Experimental Electron Density Distribution in Two Cocrystals of Betaines with p-Hydroxybenzoic Acid



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Reduction
2.2. IAM Model Refinement
2.3. Multipolar Modeling
2.4. Quality of the Model
3. Results and Discussion
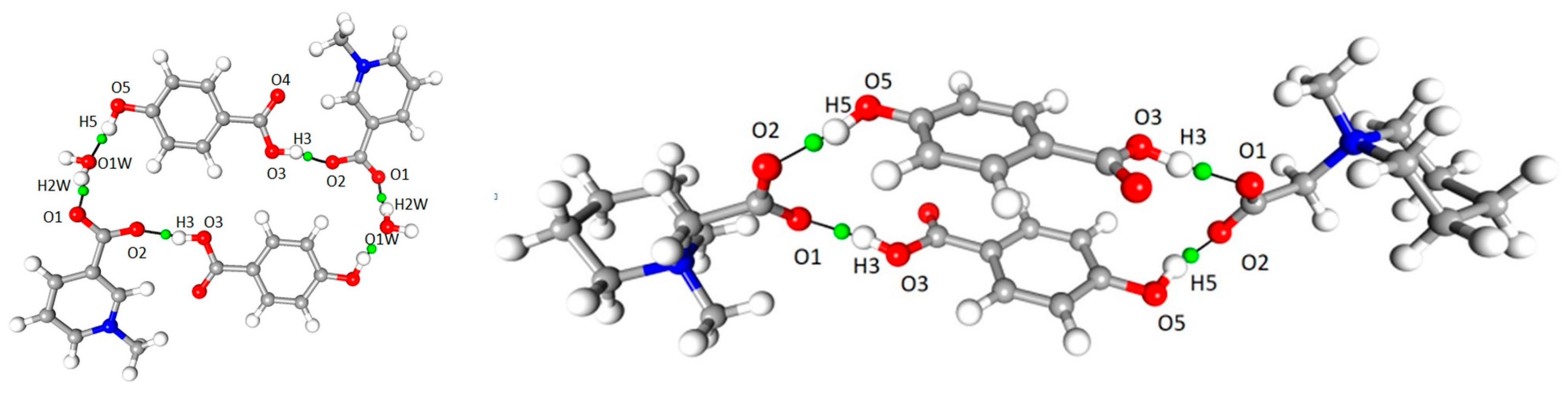
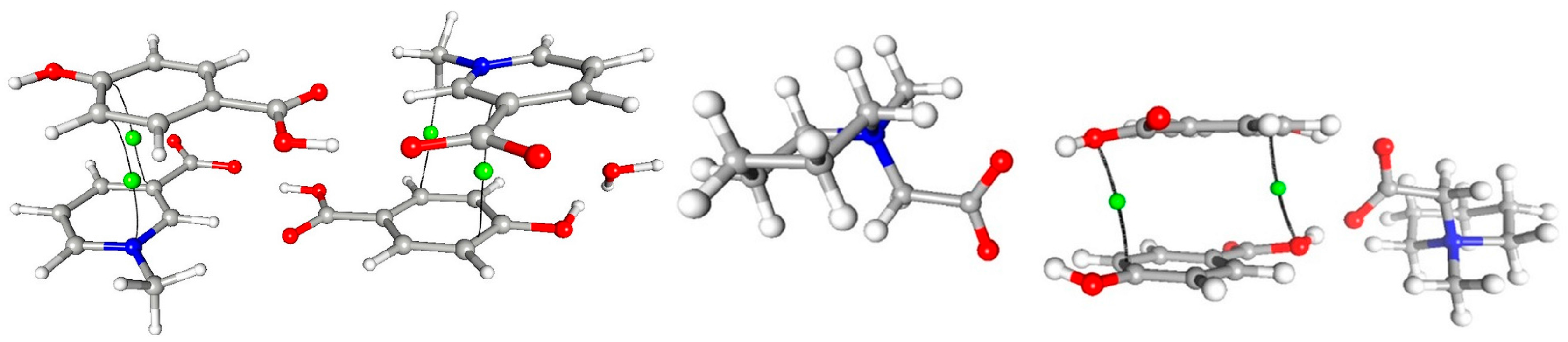
3.1. Molecular and Crystal Structure
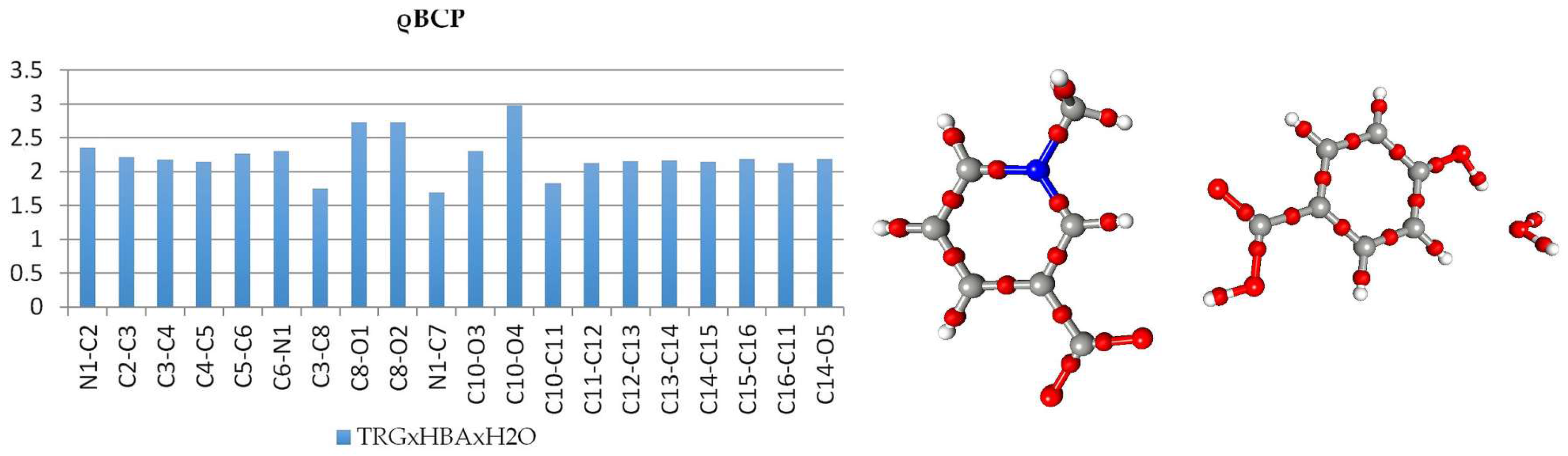
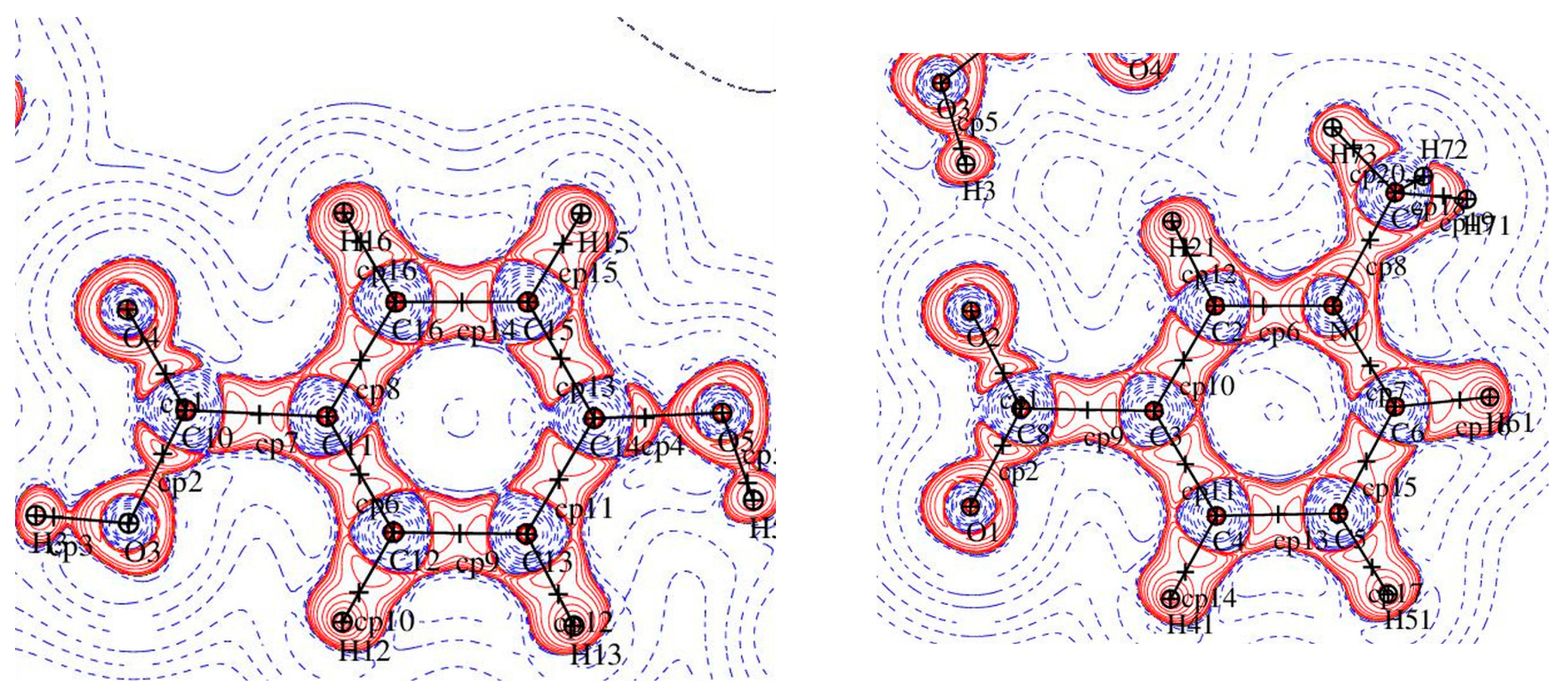
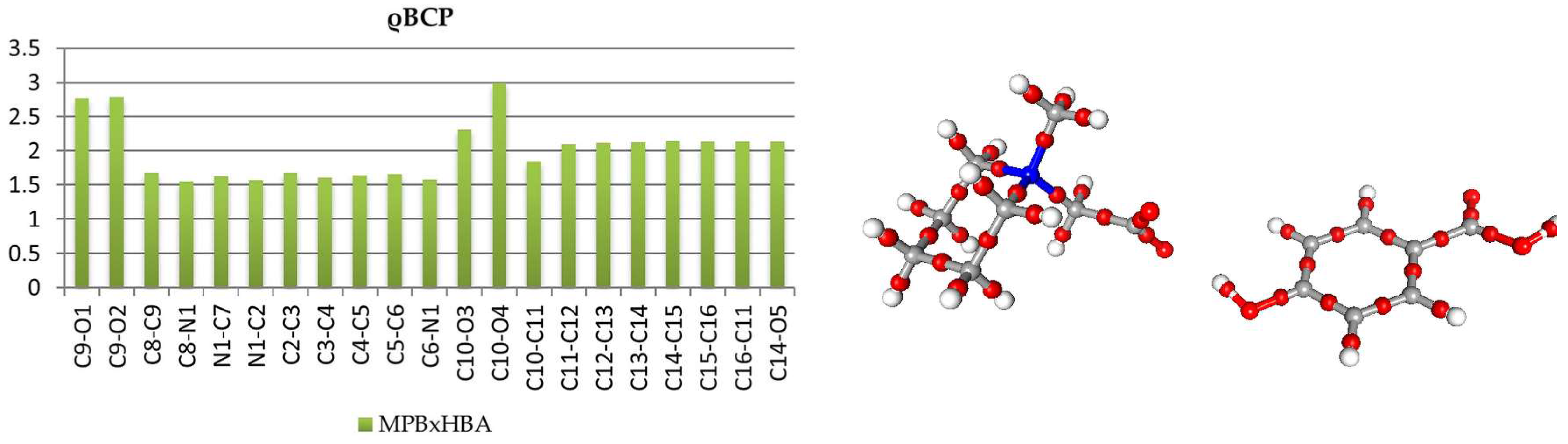
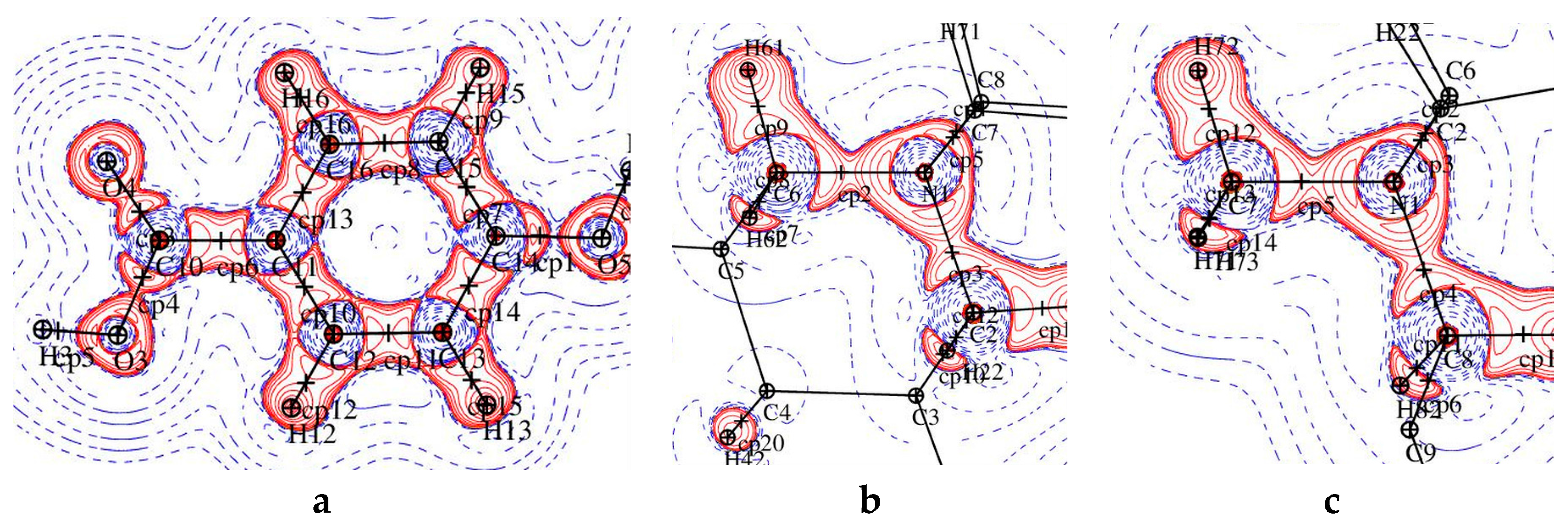
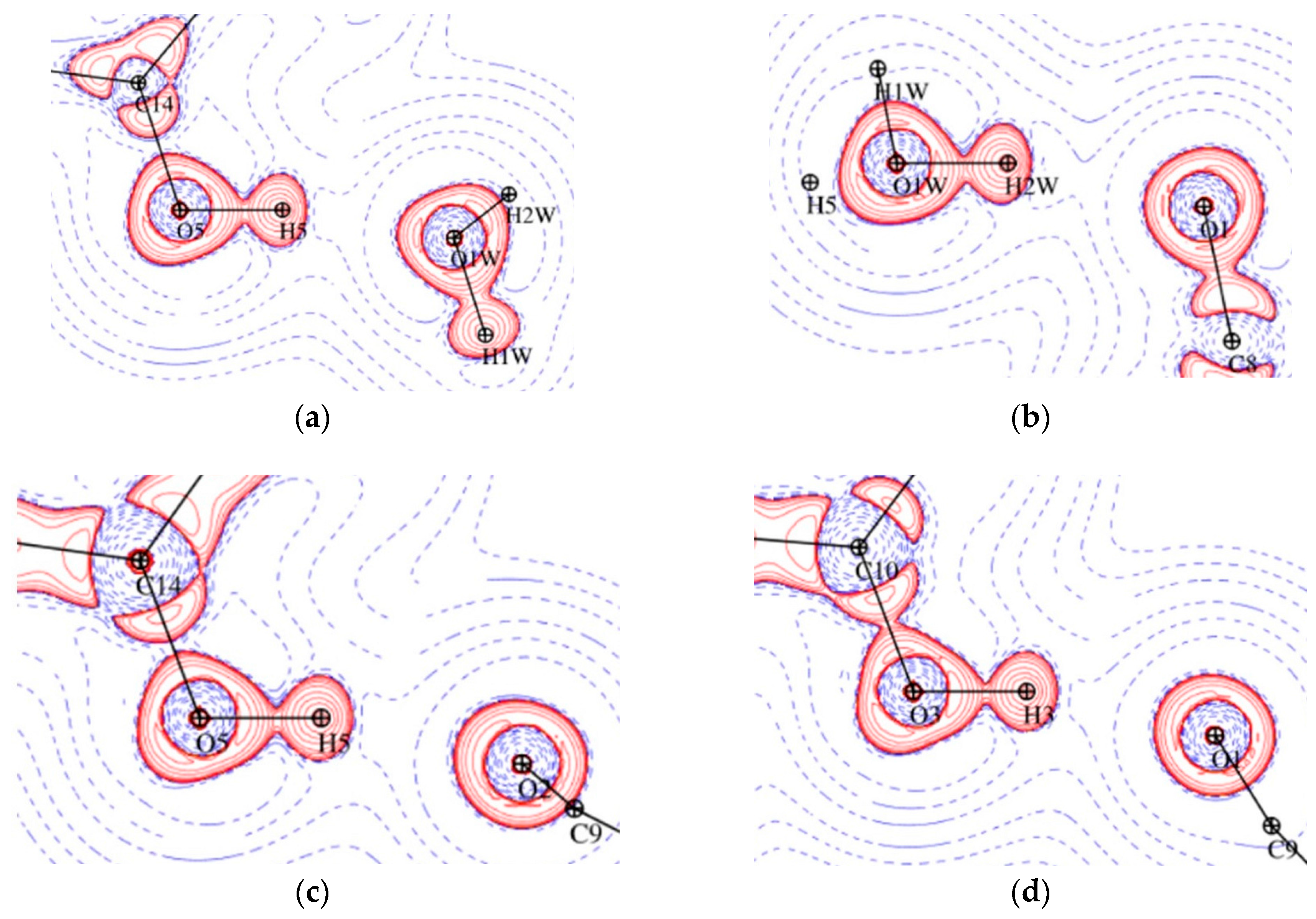
3.2. Topological Analysis of Covalent Bonds
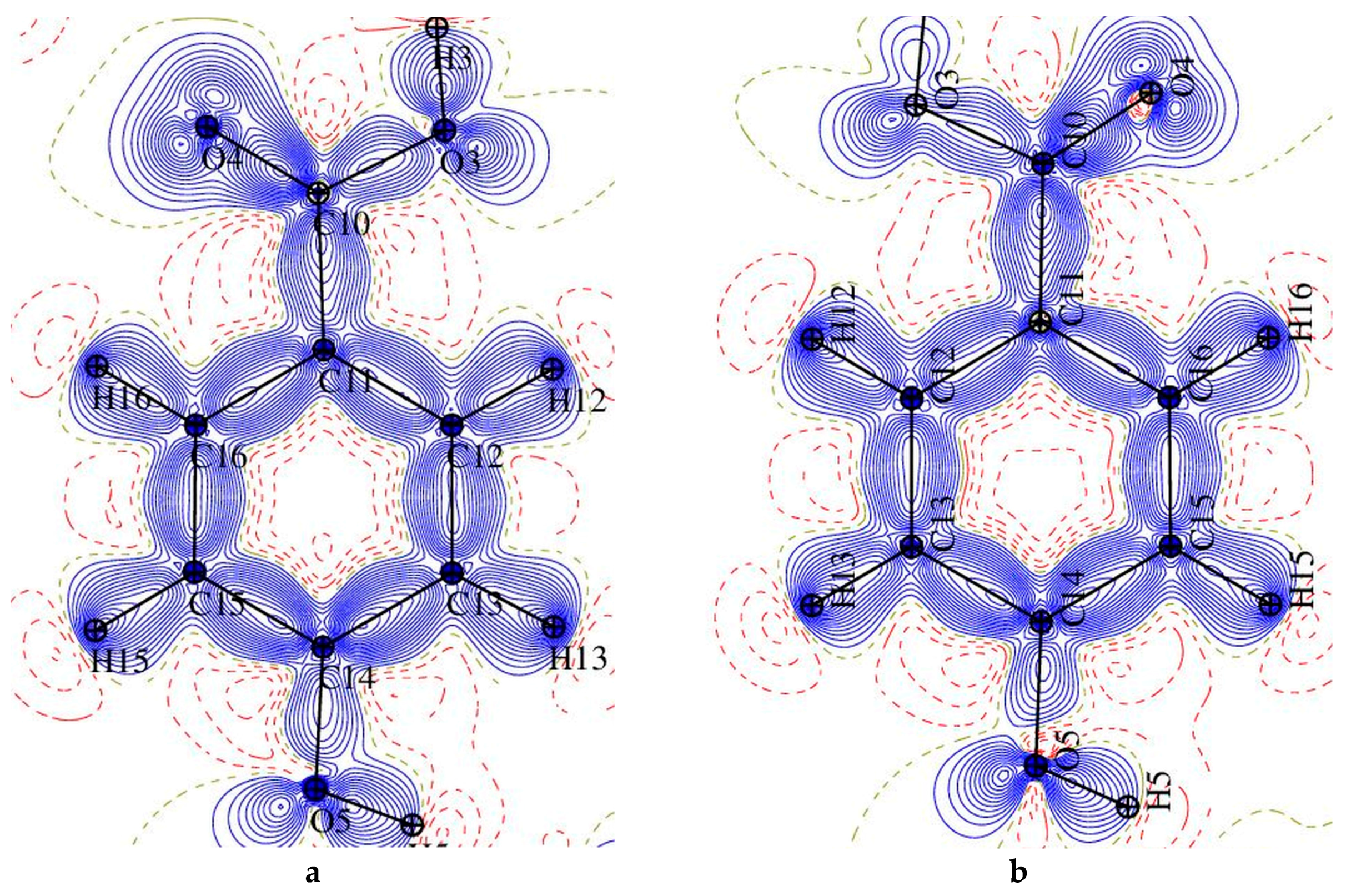
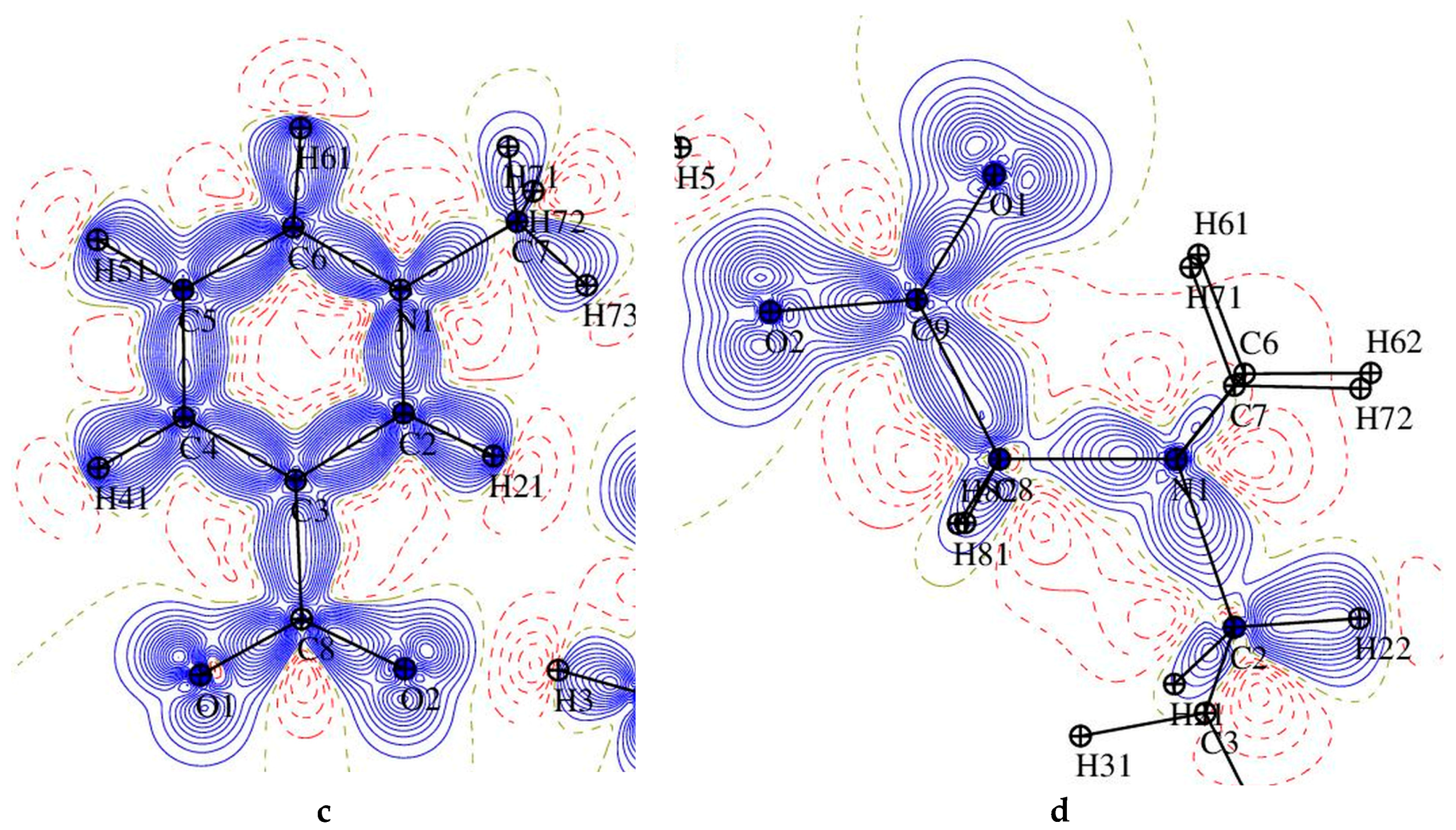
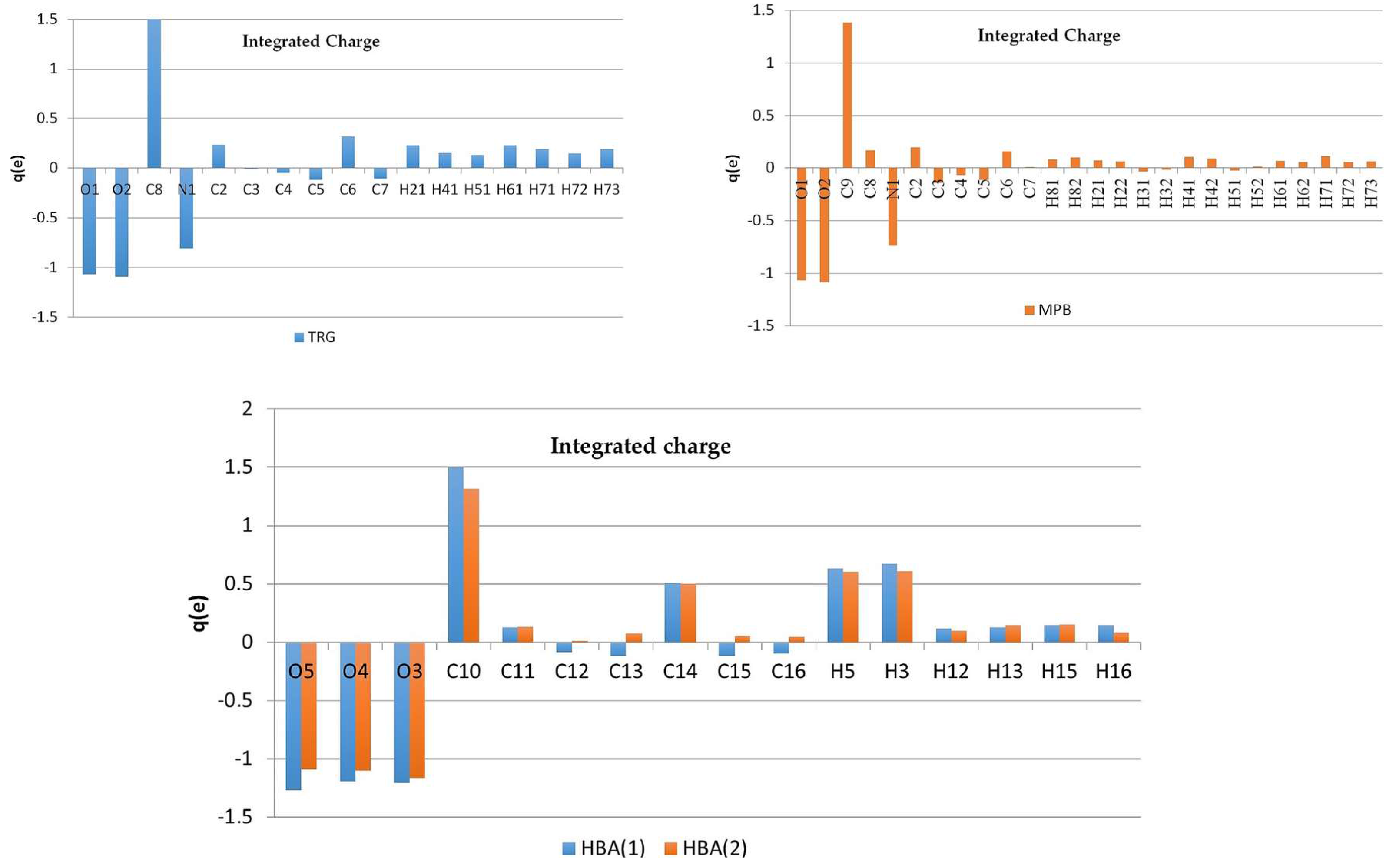
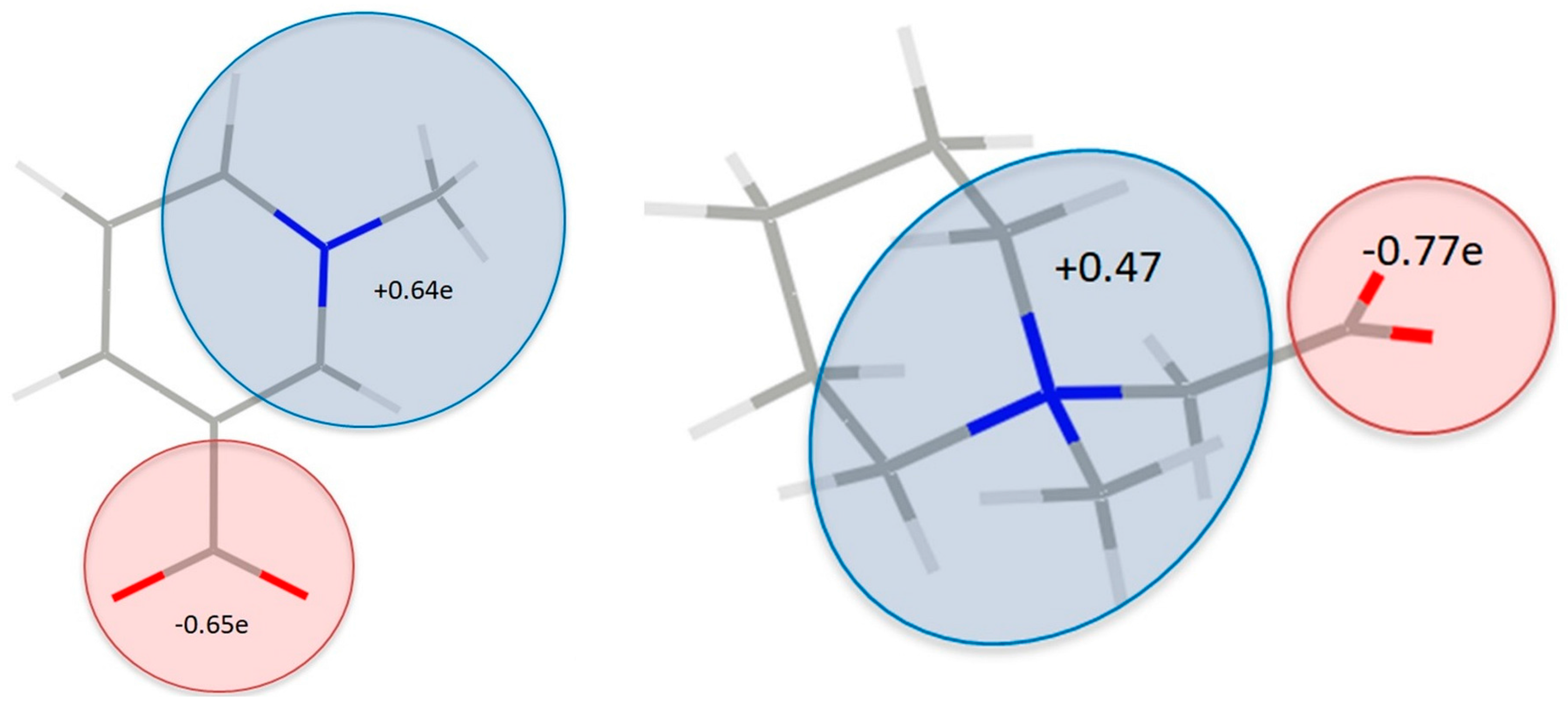
3.3. Experimental Deformation Electron Density and Atomic Charges
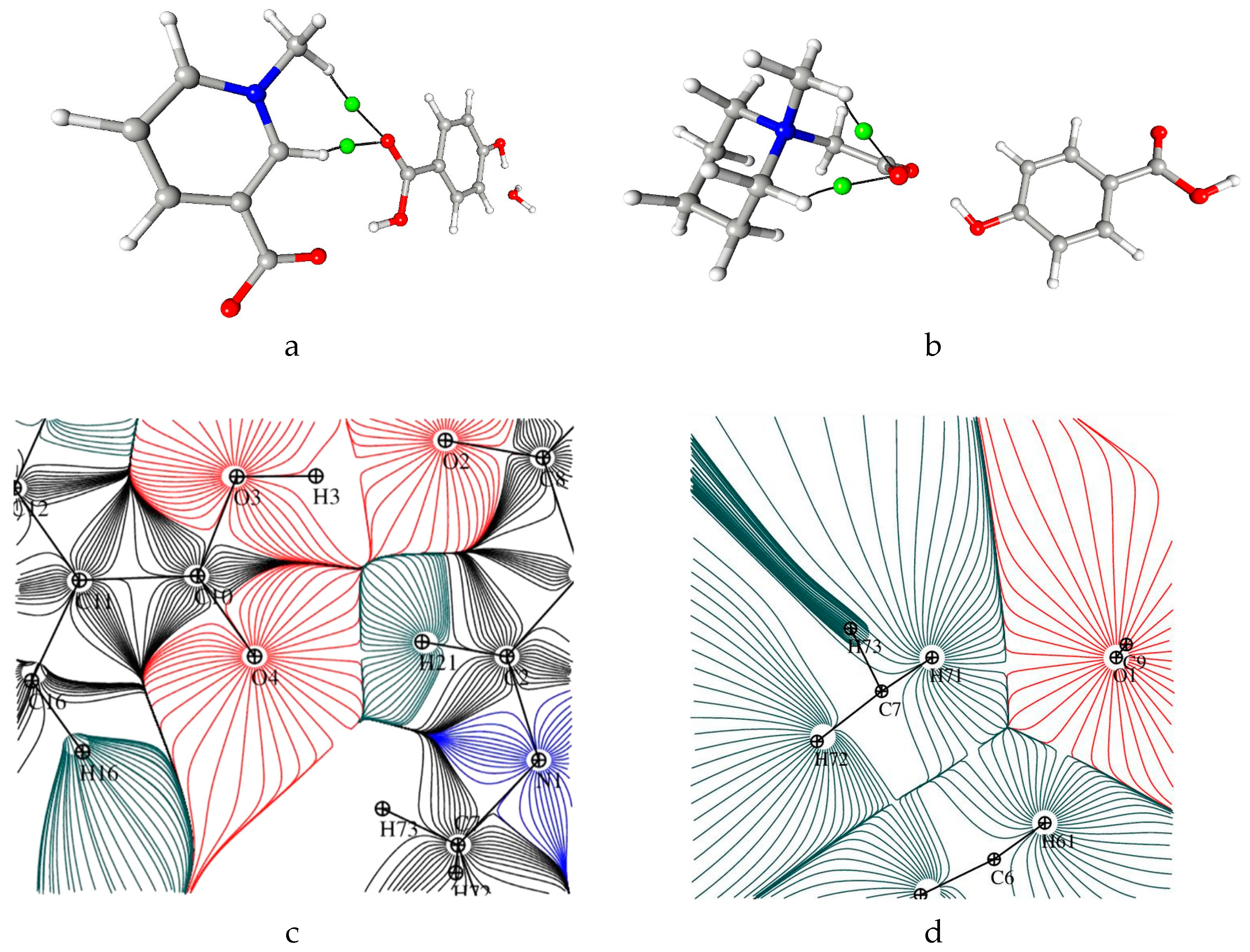
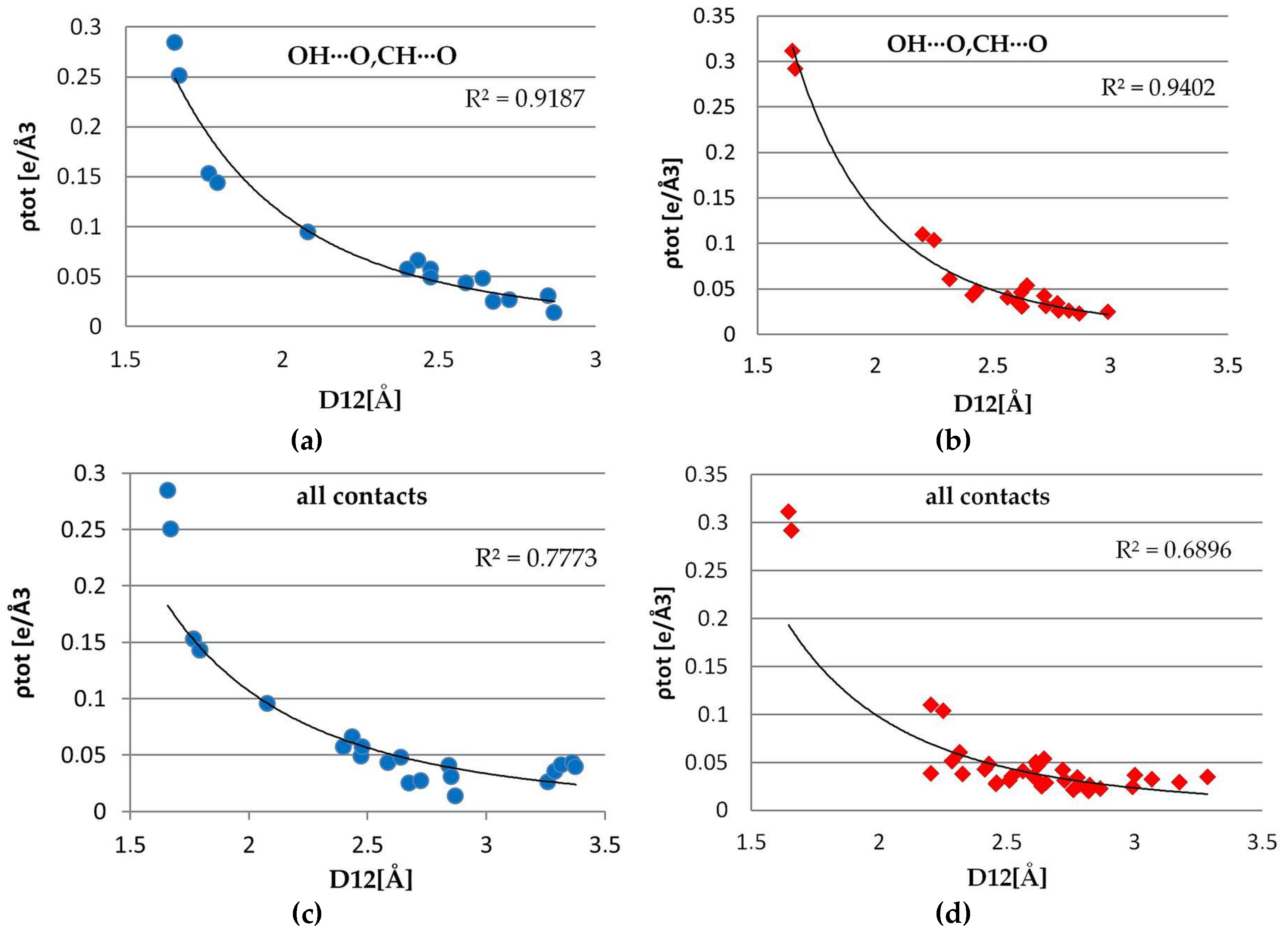
3.4. Non-Covalent Interactions
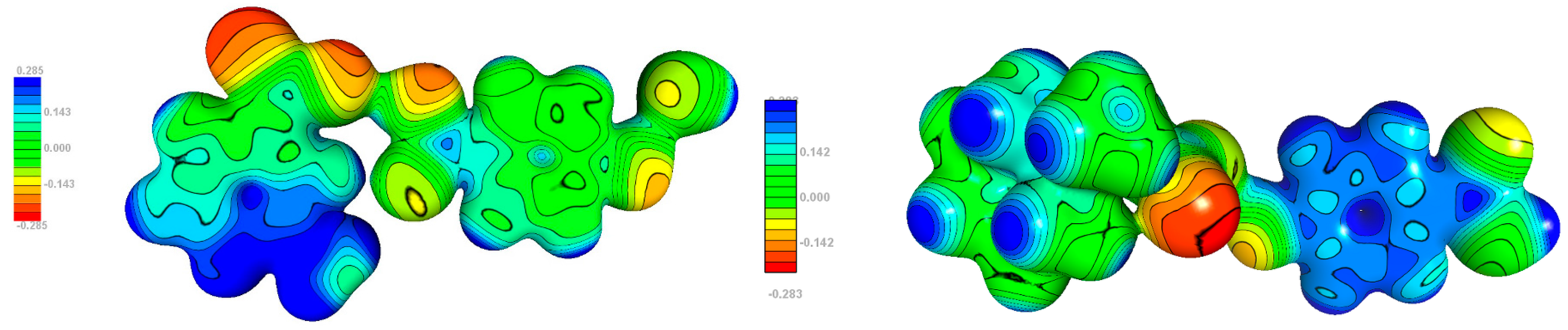
3.5. Electrostatic Potential
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Koritsanszky, T.; Coppens, P. Chemical applications of X-ray charge-density analysis. Chem. Rev. 2001, 101, 1583–1627. [Google Scholar] [CrossRef] [PubMed]
- Stalke, D. Electron Density and Chemical Bonding I: Experimental Charge Density Studies. Struct. Bond. 2012, 146. [Google Scholar] [CrossRef]
