase separation that appears on the right of (13) or on the left of (15) may be realized through specific morphologies such as core-shell, core-satellite, or Janus, with shifted free energy with respect to bulk alloys (Figure 5). This potential route to thermodynamic tuning, which requires advances in the calculations of nanoalloys phase diagrams, is to our knowledge yet unexplored.
2.2. Nanoscale Effects on Kinetics
- H2 dissociation/recombination at the surface
- Penetration of atomic H in subsurface layers
- H diffusion in the α-phase (usually fast) and in the β-phase (typically fast in interstitial hydrides, but slow in ionic and covalent hydrides)
- Possible nucleation of the new phase (e.g., β within α or vice versa)
- Motion of the α/β interface
- Hydrogen has to diffuse over a length similar to the system size to complete the transformation. Since the diffusion time is proportional to the square of the diffusion length, a size reduction by three orders of magnitude (i.e., from ≈50 µm down to ≈50 nm) implies a shortening of the diffusion time by six orders of magnitudes. This is particularly relevant for the non-interstitial hydrides, in which hydrogen diffusion is typically slow.
- The interfaces are short-circuit diffusion paths that further speed up hydrogen transport in and out of the material compared to hydrogen diffusion in the crystal. This argument applies both to homophasic interfaces such as grain boundaries in a nanocrystalline material and to heterophasic interfaces that are present in nanocomposites.
- The surfaces/interfaces can be preferential sites for heterogeneous nucleation of the new phase.
- If the surface of the host material is catalytically active, the rate of H2 dissociation/recombination is proportional to the specific surface area. If the surface is poorly active, as for pure Mg, suitable catalytic additives must be dispersed at the nanoscale using techniques such as ball milling, wet chemistry, and vapor deposition [11,26].
3. Results and Discussion
3.1. The Mg+H2↔MgH2 Transformation at the Nanoscale
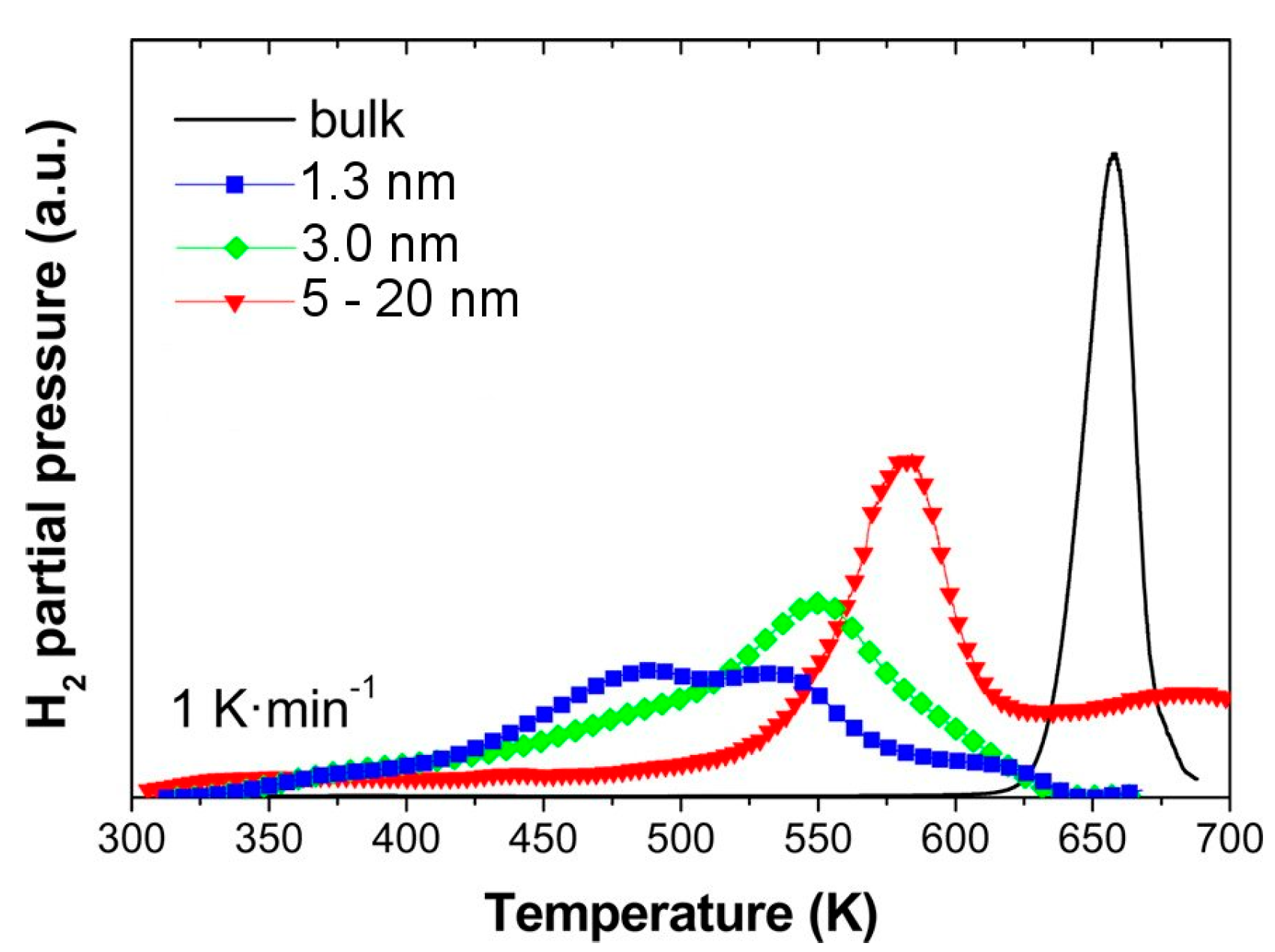
3.1.1. Nanoparticles (Freestanding)
3.1.2. Thin Films
3.1.3. Nanoparticles (Supported or Embedded)
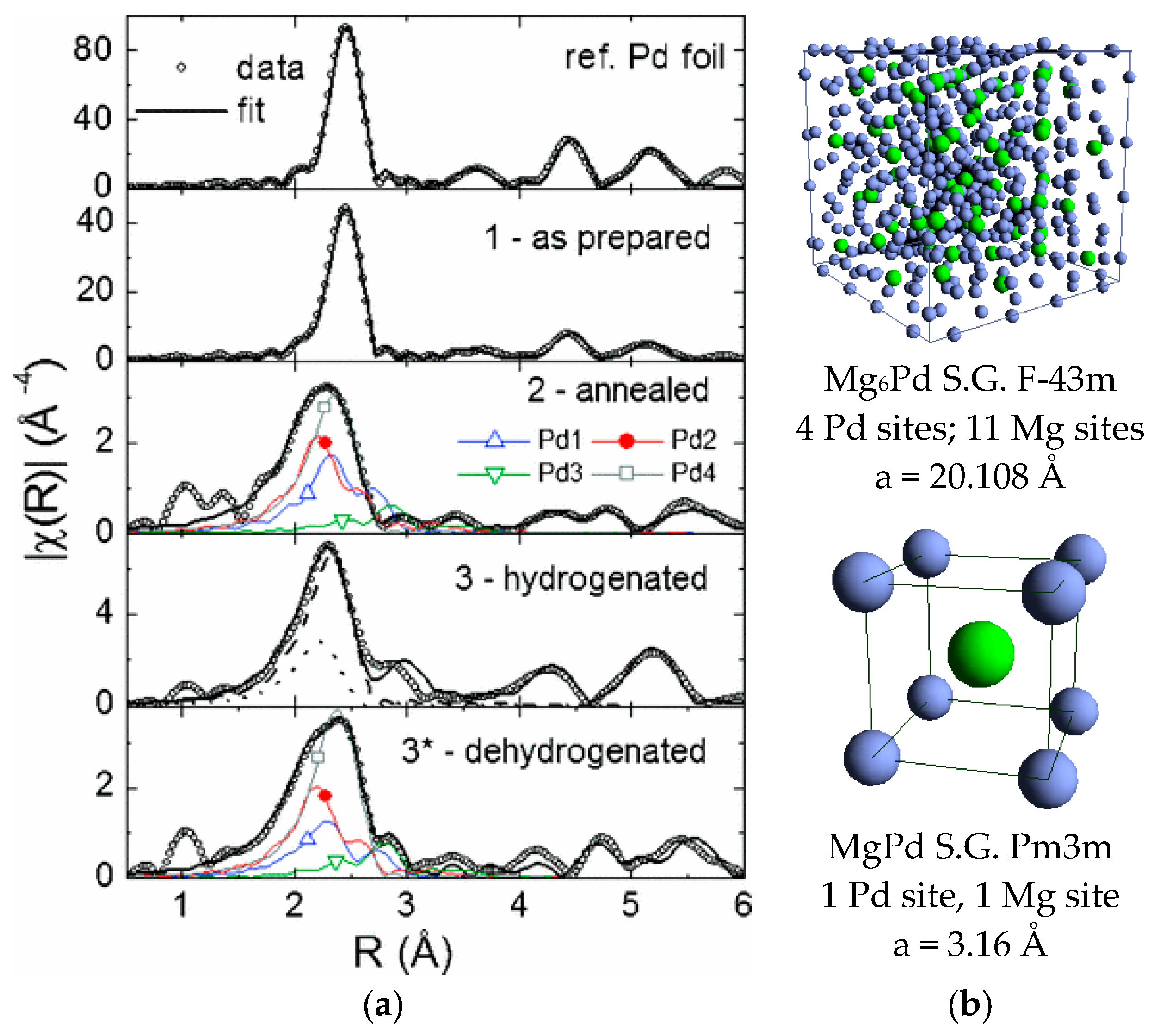
3.2. Mg-Pd Nanomaterials
- The Mg-Pd binary phase diagram is a rather complex one. Nine intermetallic compounds, with increasing Pd content from Mg6Pd to MgPd, populate the Mg-rich region (<50 at.% Pd) [63].
