(BEDT-TTF)2Cu2(CN)3 Spin Liquid: Beyond the Average Structure

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
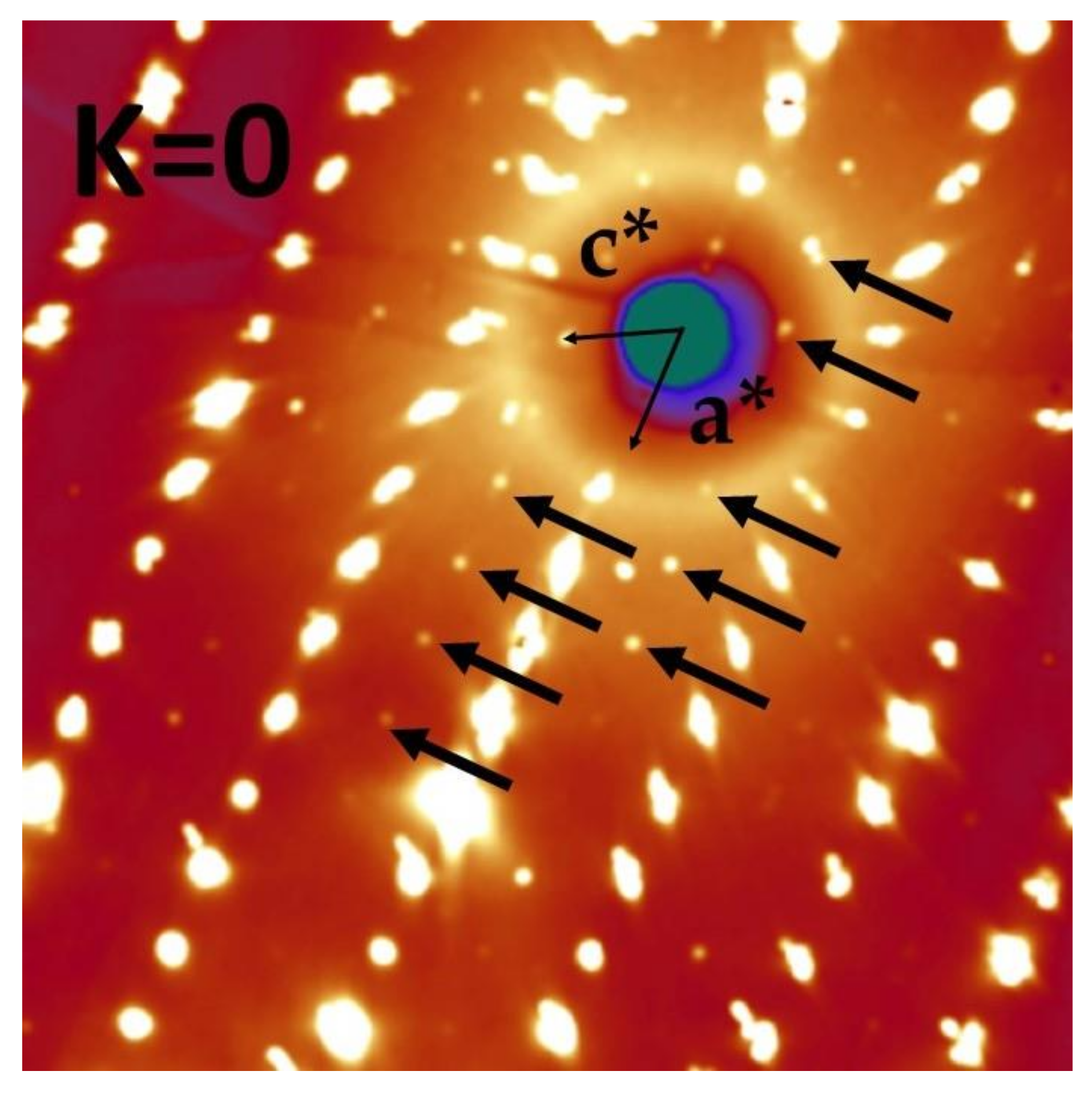
3.1. Evidence for a P21/c Symmetry Breaking
3.2. Structure at 300 K
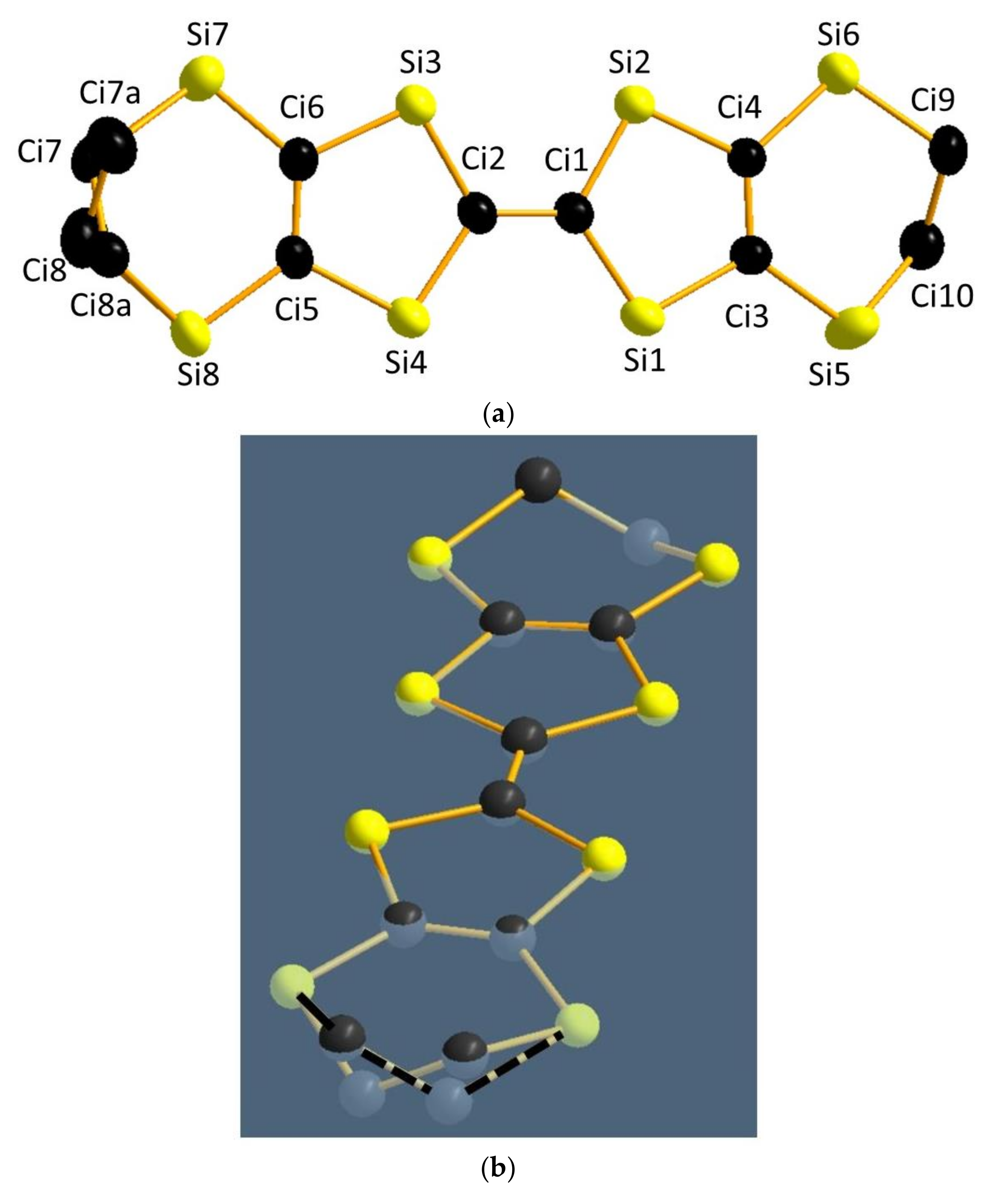
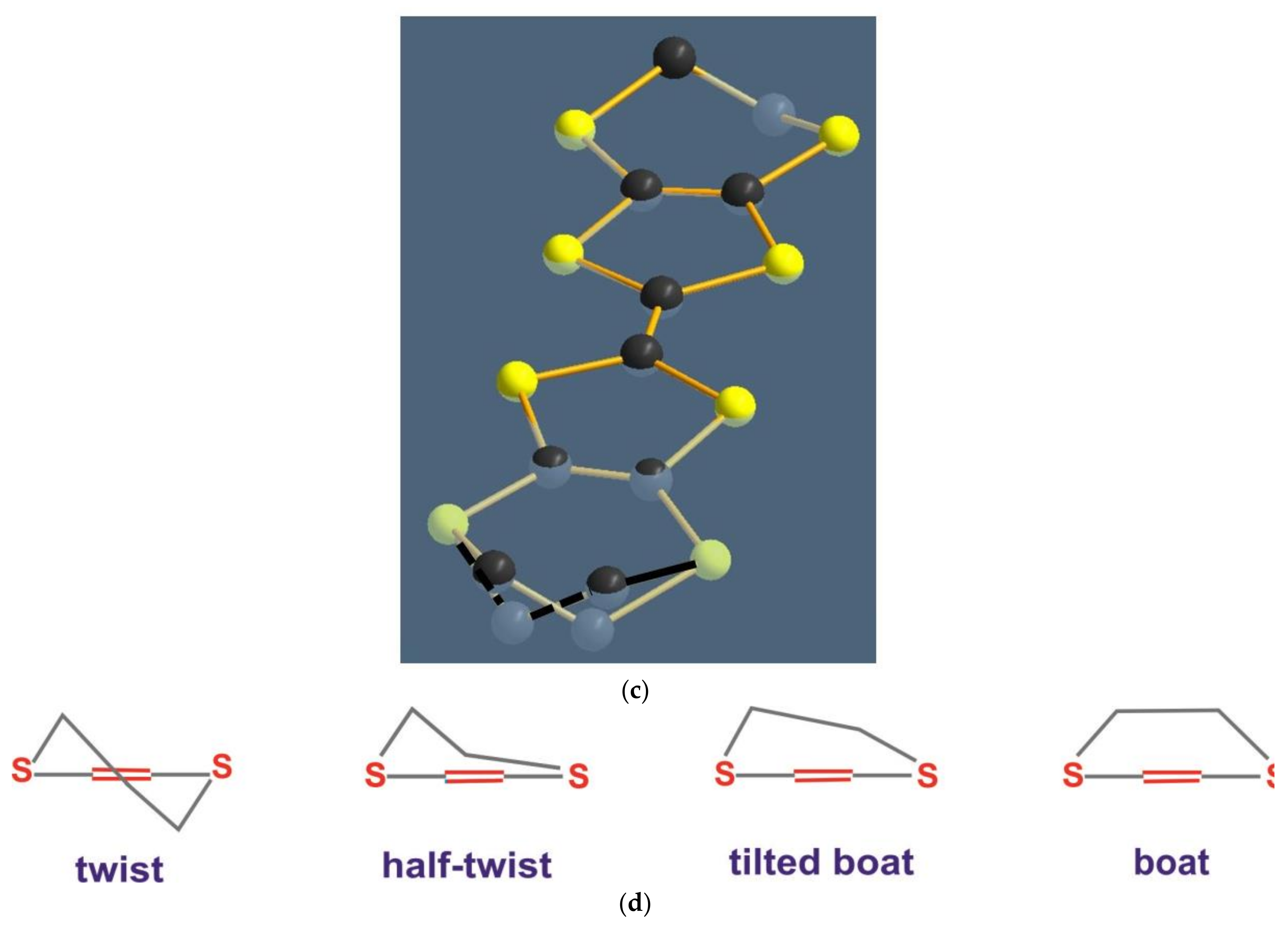
3.2.1. Modeling of the Disorder
3.2.2. Result of the Refinement
3.3. Evolution of the Structure as a Function of Temperature
4. Discussion
4.1. Inter-Dimer Charge Disproportionation
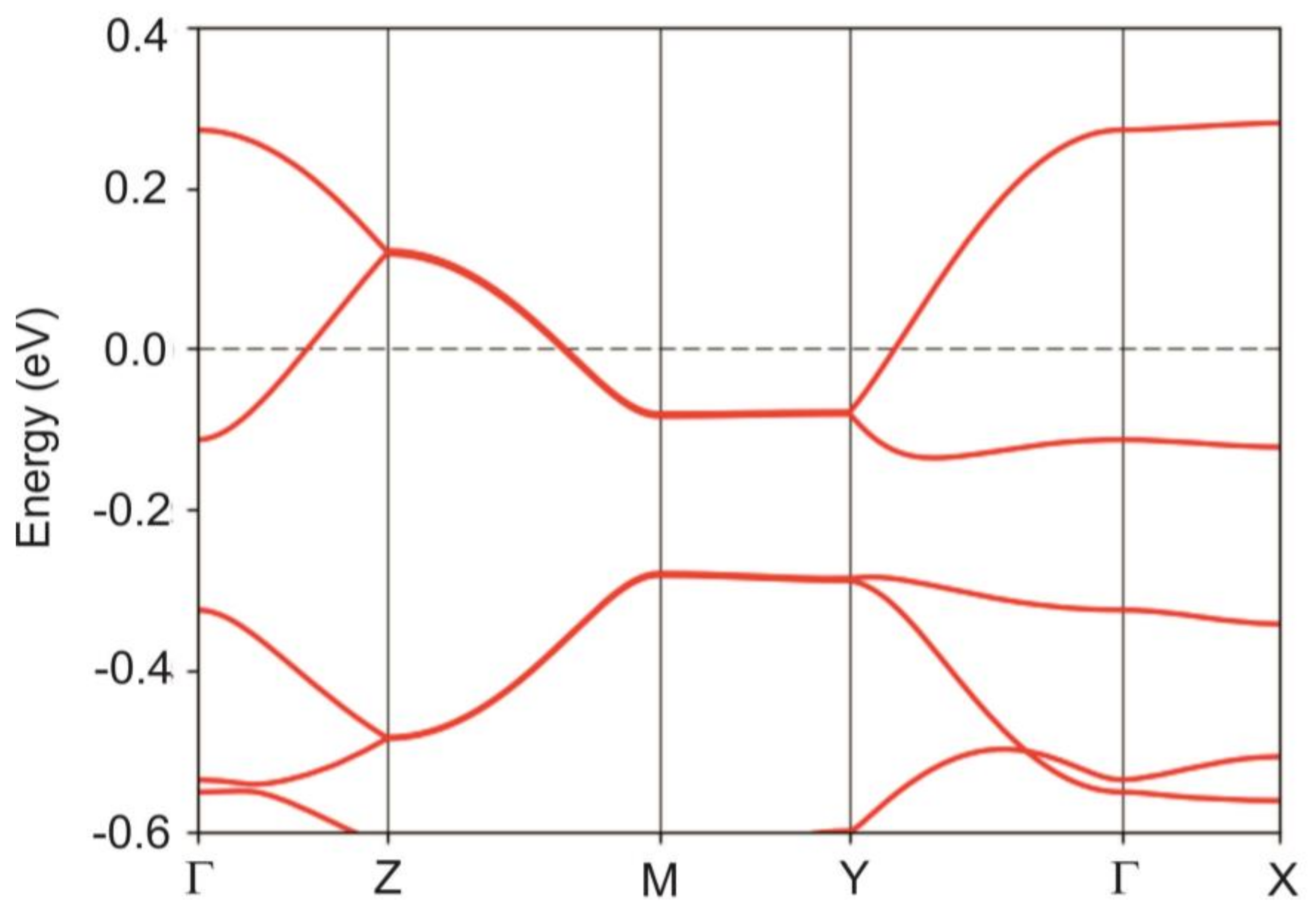
4.2. DFT Electronic Structure Calculation
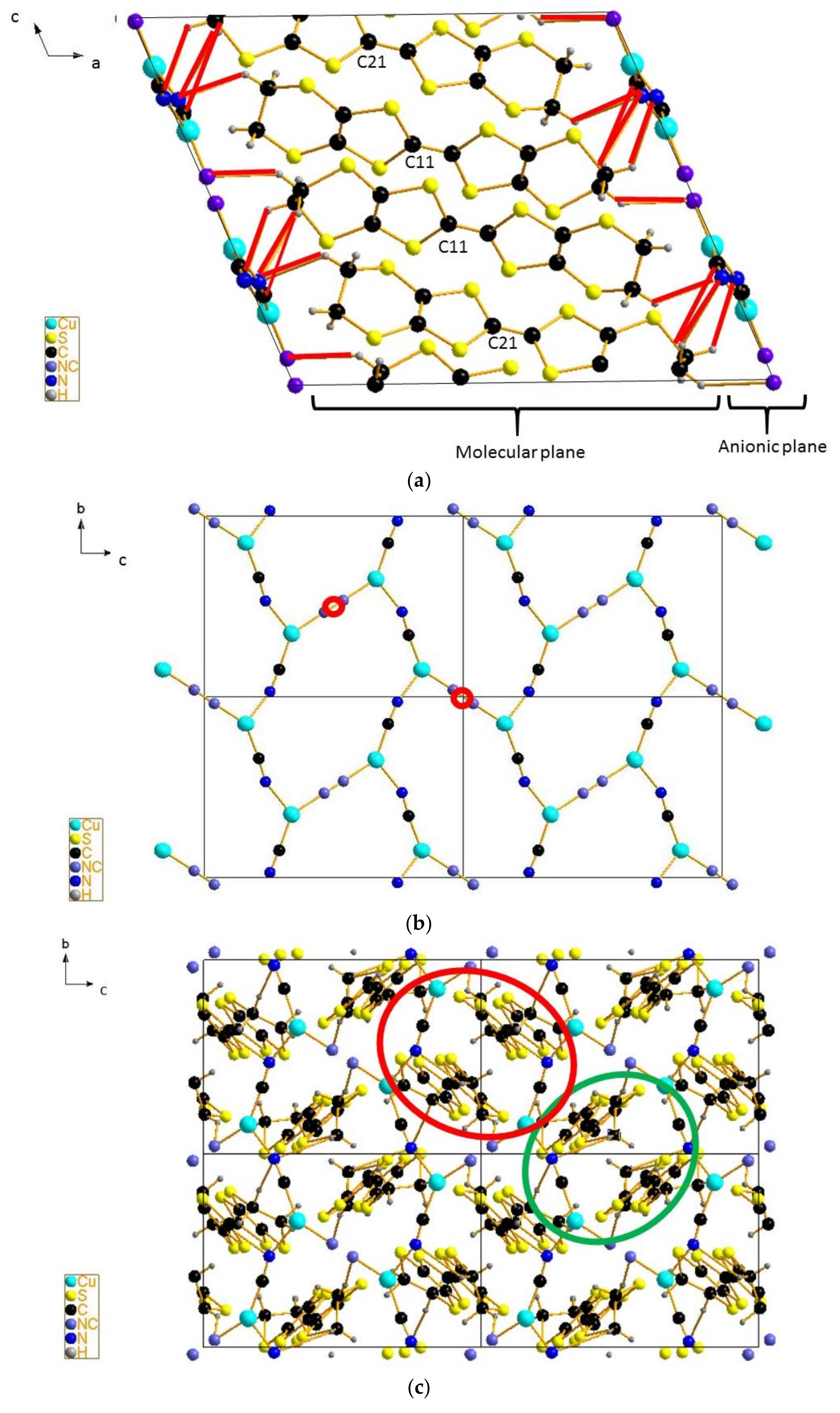
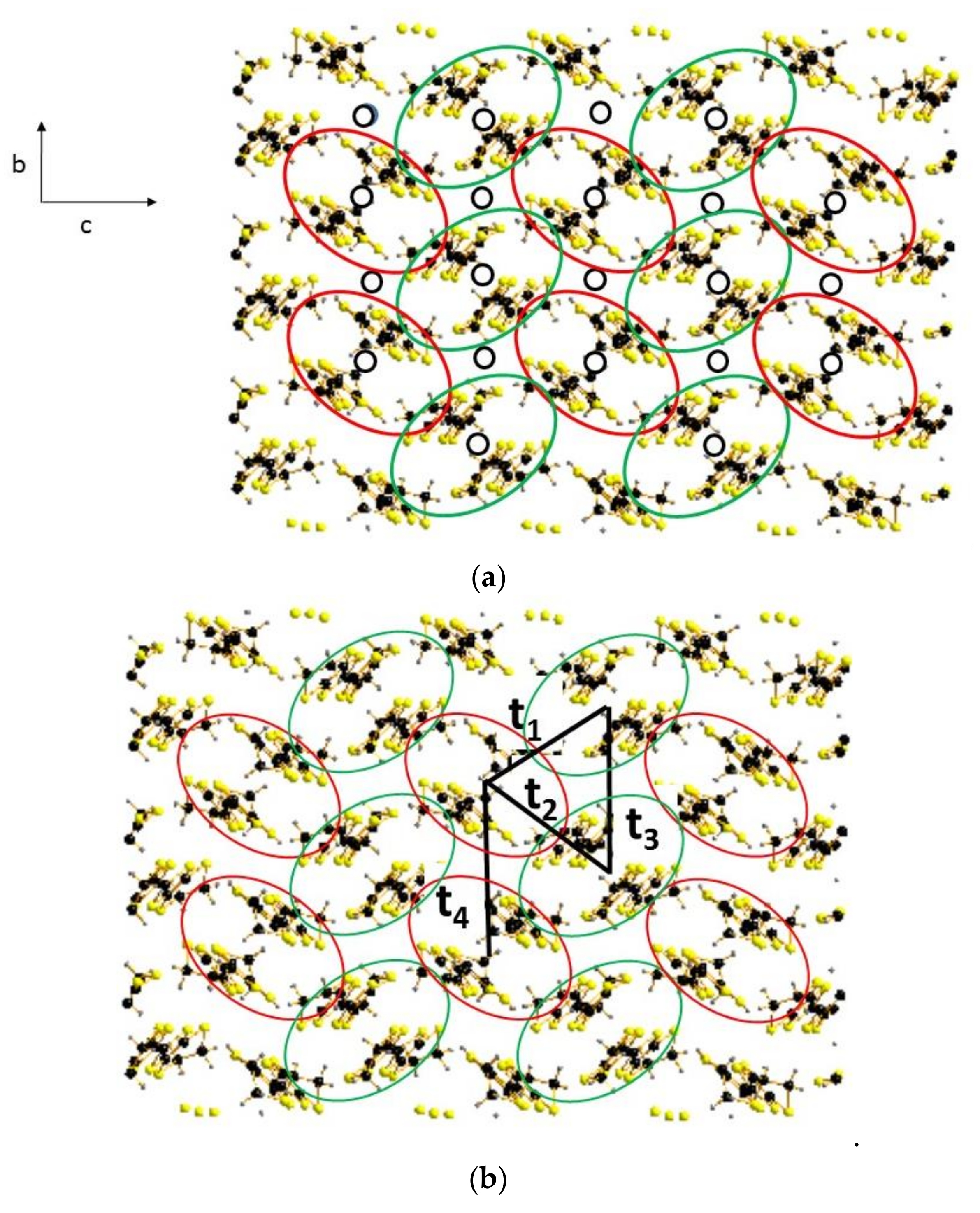
4.3. Inter-Dimer Charge Pattern
4.4. Possible Intra-Dimer Charge Disproportionation
4.5. Bringing Back Together the Puzzling Physical Behaviors
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Powell, B.J.; McKenzie, R.H. Quantum frustration in organic Mott insulators: From spin liquids to unconventional superconductors. Rep. Prog. Phys. 2011, 74, 056501. [Google Scholar] [CrossRef]
- Balents, L. Spin liquids in frustrated magnets. Nature 2010, 464, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.W. Resonating valence bonds: A new kind of insulator? Mater. Res. Bull. 1973, 8, 153–160. [Google Scholar] [CrossRef]