- Gatti, C.; Macchi, P. Modern Charge-Density Analysis; Springer: New York, NY, USA, 2012; ISBN 978-9048138357. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK; New York, NY, USA, 1999; ISBN 0-19-850252-4. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK; New York, NY, USA, 1990; ISBN 0198558651. [Google Scholar]
- Dega-Szafran, Z.; Dutkiewicz, G.; Kosturkiewicz, Z.; Szafran, M. Crystal structure and spectroscopic properties of the complex of trigonelline hydrate with p-hydroxybenzoic acid. J. Mol. Struct. 2011, 985, 219–226. [Google Scholar] [CrossRef]
- Dega-Szafran, Z.; Dutkiewicz, G.; Kosturkiewicz, Z.; Szafran, M. Structure of complex of N-methylpiperidine betaine with p-hydroxybenzoic acid studied by X-ray, FT-IR and DFT methods. J. Mol. Struct. 2008, 875, 346–353. [Google Scholar] [CrossRef]
- Allred, K.F.; Yackley, K.M.; Vanamala, J.; Allred, C.D. Trigonelline is a novel phytoestrogen in coffee beans. J. Nutr. 2009, 139, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chan, L.; Zhou, S. Trigonelline: A plant alkaloid with therapeutic potential for diabetes and central nervous system disease. Curr. Med. Chem. 2012, 19, 3523–3531. [Google Scholar] [CrossRef] [PubMed]