- Both Mg and Pd form a binary hydride. PdH0.6 is the prototypical interstitial metallic hydride: its enthalpy of formation is −37 kJ/mol H2 (i.e., much less stable than MgH2) and its H-sorption kinetics are extremely fast also at room temperature.
- There is no evidence of ternary Mg-Pd-H phases.
3.2.1. Interfacial Mg-Pd Alloys in Pd-Catalyzed Mg Thin Films and NPs
3.2.2. Nanoconfined Mg-Pd Compounds
3.3. Mg-Ni Nanomaterials
- Mg and Ni form two stable line compounds, i.e., Mg2Ni and MgNi2. The solubility of Mg in Mg2Ni and of Ni in MgNi2 are negligible.
- MgNi2 can form the hydride MgNi2H3 only at very high pressure, i.e., 28 kbar at 300 °C [12].
- Mg2Ni forms the complex ternary hydride Mg2NiH4, in which covalently bonded [NiH4]4− complexes are ionically bonded to Mg2+. The formation enthalpy is −64 kJ/mol H2, still quite high but more favorable than MgH2 as far as hydrogen desorption is concerned.
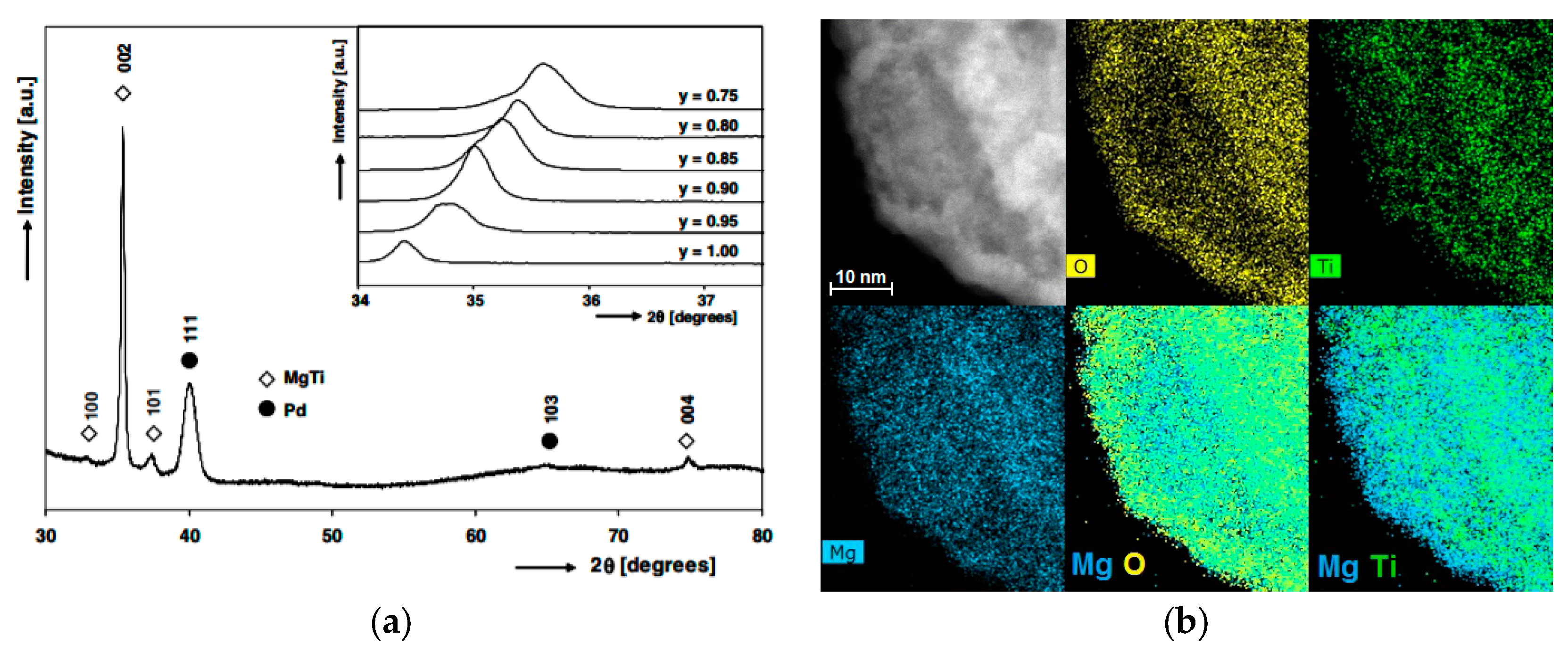
3.4. Mg-Ti Nanomaterials
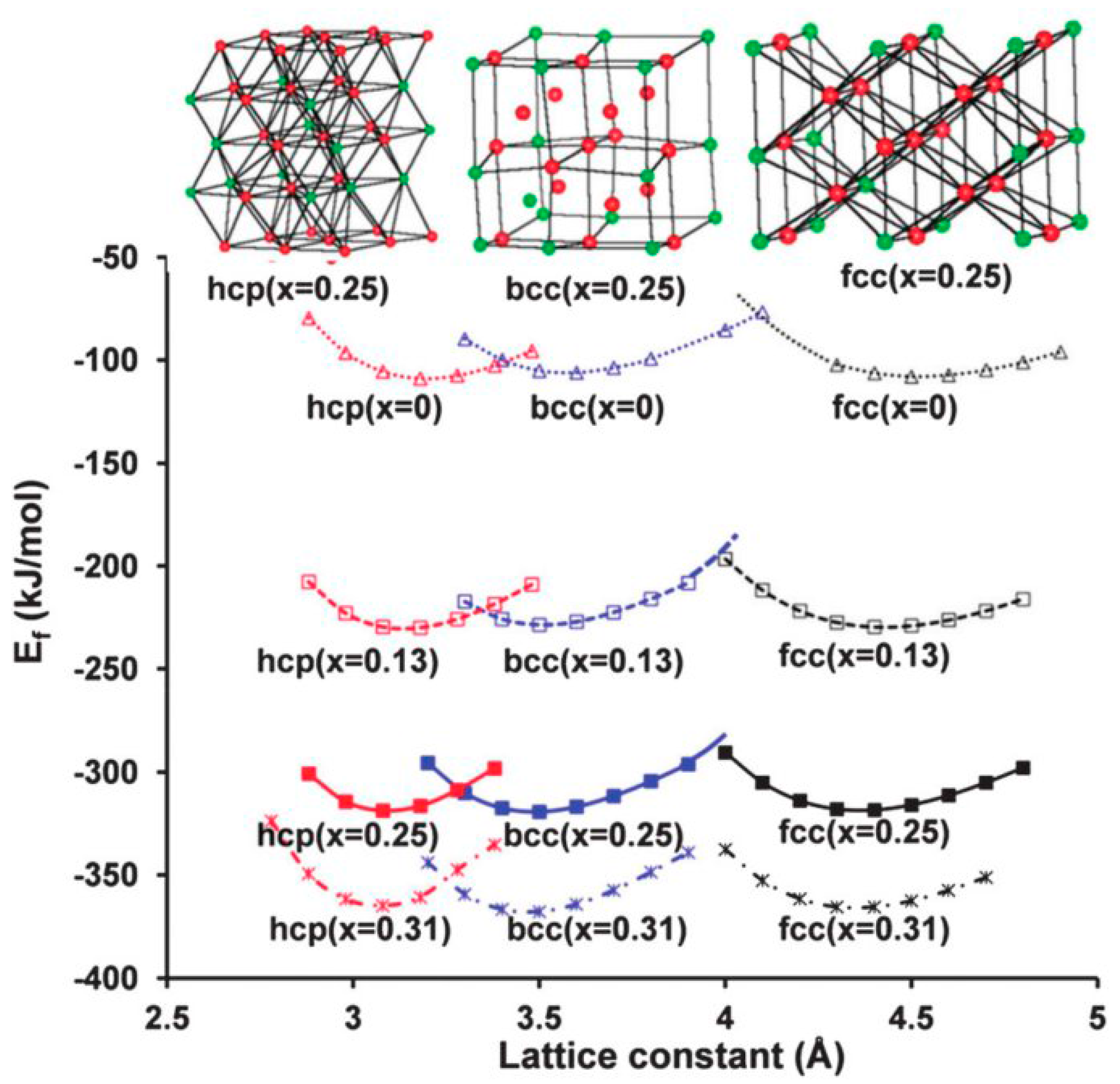
- Mg and Ti are completely immiscible in the solid-state; there are no stable compounds with intermediate stoichiometry, although metastable Mg-Ti solid solutions and compounds have been obtained by ball milling [85,86] and by physical co-deposition of mixed Mg-Ti vapors from two independent sources [87,88,89,90];
- Ti forms the highly stable TiH2 hydride (∆H0 ≈ −140 kJ/mol H2). This means that TiH2 will never decompose under the experimental conditions used to study hydrogen sorption in Mg.