- Coldea, R.; Tennant, D.A.; Tsvelik, A.M.; Tylczynski, Z. Experimental Realization of a 2D Fractional Quantum Spin Liquid. Phys. Rev. Lett. 2001, 86, 1335. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liao, H.; Zhang, Z.; Li, S.; Jin, F.; Ling, L.; Zhang, L.; Zou, Y.; Pi, L.; Yang, Z.; et al. Gapless quantum spin liquid ground state in the two-dimensional spin-1/2 triangular antiferromagnet YbMgGaO4. Sci. Rep. 2015, 5, 16419. [Google Scholar] [CrossRef] [PubMed]
- Khuntia, P.; Kumar, R.; Mahajan, A.V.; Baenitz, M.; Furukawa, Y. Spin liquid state in the disordered triangular lattice Sc2Ga2CuO7 revealed by NMR. Phys. Rev. B 2016, 93, 140408(R). [Google Scholar] [CrossRef]
- Klanjšek, M.; Zorko, A.; Žitko, R.; Mravlje, J.; Jagličić, Z.; Biswas, P.; Prelovšek, P.; Mihailovic, D.; Arčon, D. A high-temperature quantum spin liquid with polaron spins. Nat. Phys. 2017, 13, 1130–1134. [Google Scholar] [CrossRef]
- Kanoda, K.; Kato, R. Mott Physics in Organic Conductors with Triangular Lattices. Annu. Rev. Condens. Matter Phys. 2011, 2, 167–188. [Google Scholar] [CrossRef]
- Zhou, Y.; Kanoda, K.; Ng, T.-K. Quantum spin liquid states. Rev. Mod. Phys. 2017, 89, 025003. [Google Scholar] [CrossRef]
- Kandpal, H.C.; Opahle, I.; Zhang, Y.-Z.; Jeschke, H.O.; Valenti, R. Revision of Model Parameters for κ Type Charge Transfer Salts: An Ab Initio Study. Phys. Rev. Lett. 2009, 103, 067004. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, H.O.; de Souza, M.; Valenti, R.; Manna, R.S.; Lang, M.; Schlueter, J.A. Temperature dependence of structural and electronic properties of the spin-liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2012, 85, 035125. [Google Scholar] [CrossRef]
- Koretsune, T.; Hotta, C. Evaluating model parameters of the κ-and β′-type Mott insulating organic solids. Phys. Rev. B 2014, 89, 045102. [Google Scholar] [CrossRef]
- Miyagawa, Y.S.K.; Kanoda, K.; Maesato, M.; Saito, G. Spin Liquid State in an Organic Mott Insulator with a Triangular Lattice. Phys. Rev. Lett. 2003, 91, 107001. [Google Scholar]
- Zheng, W.; Singh, R.R.P.; McKenzie, R.H.; Coldea, R. Temperature dependence of the magnetic susceptibility for triangular-lattice antiferromagnets with spatially anisotropic exchange constants. Phys. Rev. B 2005, 71, 134422. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Nakazawa, Y.; Oguni, M.; Oshima, Y.; Jojiri, H.; Miyagawa, K.; Kanoda, K. Thermodynamic properties of a spin-1/2 spin-liquid state in a κ-type organic salt. Nat. Phys. 2008, 4, 459–462. [Google Scholar] [CrossRef]
- Pustogow, A.; Bories, M.; Löhle, A.; Rösslhuber, R.; Zhukova, E.; Gorshunov, B.; Tomić, S.; Schlueter, J.A.; Hübner, R.; Hiramatsu, T.; et al. Quantum Spin Liquids Unveil the Genuine Mott State. arXiv, 2017; arXiv:1710.07241. [Google Scholar]