- Tohda, C.; Kuboyama, T.; Komatsu, K. Search for natural products related to regeneration of the neuronal network. NeuroSignals 2005, 14, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Makowska, J.; Szczesny, D. Preliminary studies on trigonelline as potential anti-Alzheimer disease agent: Determination by hydrophilic interaction liquid chromatography and modeling of interactions with beta-amyloid. J. Chromatogr. B 2014, 968, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Blessing, R.H. DREADD–data reduction and error analysis for single-crystal diffractometer data. J. Appl. Crystallogr. 1989, 22, 396–397. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO; Agilent Technologies Ltd.: Yarnton, UK, 2014. [Google Scholar]
- Blessing, R.H. Data reduction and error analysis for accurate single crystal diffraction intensities. Crystallogr. Rev. 1987, 1, 3–58. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Cryst. 1987, 34, 909–992. [Google Scholar] [CrossRef]
- Guillot, B.; Viry, L.; Guillot, R.; Lecomte, C.; Jelsch, C. Refinement of proteins at subatomic resolution with MoPro. J. Appl. Crystallogr. 2001, 34, 214–223. [Google Scholar] [CrossRef]
- Jelsch, C.; Guillot, B.; Lagoutte, A.; Lecomte, C. Advances in proteins and small molecules charge density refinement methods using software MoPro. J. Appl. Crystallogr. 2005, 38, 38–54. [Google Scholar] [CrossRef]