3.5. Other Mg-X Nanomaterials
4. Conclusions
- Theoretical works predicted that increases with decreasing size of MgH2 NPs, reaching ≈5 kJ/mol H2 for a radius of 1 nm [27]. For even smaller MgH2 clusters with less than 10 Mg atoms, contrasting calculations were published: the most recent ones suggest that a stabilizing reconstruction takes place [28,29,30]. However, these ultra-small sizes have eluded experimental verification and are of little practical interest.
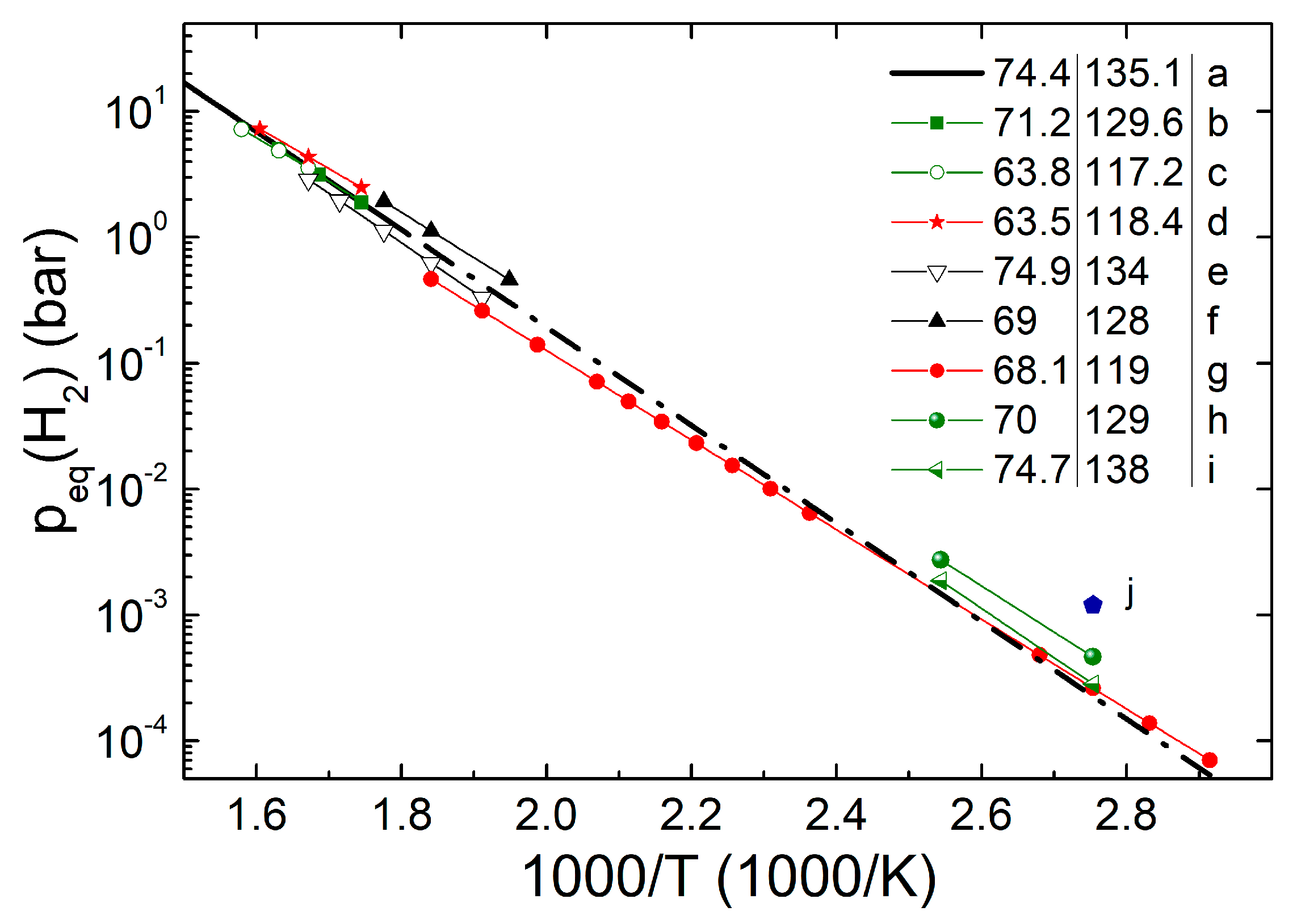
- Experiments on larger NPs (up to 15 nm diameter) sometimes yielded kJ/mol H2, far above the theoretical predictions [43,45,99]. These large values came only from measurements carried out in a narrow temperature range above 300 °C where the NPs were not stable. Moreover, sometimes only absorption data were reported. A true destabilization was not obtained because the equilibrium temperature for p(H2) = 1 bar remained very close to the value of bulk Mg (≈283 °C). The authors attributed this to an entropy compensation effect, J/K mol H2. However, it is well known that van ‘t Hoff analysis on a narrow temperature range can give rise to phantom enthalpy–entropy correlations.
- Our recent measurements down to 70 °C on MgH2/TiH2 composite NPs showed tiny variations of the equilibrium pressure compared to the extrapolated values of bulk Mg. We obtained a value kJ/mol H2 that is compatible with the calculations. These experiments ruled out the presence of a large enthalpy bias in Mg NPs with a diameter nm.