- Seo, H.; Merino, J.; Yoshioka, H.; Ogata, M. Theoretical Aspects of Charge Ordering in Molecular Conductors. J. Phys. Soc. Jpn. 2006, 75, 051009. [Google Scholar] [CrossRef]
- Lunkenheimer, P.; Müller, J.; Krohns, S.; Schrettle, F.; Loidl, A.; Hartmann, B.; Rommel, R.; de Souza, M.; Hotta, C.; Schlueter, J.A.; et al. Multiferroicity in an organic charge-transfer salt: Electric-dipole-driven magnetism. Nat. Mater. 2012, 11, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Hotta, C. Theories on Frustrated Electrons in Two-Dimensional Organic Solids. Crystals 2012, 2, 1155–1200. [Google Scholar] [CrossRef]
- Naka, M.; Ishihara, S. Electronic Ferroelectricity in a Dimer Mott Insulator. J. Phys. Soc. Jpn. 2010, 79, 063707. [Google Scholar] [CrossRef]
- Li, H.; Clay, R.T.; Mazumdar, S. The paired-electron crystal in the two-dimensional frustrated quarter-filled band. J. Phys. Condens. Matter 2010, 22, 272201. [Google Scholar] [CrossRef] [PubMed]
- Gomi, H.; Imai, T.; Takahashi, A.; Aihara, M. Purely electronic terahertz polarization in dimer Mott insulators. Phys. Rev. B 2010, 82, 035101. [Google Scholar] [CrossRef]
- Gomi, H.; Ikenaga, M.; Hiragi, Y.; Segawa, D.; Takahashi, A.; Inagaki, T.J.; Aihara, M. Ferroelectric states induced by dimer lattice disorder in dimer Mott insulators. Phys. Rev. B 2013, 87, 195126. [Google Scholar] [CrossRef]