- Clementi, E.; Roetti, C. Roothaan-Hartree-Fock atomic wavefunctions: Basis functions and their coefficients for ground and certain excited states of neutral and ionized atoms, Z ≤ 54. At. Data Nucl. Data Tables 1974, 14, 177–478. [Google Scholar] [CrossRef]
- Madsen, A.O. SHADE web server for estimation of hydrogen anisotropic displacement parameters. J. Appl. Crystallogr. 2006, 39, 757–758. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Can X-ray data distinguish bonding effects from vibrational smearing? Acta Cryst. 1976, 32, 239–244. [Google Scholar] [CrossRef]
- Meindl, K.; Henn, J. Foundations of residual-density analysis. Acta Cryst. 2008, 64, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Du, J.J.; Váradi, L.; Williams, P.A.; Groundwater, P.W.; Overgaard, J.; Platts, J.A.; Hibbs, D.E. An analysis of the experimental and theoretical charge density distributions of the piroxicam–saccharin co-crystal and its constituents. RSC Adv. 2016, 6, 81578–81590. [Google Scholar] [CrossRef]
- Munshi, P.; Guru Row, T.N. Exploring the lower limit in hydrogen bonds: Analysis of weak C-H···O and C-H···pi interactions in substituted coumarins from charge density analysis. J. Phys. Chem. A 2005, 109, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Abramov, Y.A. On the Possibility of Kinetic Energy Density Evaluation from the Experimental Electron-Density Distribution. Acta Cryst. A 1997, 53, 264–272. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical bonds without bonding electron density—Does the difference electron-density analysis suffice for a description of the chemical bond? Angew. Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Baryshnikov, G.; Minaev, B.; Minaeva, V.A.; Nenajdenko, V. Single crystal architecture and absorption spectra of octathio[8]circulene and sym-tetraselenatetrathio[8]circulene: QTAIM and TD-DFT approach. J. Mol. Model. 2013, 19, 4511–4519. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikov, G.V.; Minaev, B.F.; Minaeva, V.A.; Baryshnikova, A.T.; Pittelkow, M. DFT and QTAIM study of the tetra-tert-butyltetraoxa[8]circulene regioisomers structure. J. Mol. Struct. 2012, 1026, 127–132. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 : TRG·HBA·H2O | 2 : MPB·HBA | |
|---|---|---|
| Formula | C7H7NO2·C7H6O3·H2O | C8H15NO2·C7H6O3 |
| Formula weight | 293.27 | 295.32 |
| Crystal system | monoclinic | triclinic |
| Space group | P21/c | P-1 |
| Temperature (K) | 100(1) | 100(1) |
| a (Å) | 14.2084(2) | 6.0511(1) |
| b (Å) | 6.63080(10) | 10.5600(2) |
| c (Å) | 14.5338(2) | 11.9819 |
| α (°) | 90 | 109.277 |
| β (°) | 102.3460(10) | 95.580 |
| γ(°) | 90 | 99.312 |
| V(A−3) | 1337.61(3) | 703.80(2) |