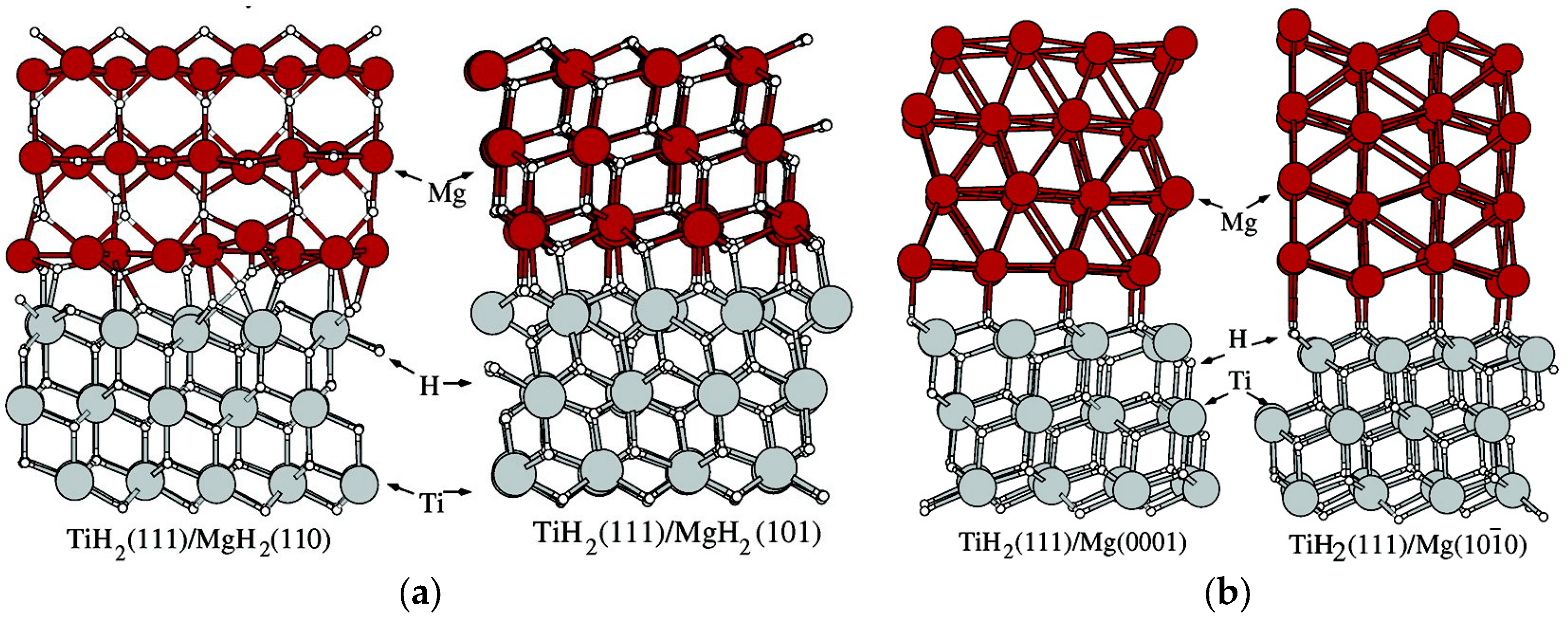
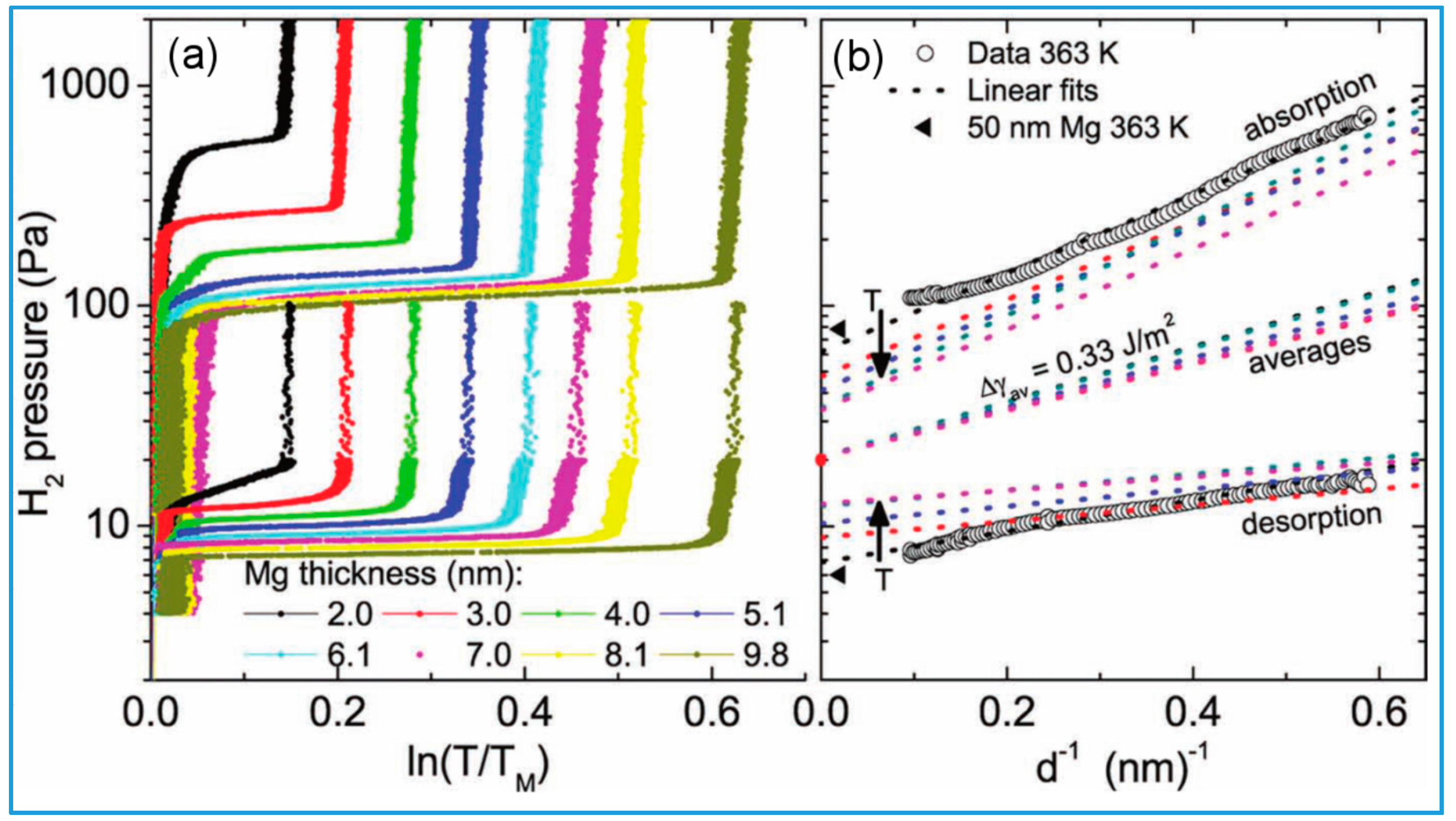
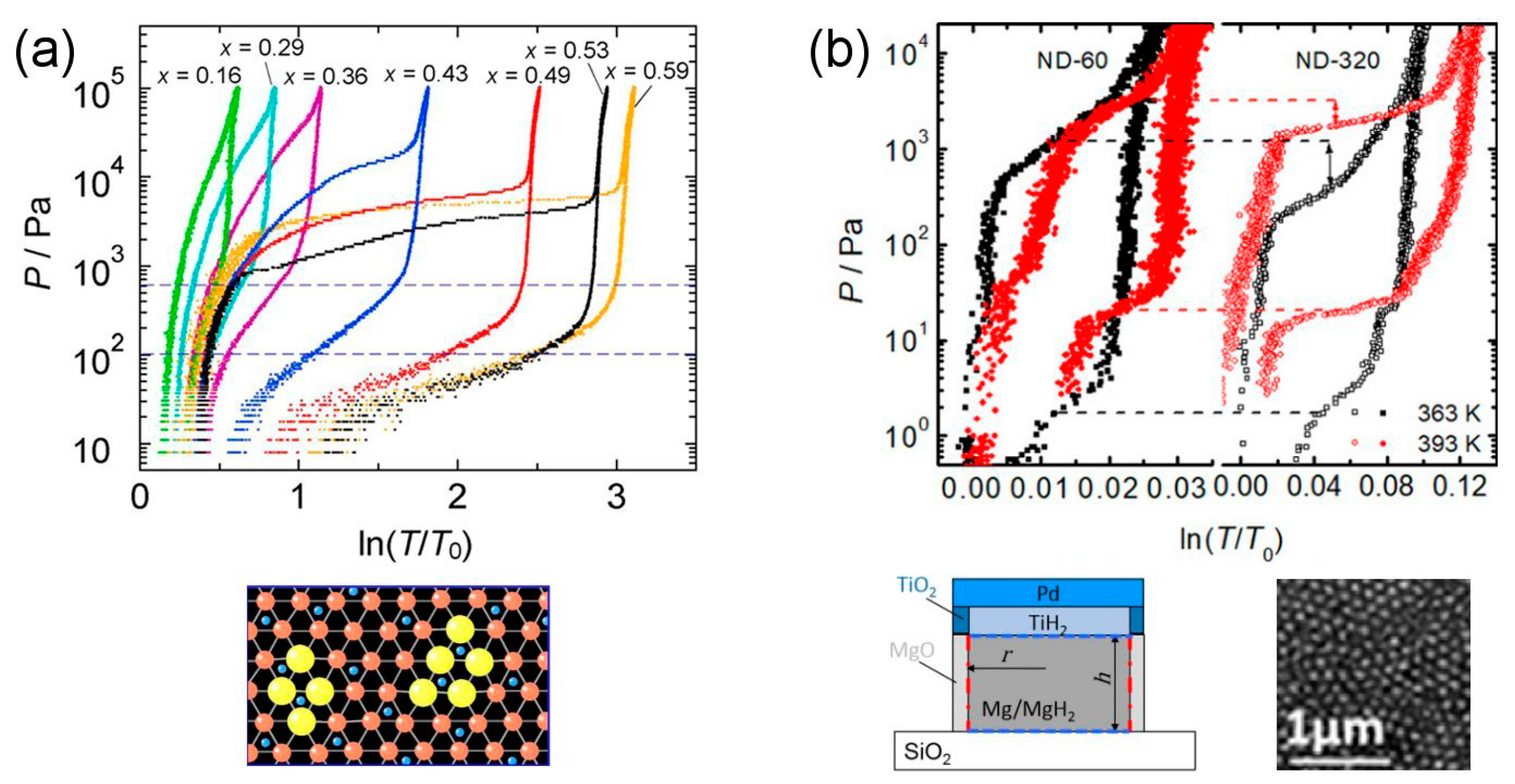
- Low temperature experiments (90–200 °C) on Mg thin films are in good agreement with the interface destabilization model described by Equations (9) and (10), the essential parameter of which is the difference ∆γ between the specific interface free energy in the hydride and in the metal phase [49]. The experimental value for Mg/MgH2 films sandwiched within TiH2 is ∆γ =0.33 J/m2, while calculations give ∆γ (0 K) = ∆h 0.5–0.6 J/m2. The difference may be due to the negative entropic contribution −T∆σ. At 90 °C, in a 2 nm film, the measured thermodynamical bias was = 4.6 kJ/mol H2, corresponding to a 5-fold pressure increase. A similar effect was observed in MgH2 clusters embedded in a TiH2 matrix.
- Calculations and experimental data on the interface/surface entropy are lacking. Knowledge of the ∆σ of interfaces between metal hydrides and different materials may prove useful for the tailoring of their thermodynamics. I suggest that this may be a prolific research direction both to improve the current understanding of nanoscale thermodynamics and to better control hydride properties towards applications.