- Gomi, H.; Inagaki, T.J.; Takahashi, A. A ferroelectric charge order enhanced by magnetic frustration in dimer Mott insulators. Phys. Rev. B 2016, 93, 035105. [Google Scholar] [CrossRef]
- Manna, R.S.; de Souza, M.; Brűhl, A.; Schlueter, J.A.; Lang, M. Lattice Effects and Entropy Release at the Low-Temperature Phase Transition in the Spin-Liquid Candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. Lett. 2010, 104, 016403. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.; de Lafontaine, M.; Miyagawa, K.; Kanoda, K.; Shimizu, Y. Ultrasonic investigation of the transition at 6 K in the spin-liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2014, 89, 045138. [Google Scholar] [CrossRef]
- Ilakovac, V.; Carniato, S.; Foury-Leylekian, P.; Tomić, S.; Pouget, J.-P.; Lazić, P.; Joly, Y.; Miyagawa, K.; Kanoda, K.; Nicolaou, A. Resonant inelastic x-ray scattering probes the electron-phonon coupling in the spin liquid κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2017, 96, 184303. [Google Scholar] [CrossRef]
- Kurosaki, Y.; Shimizu, Y.; Miyagawa, K.; Kanoda, K.; Saito, G. Mott Transition from a Spin Liquid to a Fermi Liquid in the Spin-Frustrated Organic Conductor κ-(ET)2Cu2(CN)3. Phys. Rev. Lett. 2005, 95, 177001. [Google Scholar] [CrossRef] [PubMed]