| Z | 4 | 2 |
| dx(g cm3) | 1.456 | 1.394 |
| μ(mm−1) | 0.115 | 0.104 |
| Resolution range (Å−1) | 0.083–1.13 | 0.044–1.135 |
| Reflections no. | 130790 | 88546 |
| Unique reflections (Rint) | 16,121/0.039 | 17,111/0.031 |
| IAM Refinement | ||
| No. of parameter | 250 | 274 |
| R1 [I > 2σ(I)] | 0.0358 | 0.0378 |
| wR2(F2) [I > 2σ(I)] | 0.01049 | 0.01034 |
| S | 1.057 | 1.105 |
| Δρmax, Δρmin (e Å−3) | −0.27/0.62 | −0.32/0.66 |
| Multipolar Refinement | ||
| No. of parameter | 657 | 652 |
| R1 [I > 2σ(I)] | 0.0211 | 0.0268 |
| wR2(F2) [I > 2σ(I)] | 0.0415 | 0.0561 |
| S | 1.018 | 1.26 |
| Δρmax/Δρmin (e Å−3) | −0.27/0.30 | −0.30/0.30 |
| Atom1 | Atom2 | Symmetry | G(rCP) | V(rCP) | D12 [Å] | D1cp [Å] | D2cp [Å] | ρtot [e/Å3] | ∇2ρ [e/Å5] | Type |
|---|---|---|---|---|---|---|---|---|---|---|
| TRG·HBA | ||||||||||
| O1W | H5 | x, y, z | 124.16 | −119.75 | 1.658 | 1.118 | 0.5415 | 0.2843 | 4.72 | OH···O |
| O2 | H3 | x, y, z | 110.91 | −102.23 | 1.6714 | 1.1206 | 0.5544 | 0.2507 | 4.39 | OH···O |
| O1 | H2W | 1−x, 1−y, 1−z | 91.07 | −66.07 | 1.7664 | 1.1893 | 0.5958 | 0.153 | 4.26 | OH···O |
| O1 | H1W | 1+x, −½+y, ½+z | 86.45 | −61.74 | 1.7917 | 1.1998 | 0.6009 | 0.1438 | 4.08 | OH···O |
| O4 | H21 | x, y, z | 41.83 | −30.24 | 2.0795 | 1.3177 | 0.7708 | 0.0953 | 1.96 | CH···O |