- In order to prove the destabilization of the hydride it is necessary to measure the desorption plateau. In a truly destabilized hydride, an upward shift should be observed. Conversely, in Mg nanostructures, an upward shift of only the absorption plateau was frequently reported [33,34,35]. This may simply be caused by a large pressure hysteresis. Any destabilization claims and any enthalpy/entropy results based solely on the absorption plateau pressures are unreliable.
- In elastically constrained systems, the stresses that build up during volume expansion exceed the yield point of the material, leading to plastic rather than elastic strain. Plasticity is coupled with a large pressure hysteresis. The bias associated with the small elastic strain induces only a tiny shift of the average equilibrium pressure [35,68]. For this reason, a practical destabilization of MgH2 by elastic constraint has not yet been achieved.
- The spatial distribution of phases in small nanoparticles that individually undergo reaction schemes (13)–(15) is relatively unexplored. I believe that this may be a fruitful subject for future structural/thermodynamic calculations and experiments.
- TiH2 is an excellent partner of Mg/MgH2, making it possible to grow quasi-free films and composite NPs with very small hysteresis. Furthermore, it greatly speeds up the hydrogen sorption kinetics. Further studies are needed in order to unravel the detailed microscopic mechanisms behind its “catalytic” action.
Acknowledgments
Conflicts of Interest
References
- Takagi, S.; Orimo, S.-I. Recent progress in hydrogen-rich materials from the perspective of bonding flexibility of hydrogen. Scr. Mater. 2015, 109, 1–5. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and physical solutions for hydrogen storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Langhammer, C.; Dam, B. Metal hydrides for smart window and sensor applications. MRS Bull. 2013, 38, 495–503. [Google Scholar] [CrossRef]
- Lototskyy, M.V.; Yartys, V.A.; Pollet, B.G.; Bowman, R.C. Metal hydride hydrogen compressors: A review. Int. J. Hydrog. Energy 2014, 39, 5818–5851. [Google Scholar] [CrossRef]
- Felderhoff, M.; Bogdanović, B. High temperature metal hydrides as heat storage materials for solar and related applications. Int. J. Mol. Sci. 2009, 10, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Mohtadi, R.; Orimo, S. The renaissance of hydrides as energy materials. Nat. Rev. Mater. 2016, 2, 16091. [Google Scholar] [CrossRef]
- Unemoto, A.; Matsuo, M.; Orimo, S. Complex hydrides for electrochemical energy storage. Adv. Funct. Mater. 2014, 24, 2267–2279. [Google Scholar] [CrossRef]
- Oumellal, Y.; Rougier, A.; Nazri, G.A.; Tarascon, J.-M.; Aymard, L. Metal hydrides for lithium-ion batteries. Nat. Mater. 2008, 7, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.P.; Lal, C.; Jain, A. Hydrogen storage in Mg: A most promising material. Int. J. Hydrog. Energy 2010, 35, 5133–5144. [Google Scholar] [CrossRef]
- Crivello, J.-C.; Dam, B.; Denys, R.V.; Dornheim, M.; Grant, D.M.; Huot, J.; Jensen, T.R.; de Jongh, P.; Latroche, M.; Milanese, C.; Milčius, D.; et al. Review of magnesium hydride-based materials: Development and optimisation. Appl. Phys. A Mater. Sci. Process. 2016, 122. [Google Scholar] [CrossRef]
- Crivello, J.-C.; Denys, R.V.; Dornheim, M.; Felderhoff, M.; Grant, D.M.; Huot, J.; Jensen, T.R.; de Jongh, P.; Latroche, M.; Walker, G.S.; Webb, C.J.; et al. Mg-based compounds for hydrogen and energy storage. Appl. Phys. A 2016, 122, 85. [Google Scholar] [CrossRef]
- Stampfer, J.F.; Holley, C.E.; Suttle, J.F. The magnesium-hydrogen system. J. Am. Chem. Soc. 1960, 82, 3504–3508. [Google Scholar] [CrossRef]
- Paskevicius, M.; Sheppard, D.A.; Buckley, C.E. Thermodynamic Changes in Mechanochemically Synthesized Magnesium Hydride Nanoparticles. J. Am. Chem. Soc. 2010, 6, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Lototskyy, M.V.; Tolj, I.; Pickering, L.; Sita, C.; Barbir, F.; Yartys, V. The use of metal hydrides in fuel cell applications. Prog. Nat. Sci. Mater. Int. 2017, 27, 3–20. [Google Scholar] [CrossRef]
- Gremaud, R.; Broedersz, C.P.; Borsa, D.M.; Borgschulte, A.; Mauron, P.; Schreuders, H.; Rector, J.H.; Dam, B.; Griessen, R. Hydrogenography: An optical combinatorial method to find new light-weight hydrogen-storage materials. Adv. Mater. 2007, 19, 2813–2817. [Google Scholar] [CrossRef]
- Buschow, K.H.J.; Bouten, P.C.P.; Miedema, A.R. Hydrides formed from intermetallic compounds of two transition metals: A special class of ternary alloys. Rep. Prog. Phys. 2000, 45, 937–1039. [Google Scholar] [CrossRef]
- Callini, E.; Aguey-Zinsou, K.-F.; Ahuja, R.; Ares, J.R.; Bals, S.; Biliškov, N.; Chakraborty, S.; Charalambopoulou, G.; Chaudhary, A.-L.; Cuevas, F.; Dam, B.; et al. Nanostructured materials for solid-state hydrogen storage: A review of the achievement of COST Action MP1103. Int. J. Hydrog. Energy 2016, 41, 14404–14428. [Google Scholar] [CrossRef]
- Van Wees, B.J.; van Houten, H.; Beenakker, C.W.J.; Williamson, J.G.; Kouwenhoven, L.P.; van der Marel, D.; Foxon, C.T. Quantized conductance of point contacts in a two-dimensional electron gas. Phys. Rev. Lett. 1988, 60, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, L.; Rempel, A.A.; Würschum, R.; Reimann, K.; Müller, M.A.; Fultz, B.; Schaefer, H.-E. Thermal vacancy formation and D03 ordering in nanocrystalline intermetallic (Fe3Si) 95Nb5. Phys. Rev. B Condens. Matter Mater. Phys. 2001, 63. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Pasquini, L.; Barla, A.; Chumakov, A.I.; Leupold, O.; Rüffer, R.; Deriu, A.; Bonetti, E. Size and oxidation effects on the vibrational properties of nanocrystalline α-Fe. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 66. [Google Scholar] [CrossRef]
- Calvo, F. Thermodynamics of nanoalloys. Phys. Chem. Chem. Phys. 2015, 17, 27922–27939. [Google Scholar] [CrossRef] [PubMed]
- Bérubé, V.; Radtke, G.; Dresselhaus, M.; Chen, G. Size effects on the hydrogen storage properties of nanostructured metal hydrides: A. review. Int. J. Energy Res. 2007, 31, 637–663. [Google Scholar] [CrossRef]
- De Jongh, P.E.; Adelhelm, P. Nanosizing and nanoconfinement: New strategies towards meeting hydrogen storage goals. ChemSusChem 2010, 3, 1332–1348. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shen, C.; Lai, Q.; Liu, W.; Wang, D.-W.; Aguey-Zinsou, K.-F. Tailoring magnesium based materials for hydrogen storage through synthesis: Current state of the art. Energy Storage Mater. 2018, 10, 168–198. [Google Scholar] [CrossRef]