- Geiser, U.; Wang, H.H.; Carlson, K.D.; Williams, J.M.; Charlier, H.A.; Heindl, J.E.; Yaconi, G.A.; Love, B.J.; Lathrop, M.W.; Schirber, J.E.; et al. Superconductivity at 2.8 K and 1.5 kbar in K-(BEDT-TTF)2Cu2(CN)3: The first organic superconductor containing a polymeric copper cyanide anion. Inorg. Chem. 1991, 30, 2586–2588. [Google Scholar] [CrossRef]
- Bu, X.; Frost-Jensen, A.; Allendoerfer, R.; Coppens, P.; Lederle, B.; Naughton, M.J. Structure and properties of a new κ-phase organic metal: (BEDT-TTF)2Cu2(CN)3. Solid State Commun. 1991, 79, 1053–1057. [Google Scholar] [CrossRef]
- Bu, X.; Coppens, P. Zeitschrift für Kristallographie. New Cryst. Struct. 1997, 212, 103–104. [Google Scholar]
- Yamochi, H.; Nakamura, T.; Komatsu, T.; Matsukawa, N.; Inoue, T.; Saito, G. Crystal and electronic structures of the organic superconductors, ϰ-(BEDT-TTF)2Cu (CN)[N(CN)2] and ϰ′-(BEDT-TTF)2Cu2(CN)3. Solid State Commun. 1992, 82, 101–105. [Google Scholar] [CrossRef]
- Papavassiliou, G.C.; Lagouvardos, D.J.; Terzis, A.; Hountas, A.; Hilti, B.; Zambounis, J.S.; Mayer, C.W.; Pfeiffer, J.; Hofherr, W.; Delhaes, P.; et al. Physical properties in the normal state of the molecular superconductor κ-(BEDT)2Cu2(CN)3. Synth. Met. 1993, 61, 267–273. [Google Scholar] [CrossRef]