| O2 | H61 | x, 3/2−y, ½+z | 21.11 | −15.61 | 2.434 | 1.4289 | 1.0379 | 0.066 | 0.98 | CH···O |
| O3 | H71 | x, 3/2−y, ½+z | 18.54 | −13.26 | 2.3997 | 1.4565 | 0.9597 | 0.0573 | 0.87 | CH···O |
| O4 | H73 | 1−x, −½+y, ½+z | 15.48 | −10.82 | 2.4728 | 1.4745 | 1.0282 | 0.049 | 0.74 | CH···O |
| O1 | H61 | x, 3/2−y, ½+z | 15.2 | −10.6 | 2.639 | 1.5289 | 1.1303 | 0.0482 | 0.73 | CH···O |
| MPB·HBA | ||||||||||
| O2 | H5 | x, y, z | 133.68 | −133.94 | 1.6474 | 1.111 | 0.5371 | 0.3113 | 4.9 | OH···O |
| O1 | H3 | 1−x, 1−y, 2−z | 122.85 | −121.63 | 1.6591 | 1.1057 | 0.5541 | 0.2917 | 4.55 | OH···O |
| O1 | H61 | x, y, x | 38.36 | −30.99 | 2.2026 | 1.3184 | 0.9141 | 0.1097 | 1.68 | CH···O |
| O1 | H71 | x, y, z | 35.39 | −28.57 | 2.2513 | 1.3394 | 0.9383 | 0.1036 | 1.57 | CH···O |
| O5 | H82 | −1+x, y, z | 23.15 | −15.95 | 2.3161 | 1.429 | 0.9132 | 0.0605 | 1.11 | CH···O |
| O4 | H73 | 2−x, 1−y, 2−z | 17.19 | −12.16 | 2.6461 | 1.4505 | 1.2317 | 0.0535 | 0.82 | CH···O |
| Atom1 | Atom2 | Symmetry | G(rCP) | V(rCP) | D12 [Å] | D1cp [Å] | D2cp [Å] | ρtot [e/Å3] | ∇2 [e/Å5] | Type |
|---|---|---|---|---|---|---|---|---|---|---|
| TRG·HBA | ||||||||||
| C3 | C15 | 1−x, 1/2+y, 3/2−z | 9.96 | −7.21 | 3.3639 | 1.7209 | 1.7724 | 0.0434 | 0.48 | π∙∙∙π |
| N1 | C13 | 1−x, 1/2+y, 3/2−z | 10.69 | −7.71 | 3.3188 | 1.6661 | 1.7204 | 0.0418 | 0.5 | π∙∙∙π |