- Kim, K.C.; Dai, B.; Karl Johnson, J.; Sholl, D.S. Assessing nanoparticle size effects on metal hydride thermodynamics using the Wulff construction. Nanotechnology 2009, 20, 204001. [Google Scholar] [CrossRef] [PubMed]
- Wagemans, R.W.P.; Van Lenthe, J.H.; De Jongh, P.E.; Van Dillen, A.J.; De Jong, K.P. Hydrogen storage in magnesium clusters: Quantum chemical study. J. Am. Chem. Soc. 2005, 127, 16675–16680. [Google Scholar] [CrossRef] [PubMed]
- Shevlin, S.A.; Guo, Z.X. MgH2 dehydrogenation thermodynamics: Nanostructuring and transition metal doping. J. Phys. Chem. C 2013, 117, 10883–10891. [Google Scholar] [CrossRef]
- Wu, Z.; Allendorf, M.D.; Grossman, J.C. Quantum Monte Carlo Simulation of Nanoscale MgH2 Cluster Thermodynamics. J. Am. Chem. Soc. 2009, 131, 13918–13919. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.X.; Notten, P.H.L.; Van Santen, R.A.; Jansen, A.P.J. Fluorite transition metal hydride induced destabilization of the MgH2 system in MgH2/TMH2 multilayers (TM = Sc, Ti, V, Cr, Y, Zr, Nb, La, Hf). Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 1–5. [Google Scholar] [CrossRef]
- Hao, S.Q.; Sholl, D.S. Effect of TiH2-induced strain on thermodynamics of hydrogen release from MgH2. J. Phys. Chem. C 2012, 116, 2045–2050. [Google Scholar] [CrossRef]
- Pasquini, L.; Sacchi, M.; Brighi, M.; Boelsma, C.; Bals, S.; Perkisas, T.; Dam, B. Hydride destabilization in core-shell nanoparticles. Int. J. Hydrog. Energy 2014, 39. [Google Scholar] [CrossRef]
- Baldi, A.; Gonzalez-Silveira, M.; Palmisano, V.; Dam, B.; Griessen, R. Destabilization of the Mg-H system through elastic constraints. Phys. Rev. Lett. 2009, 102, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Molinari, A.; D’Amico, F.; Calizzi, M.; Zheng, Y.; Boelsma, C.; Mooij, L.; Lei, Y.; Hahn, H.; Dam, B.; Pasquini, L. Interface and strain effects on the H-sorption thermodynamics of size-selected Mg nanodots. Int. J. Hydrog. Energy 2016, 41, 9841–9851. [Google Scholar] [CrossRef]
- Cleri, F.; Celino, M.; Montone, A.; Bonetti, E.; Pasquini, L. Experimental and theoretical characterization of the 3D-dopants bias on the H desorption of Mg hydrides. Mater. Sci. Forum 2007, 555, 349–354. [Google Scholar] [CrossRef]
- Hussain, T.; Maark, T.A.; Chakraborty, S.; Ahuja, R. Improvement in hydrogen desorption from β- and γ-MgH2 upon transition-metal doping. ChemPhysChem 2015, 16, 2557–2561. [Google Scholar] [CrossRef] [PubMed]
- Paskaš Mamula, B.; Grbović Novaković, J.; Radisavljević, I.; Ivanović, N.; Novaković, N. Electronic structure and charge distribution topology of MgH2 doped with 3D transition metals. Int. J. Hydrog. Energy 2014, 39, 5874–5887. [Google Scholar] [CrossRef]
- Friedrichs, O.; Kolodziejczyk, L.; Sanchez-Lopez, J.C.; Fernandez, A.; Lyubenova, L.; Zander, D.; Köster, U.; Aguey-Zinsou, K.-F.; Klassen, T.; Bormann, R. Influence of particle size on electrochemical and gas-phase hydrogen storage in nanocrystalline Mg. J. Alloys Compd. 2008, 463, 539–545. [Google Scholar] [CrossRef]
- Pasquini, L.; Callini, E.; Piscopiello, E.; Montone, A.; Antisari, M.V.; Bonetti, E. Metal-hydride transformation kinetics in Mg nanoparticles. Appl. Phys. Lett. 2009, 94. [Google Scholar] [CrossRef]
- Krishnan, G.; Kooi, B.J.; Palasantzas, G.; Pivak, Y.; Dam, B. Thermal stability of gas phase magnesium nanoparticles. J. Appl. Phys. 2010, 107, 1–7. [Google Scholar] [CrossRef]
- Norberg, N.S.; Arthur, T.S.; Fredrick, S.J.; Prieto, A.L. Size-Dependent Hydrogen Storage Properties of Mg Nanocrystals Prepared from Solution. J. Am. Chem. Soc. 2011, 133, 10679–10681. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Aguey-Zinsou, K.-F. Size effects and hydrogen storage properties of Mg nanoparticles synthesised by an electroless reduction method. J. Mater. Chem. A 2014, 2, 9718. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Enthalpy-entropy compensation: A phantom phenomenon. J. Biosci. 2002, 27, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Zhao-Karger, Z.; Hu, J.; Roth, A.; Wang, D.; Kübel, C.; Lohstroh, W.; Fichtner, M. Altered thermodynamic and kinetic properties of MgH(2) infiltrated in microporous scaffold. Chem. Commun. (Camb.) 2010, 46, 8353–8355. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, F.; Korablov, D.; Latroche, M. Synthesis, structural and hydrogenation properties of Mg-rich MgH2–TiH2 nanocomposites prepared by reactive ball milling under hydrogen gas. Phys. Chem. Chem. Phys. 2012, 14, 1200. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Choi, Y.J.; Fang, Z.Z.; Sohn, H.Y.; Rönnebro, E. Hydrogen storage properties of nanosized MgH2-0.1TiH2 prepared by ultrahigh-energy-high-pressure milling. J. Am. Chem. Soc. 2009, 131, 15843–15852. [Google Scholar] [CrossRef] [PubMed]
- Patelli, N.; Calizzi, M.; Migliori, A.; Morandi, V.; Pasquini, L. Hydrogen Desorption Below 150 °C in MgH2–TiH2 Composite Nanoparticles: Equilibrium and Kinetic Properties. J. Phys. Chem. C 2017, 121, 11166–11177. [Google Scholar] [CrossRef]
- Mooij, L.P.A.; Baldi, A.; Boelsma, C.; Shen, K.; Wagemaker, M.; Pivak, Y.; Schreuders, H.; Griessen, R.; Dam, B. Interface energy controlled thermodynamics of nanoscale metal hydrides. Adv. Energy Mater. 2011, 1, 754–758. [Google Scholar] [CrossRef]
- Patelli, N.; Calizzi, M.; Pasquini, L. Interface enthalpy-entropy competition in nanoscale metal hydrides. Inorganics 2018, 6, 13. [Google Scholar] [CrossRef]
- Baldi, A.; Palmisano, V.; Gonzalez-Silveira, M.; Pivak, Y.; Slaman, M.; Schreuders, H.; Dam, B.; Griessen, R. Quasifree Mg-H thin films. Appl. Phys. Lett. 2009, 95, 20–22. [Google Scholar] [CrossRef]
- Pivak, Y.; Schreuders, H.; Dam, B. Thermodynamic Properties, Hysteresis behavior and stress-strain analysis of MgH2 thin films, studied over a wide temperature range. Crystals 2012, 2, 710–729. [Google Scholar] [CrossRef]
- Kalisvaart, P.; Shalchi-Amirkhiz, B.; Zahiri, R.; Zahiri, B.; Tan, X.; Danaie, M.; Botton, G.; Mitlin, D. Thermodynamically destabilized hydride formation in “bulk” Mg–AlTi multilayers for hydrogen storage. Phys. Chem. Chem. Phys. 2013, 15, 16432–16436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gross, A.F.; Van Atta, S.L.; Lopez, M.; Liu, P.; Ahn, C.C.; Vajo, J.J.; Jensen, C.M. The synthesis and hydrogen storage properties of a MgH(2) incorporated carbon aerogel scaffold. Nanotechnology 2009, 20, 204027. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.K.; Besenbacher, F.; Jensen, T.R. Nanoconfined hydrides for energy storage. Nanoscale 2011, 3, 2086–2098. [Google Scholar] [CrossRef] [PubMed]