- Hiramatsu, T.; Yoshida, Y.; Saito, G.; Otsuka, A.; Yamochi, H.; Maesato, M.; Shimizu, Y.; Ito, H.; Kishida, H. Quantum spin liquid: Design of a quantum spin liquid next to a superconducting state based on a dimer-type ET Mott insulator. J. Mater. Chem. C 2015, 3, 1378–1388. [Google Scholar] [CrossRef]
- Sedlmeier, K.; Elsässer, S.; Neubauer, D.; Beyer, R.; Wu, D.; Ivek, T.; Tomić, S.; Schlueter, J.A.; Dressel, M. Absence of charge order in the dimerized κ-phase BEDT-TTF salts. Phys. Rev. B 2012, 86, 245103. [Google Scholar] [CrossRef]
- Abdel-Jawad, M.; Terasaki, I.; Sasaki, T.; Yoneyama, N.; Kobayashi, N.; Uesu, Y.; Hotta, C. Anomalous dielectric response in the dimer Mott insulator κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2010, 82, 125119. [Google Scholar] [CrossRef]
- Pinterić, M.; Čulo, M.; Milat, O.; Basletić, M.; Korin-Hamzić, B.; Tafra, E.; Hamzić, A.; Ivek, T.; Peterseim, T.; Miyagawa, K.; et al. Anisotropic charge dynamics in the quantum spin-liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2014, 90, 195139. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlis PRO; Rigaku Oxford Diffraction: Yarnton, UK, 2015. [Google Scholar]
- Petricek, V.; Dusek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Kristallogr. 2014, 229, 345–352. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter. 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- SIESTA Code. Available online: http://departments.icmab.es/leem/siesta/ (accessed on 25 March 2018).