| C12 | H72 | 1−x, 1/2+y, 3/2−z | 9.96 | −7.21 | 2.8404 | 1.6590 | 1.1877 | 0.0403 | 0.47 | CH∙∙∙π |
| C3 | C14 | 1−x, 1/2+y, 3/2−z | 9.52 | −6.91 | 3.3768 | 1.6953 | 1.6859 | 0.0395 | 0.45 | π∙∙∙π |
| MPB·HBA | ||||||||||
| H16 | H52 | x, y, z+1 | 13.66 | −10.16 | 2.2868 | 1.2019 | 1.0928 | 0.0513 | 0.63 | H∙∙∙H |
| C13 | H31 | 1−x, −y, 1−z | 14.06 | −10.19 | 2.6137 | 1.5459 | 1.0858 | 0.0498 | 0.66 | CH∙∙∙C |
| C15 | O3 | 1−x, 1−y, 2−z | 9.57 | −6.51 | 3.2854 | 1.6220 | 1.7682 | 0.0346 | 0.46 | C∙∙∙O |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owczarzak, A.; Kubicki, M. Experimental Electron Density Distribution in Two Cocrystals of Betaines with p-Hydroxybenzoic Acid. Crystals 2018, 8, 132. https://doi.org/10.3390/cryst8030132
Owczarzak A, Kubicki M. Experimental Electron Density Distribution in Two Cocrystals of Betaines with p-Hydroxybenzoic Acid. Crystals. 2018; 8(3):132. https://doi.org/10.3390/cryst8030132
Chicago/Turabian StyleOwczarzak, Agata, and Maciej Kubicki. 2018. "Experimental Electron Density Distribution in Two Cocrystals of Betaines with p-Hydroxybenzoic Acid" Crystals 8, no. 3: 132. https://doi.org/10.3390/cryst8030132
APA StyleOwczarzak, A., & Kubicki, M. (2018). Experimental Electron Density Distribution in Two Cocrystals of Betaines with p-Hydroxybenzoic Acid. Crystals, 8(3), 132. https://doi.org/10.3390/cryst8030132