- Zlotea, C.; Oumellal, Y.; Hwang, S.-J.; Ghimbeu, C.M.; de Jongh, P.E.; Latroche, M. Ultrasmall MgH2 nanoparticles embedded in an ordered microporous carbon exhibiting rapid hydrogen sorption kinetics. J. Phys. Chem. C 2015, 119, 18091–18098. [Google Scholar] [CrossRef]
- De Jongh, P.E.; Allendorf, M.; Vajo, J.J.; Zlotea, C. Nanoconfined light metal hydrides for reversible hydrogen storage. MRS Bull. 2013, 38, 488–494. [Google Scholar] [CrossRef]
- Xia, G.; Tan, Y.; Chen, X.; Sun, D.; Guo, Z.; Liu, H.; Ouyang, L.; Zhu, M.; Yu, X. Monodisperse magnesium hydride nanoparticles uniformly self-assembled on graphene. Adv. Mater. 2015, 27, 5981–5988. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.-J.; Moon, H.R.; Ruminski, A.M.; Jiang, B.; Kisielowski, C.; Bardhan, R.; Urban, J.J. Air-stable magnesium nanocomposites provide rapid and high-capacity hydrogen storage without using heavy-metal catalysts. Nat. Mater. 2011, 10, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Westerwaal, R.J.; Anastasopol, A.; Mooij, L.P.A.; Boelsma, C.; Ngene, P.; Schreuders, H.; Eijt, S.W.H.; Dam, B. Destabilization of Mg Hydride by Self-Organized Nanoclusters in the Immiscible Mg-Ti System. J. Phys. Chem. C 2015, 119, 12157–12164. [Google Scholar] [CrossRef]
- Shegai, T.; Langhammer, C. Hydride formation in single palladium and magnesium nanoparticles studied by nanoplasmonic dark-field scattering spectroscopy. Adv. Mater. 2011, 23, 4409–4414. [Google Scholar] [CrossRef] [PubMed]
- Syrenova, S.; Langhammer, C. Hydride formation thermodynamics and hysteresis in individual Pd nanocrystals with different size and shape. Nat. Mater. 2015, 14, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Makongo, J.P.A.; Prots, Y.; Burkhardt, U.; Niewa, R.; Kudla, C.; Kreiner, G. A case study of complex metallic alloy phases: Structure and disorder phenomena of Mg–Pd compounds. Philos. Mag. 2006, 86, 427–433. [Google Scholar] [CrossRef]
- Huot, J.; Yonkeu, A.; Dufour, J. Rietveld analysis of neutron powder diffraction of Mg6Pd alloy at various hydriding stages. J. Alloys Compd. 2009, 475, 168–172. [Google Scholar] [CrossRef]
- Siviero, G.; Bello, V.; Mattei, G.; Mazzoldi, P.; Battaglin, G.; Bazzanella, N.; Checchetto, R.; Miotello, A. Structural evolution of Pd-capped Mg thin films under H2 absorption and desorption cycles. Int. J. Hydrog. Energy 2009, 34, 4817–4826. [Google Scholar] [CrossRef]
- Kelekar, R.; Giffard, H.; Kelly, S.T.; Clemens, B.M. Formation and dissociation of MgH2 in epitaxial Mg thin films. J. Appl. Phys. 2007, 101, 1–7. [Google Scholar] [CrossRef]
- Krozer, A.; Kasemo, B. Hydrogen uptake by Pd-coated Mg: Absorption-decomposition isotherms and uptake kinetics. J. Less Common Met. 1990, 160, 323–342. [Google Scholar] [CrossRef]
- Chung, C.J.; Lee, S.C.; Groves, J.R.; Brower, E.N.; Sinclair, R.; Clemens, B.M. Interfacial alloy hydride destabilization in Mg/Pd thin films. Phys. Rev. Lett. 2012, 108, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Callini, E.; Pasquini, L.; Piscopiello, E.; Montone, A.; Antisari, M.V.; Bonetti, E. Hydrogen sorption in Pd-decorated Mg-MgO core-shell nanoparticles. Appl. Phys. Lett. 2009, 94. [Google Scholar] [CrossRef]
- Callini, E.; Pasquini, L.; Rude, L.H.; Nielsen, T.K.; Jensen, T.R.; Bonetti, E. Hydrogen storage and phase transformations in Mg-Pd nanoparticles. J. Appl. Phys. 2010, 108. [Google Scholar] [CrossRef]
- Pasquini, L.; Boscherini, F.; Callini, E.; Maurizio, C.; Pasquali, L.; Montecchi, M.; Bonetti, E. Local structure at interfaces between hydride-forming metals: A case study of Mg-Pd nanoparticles by X-ray spectroscopy. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 83. [Google Scholar] [CrossRef]
- Fernandez, J.F.; Ares, J.R.; Cuevas, F.; Bodega, J.; Leardini, F.; Sánchez, C. A thermodynamic study of the hydrogenation of the pseudo-binary Mg6Pd0.5Ni0.5 intermetallic compound. Intermetallics 2010, 18, 233–241. [Google Scholar] [CrossRef]
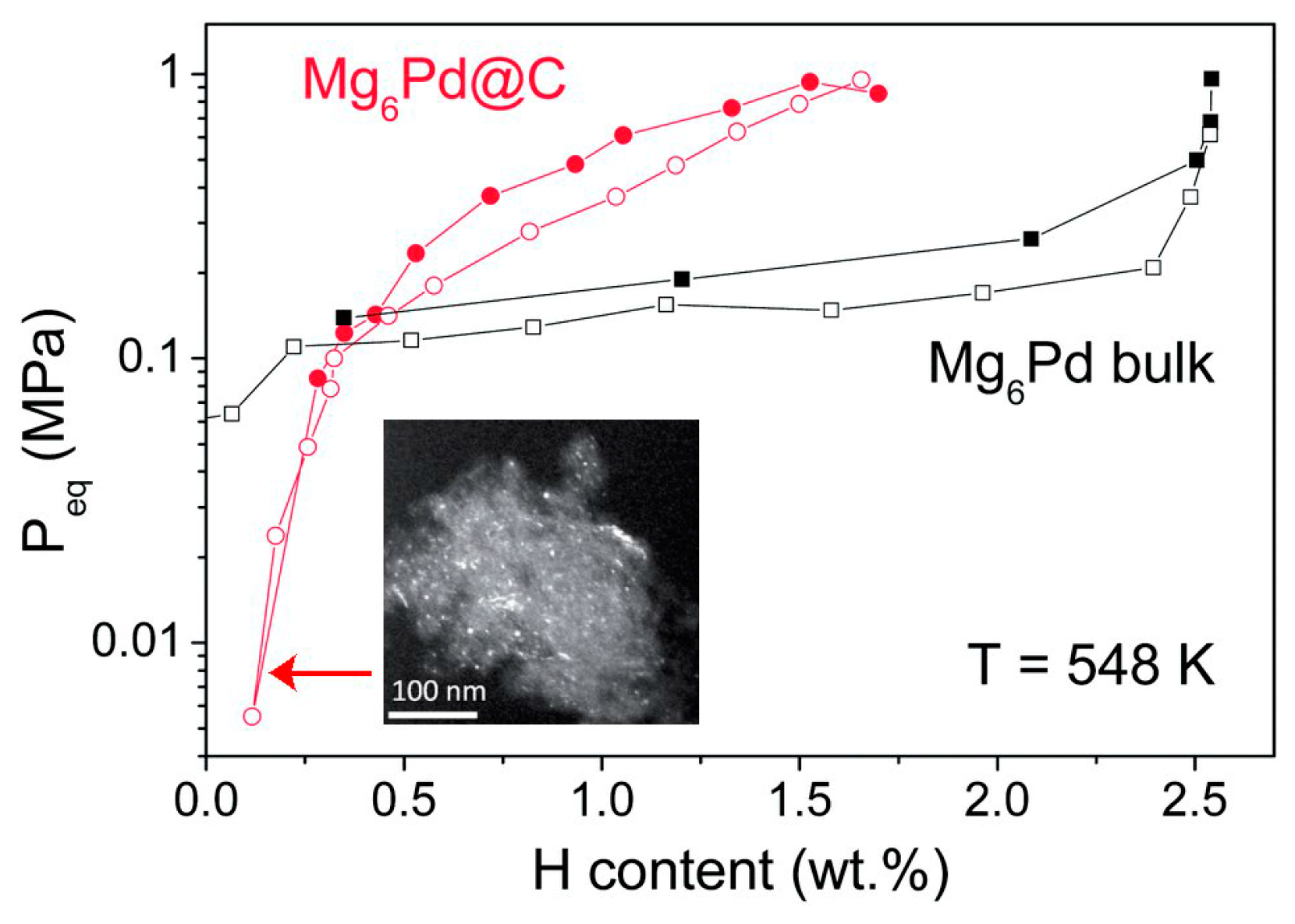
- Ponthieu, M.; Au, Y.S.; Provost, K.; Zlotea, C.; Leroy, E.; Fernández, J.F.; Latroche, M.; de Jongh, P.E.; Cuevas, F. Nanoconfinement of Mg6Pd particles in porous carbon: Size effects on structural and hydrogenation properties. J. Mater. Chem. A 2014, 2, 18444–18453. [Google Scholar] [CrossRef]
- Friedlmeier, G.; Arakawa, M.; Hirai, T.; Akiba, E. Preparation and structural, thermal and hydriding characteristics of melt-spun Mg–Ni alloys. J. Alloys Compd. 1999, 292, 107–117. [Google Scholar] [CrossRef]
- Orimo, S.-I.; Fujii, H. Materials science of Mg-Ni-based new hydrides. Appl. Phys. A 2001, 72, 167–186. [Google Scholar] [CrossRef]
- Zaluska, A.; Zaluski, L.; Ström-Olsen, J.O. Structure, catalysis and atomic reactions on the nano-scale: A systematic approach to metal hydrides for hydrogen storage. Appl. Phys. A Mater. Sci. Process. 2001, 72, 157–165. [Google Scholar] [CrossRef]
- Montone, A.; Grbovic Novakovic, J.; Vittori Antisari, M.; Bassetti, A.; Bonetti, E.; Fiorini, A.L.; Pasquini, L.; Mirenghi, L.; Rotolo, P. Nano-micro MgH2-Mg2 NiH4 composites: Tayloring a multichannel system with selected hydrogen sorption properties. Int. J. Hydrog. Energy 2007, 32. [Google Scholar] [CrossRef]
- Callini, E.; Pasquini, L.; Jensen, T.R.; Bonetti, E. Hydrogen storage properties of Mg-Ni nanoparticles. Int. J. Hydrog. Energy 2013, 38. [Google Scholar] [CrossRef]
- Gajdics, M.; Calizzi, M.; Pasquini, L.; Schafler, E.; Révész, A. Characterization of a nanocrystalline Mg-Ni alloy processed by high-pressure torsion during hydrogenation and dehydrogenation. Int. J. Hydrog. Energy 2016, 41, 9803–9809. [Google Scholar] [CrossRef]
- Zou, J.; Sun, H.; Zeng, X.; Ji, G.; Ding, W. Preparation and Hydrogen Storage Properties of Mg-Rich Mg-Ni Ultrafine Particles. J. Nanomater. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Matsuda, J.; Yoshida, K.; Sasaki, Y.; Uchiyama, N.; Akiba, E. In situ observation on hydrogenation of Mg-Ni films using environmental transmission electron microscope with aberration correction. Appl. Phys. Lett. 2014, 105, 83903. [Google Scholar] [CrossRef]
- Pasquini, L.; Callini, E.; Brighi, M.; Boscherini, F.; Montone, A.; Jensen, T.R.; Maurizio, C.; Antisari, M.V.; Bonetti, E. Magnesium nanoparticles with transition metal decoration for hydrogen storage. J. Nanopart. Res. 2011, 13. [Google Scholar] [CrossRef]
- Bogerd, R.; Adelhelm, P.; Meeldijk, J.H.; de Jong, K.P.; de Jongh, P.E. The structural characterization and H2 sorption properties of carbon-supported Mg1−x Nix nanocrystallites. Nanotechnology 2009, 20, 204019. [Google Scholar] [CrossRef] [PubMed]
- Zlotea, C.; Cuevas, F.; Andrieux, J.; Matei Ghimbeu, C.; Leroy, E.; Léonel, E.; Sengmany, S.; Vix-Guterl, C.; Gadiou, R.; Martens, T.; et al. Tunable synthesis of (Mg-Ni)-based hydrides nanoconfined in templated carbon studied by in situ synchrotron diffraction. Nano Energy 2013, 2, 12–20. [Google Scholar] [CrossRef]
- Liang, G.; Schulz, R. Synthesis of Mg-Ti alloy by mechanical alloying. J. Mater. Sci. 2003, 38, 1179–1184. [Google Scholar] [CrossRef]
- Asano, K.; Enoki, H.; Akiba, E. Synthesis of HCP, FCC and BCC structure alloys in the Mg-Ti binary system by means of ball milling. J. Alloys Compd. 2009, 480, 558–563. [Google Scholar] [CrossRef]
- Borsa, D.M.; Gremaud, R.; Baldi, A.; Schreuders, H.; Rector, J.H.; Kooi, B.; Vermeulen, P.; Notten, P.H.L.; Dam, B.; Griessen, R. Structural, optical, and electrical properties of Mgy Ti1-y Hx thin films. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 1–9. [Google Scholar] [CrossRef]
- Vermeulen, P.; Graat, P.C.J.; Wondergem, H.J.; Notten, P.H.L. Crystal structures of MgyTi100-y thin film alloys in the as-deposited and hydrogenated state. Int. J. Hydrog. Energy 2008, 33, 5646–5650. [Google Scholar] [CrossRef]
- Calizzi, M.; Venturi, F.; Ponthieu, M.; Cuevas, F.; Morandi, V.; Perkisas, T.; Bals, S.; Pasquini, L. Gas-phase synthesis of Mg–Ti nanoparticles for solid-state hydrogen storage. Phys. Chem. Chem. Phys. 2016, 18, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Anastasopol, A.; Pfeiffer, T.V.; Middelkoop, J.; Lafont, U.; Canales-Perez, R.J.; Schmidt-Ott, A.; Mulder, F.M.; Eijt, S.W.H. Reduced enthalpy of metal hydride formation for Mg-Ti nanocomposites produced by spark discharge generation. J. Am. Chem. Soc. 2013, 135, 7891–7900. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, P.; Niessen, R.A.H.; Notten, P.H.L. Hydrogen storage in metastable MgyTi(1−y) thin films. Electrochem. Commun. 2006, 8, 27–32. [Google Scholar] [CrossRef]
- Baldi, A.; Gremaud, R.; Borsa, D.M.; Baldé, C.P.; van der Eerden, A.M.J.; Kruijtzer, G.L.; de Jongh, P.E.; Dam, B.; Griessen, R. Nanoscale composition modulations in MgyTi1-yHx thin film alloys for hydrogen storage. Int. J. Hydrog. Energy 2009, 34, 1450–1457. [Google Scholar] [CrossRef]
- Ponthieu, M.; Cuevas, F.; Fernández, J.F.; Laversenne, L.; Porcher, F.; Latroche, M. Structural properties and reversible deuterium loading of MgD2–TiD2 nanocomposites. J. Phys. Chem. C 2013, 117, 18851–18862. [Google Scholar] [CrossRef]
- Calizzi, M.; Chericoni, D.; Jepsen, L.H.; Jensen, T.R.; Pasquini, L. Mg-Ti nanoparticles with superior kinetics for hydrogen storage. Int. J. Hydrog. Energy 2015. [Google Scholar] [CrossRef]
- Krishnan, G.; Negrea, R.F.; Ghica, C.; ten Brink, G.H.; Kooi, B.J.; Palasantzas, G. Synthesis and exceptional thermal stability of Mg-based bimetallic nanoparticles during hydrogenation. Nanoscale 2014, 6, 11963–11970. [Google Scholar] [CrossRef] [PubMed]
- Jangir, M.; Jain, A.; Agarwal, S.; Zhang, T.; Kumar, S.; Selvaraj, S.; Ichikawa, T.; Jain, I.P. The enhanced de/re-hydrogenation performance of MgH2 with TiH2 additive. Int. J. Energy Res. 2017, 1–9. [Google Scholar] [CrossRef]
- Shao, H.; Xin, G.; Zheng, J.; Li, X.; Akiba, E. Nanotechnology in Mg-based materials for hydrogen storage. Nano Energy 2012, 1, 590–601. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, R.; Qu, J.; Zhao, W.; Xie, L.; Tian, W.; Li, X. The synthesis and hydrogen storage properties of pure nanostructured Mg2 FeH6. Nanotechnology 2010, 21, 95706. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Setijadi, E.J.; Aguey-Zinsou, K.-F. Tuning the Thermodynamic properties of MgH2 at the nanoscale via a catalyst or destabilizing element coating strategy. J. Phys. Chem. C 2014. [Google Scholar] [CrossRef]
- Bazzanella, N.; Checchetto, R.; Miotello, A.; Sada, C.; Mazzoldi, P.; Mengucci, P. Hydrogen kinetics in magnesium hydride: On different catalytic effects of niobium. Appl. Phys. Lett. 2006, 89, 5–7. [Google Scholar] [CrossRef]
- Barkhordarian, G.; Klassen, T.; Bormann, R. Catalytic mechanism of transition-metal compounds on Mg hydrogen sorption reaction. J. Phys. Chem. B 2006, 110, 11020–11024. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.H.; Wang, L.Y.; Holt, C.M.B.; Zahiri, B.; Eikerling, M.H.; Mitlin, D. Body centered cubic magnesium niobium hydride with facile room temperature absorption and four weight percent reversible capacity. Phys. Chem. Chem. Phys. 2012, 14, 10904–10909. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, H.G.; Huot, J.; Chapon, L.C.; Tichelaar, F.D.; Mulder, F.M. Hydrogen cycling of niobium and vanadium catalyzed nanostructured magnesium. J. Am. Chem. Soc. 2005, 127, 14348–14354. [Google Scholar] [CrossRef] [PubMed]
- Milošević, S.; Kurko, S.; Pasquini, L.; Matović, L.; Vujasin, R.; Novaković, N.; Novaković, J.G. Fast hydrogen sorption from MgH2-VO2(B) composite materials. J. Power Sources 2016, 307. [Google Scholar] [CrossRef]
- Liang, G.; Huot, J.; Boily, S.; Van Neste, A.; Schulz, R. Hydrogen storage properties of the mechanically milled MgH2-V nanocomposite. J. Alloys Compd. 1999, 291, 295–299. [Google Scholar] [CrossRef]
- Kyoi, D.; Sato, T.; Rönnebro, E.; Tsuji, Y.; Kitamura, N.; Ueda, A.; Ito, M.; Katsuyama, S.; Hara, S.; Noréus, D.; Sakai, T. A novel magnesium-vanadium hydride synthesized by a gigapascal-high-pressure technique. J. Alloys Compd. 2004, 375, 253–258. [Google Scholar] [CrossRef]














© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquini, L. The Effects of Nanostructure on the Hydrogen Sorption Properties of Magnesium-Based Metallic Compounds: A Review. Crystals 2018, 8, 106. https://doi.org/10.3390/cryst8020106
Pasquini L. The Effects of Nanostructure on the Hydrogen Sorption Properties of Magnesium-Based Metallic Compounds: A Review. Crystals. 2018; 8(2):106. https://doi.org/10.3390/cryst8020106
Chicago/Turabian StylePasquini, Luca. 2018. "The Effects of Nanostructure on the Hydrogen Sorption Properties of Magnesium-Based Metallic Compounds: A Review" Crystals 8, no. 2: 106. https://doi.org/10.3390/cryst8020106
APA StylePasquini, L. (2018). The Effects of Nanostructure on the Hydrogen Sorption Properties of Magnesium-Based Metallic Compounds: A Review. Crystals, 8(2), 106. https://doi.org/10.3390/cryst8020106