- Artacho, E.; Anglada, E.; Diéguez, O.; Gale, J.D.; García, A.; Junquera, J.; Martin, R.M.; Ordejón, P.; Pruneda, J.M.; Sánchez-Portal, D.; et al. The SIESTA method; developments and applicability. J. Phys. Condens. Matter 2008, 20, 064208. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Kleinman, L.; Bylander, D.M. Efficacious Form for Model Pseudopotentials. Phys. Rev. Lett. 1982, 48, 1425. [Google Scholar] [CrossRef]
- Artacho, E.; Sánchez-Portal, D.; Ordejón, P.; García, A.; Soler, J.M. Linear-scaling ab-initio calculations for large and complex systems. Phys. Status Solidi (b) 1999, 215, 809–817. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Čulo, M.; Tafra, E.; Basletić, M.; Tomić, S.; Hamzić, A.; Korin-Hamzić, B.; Dressel, M.; Schlueter, J.A. Two-dimensional variable range hopping in the spin-liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. B 2015, 460, 208–210. [Google Scholar] [CrossRef]
- The Crystallographic Data. Available online: https://www.ccdc.cam.ac.uk (accessed on 25 March 2018).
- Guionneau, P.; Kepert, C.J.; Bravic, G.; Chasseau, D.; Truter, M.R.; Kurmoo, M.; Day, P. Determining the charge distribution in BEDT-TTF salts. Synth. Met. 1997, 86, 1973–1974. [Google Scholar] [CrossRef]
- Novoa, J.J.; Whangbo, M.-H.; Williams, J.M. Ab Initio Computational Study of the C-H…Donor and C-H…Anion Contact Interactions in Organic Donor Salts. Mol. Cryst. Liq. Cryst. 1990, 181, 25–42. [Google Scholar] [CrossRef]
- Whangbo, M.-H.; Jung, D.; Ren, J.; Evain, M.; Novoa, J.J.; Mota, F.; Alvarez, S.; Williams, J.M.; Beno, M.A.; Kini, A.M.; et al. The Physics and Chemistry of Organic Superconductors; Saito, G., Kagoshima, S., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 262–266. [Google Scholar]
- Aburto, A.; Orgaz, E. Ab initio electronic structure of the eclipsed and staggered conformations of the κ-(BEDT-TTF)2Cu[N(CN)2]Br organic superconductor. Phys. Rev. B 2008, 78, 113104. [Google Scholar] [CrossRef]
- Pouget, J.P.; Alemany, P.; Canadell, E. Donor-Anion Interactions in Quarter-Filled Low-Dimensional Organic Conductors. In Preparation.
- Pouget, J.-P.; Foury-Leylekian, P.; Alemany, P.; Canadell, E. Charge ordering in low dimensional organic conductors: Structural aspects. Phys. Status Solidi (b) 2012, 249, 937–942. [Google Scholar] [CrossRef]
- Alemany, P.; Pouget, J.-P.; Canadell, E. Essential role of anions in the charge ordering transition of α-(BEDT-TTF)2I3. Phys. Rev. B 2012, 85, 195118. [Google Scholar] [CrossRef]
- Dressel, M.; Lazić, P.; Pustogow, A.; Zhukova, E.; Gorshunov, B.; Schlueter, J.A.; Milat, O.; Gumhalter, B.; Tomić, S. Lattice vibrations of the charge-transfer salt κ-(BEDT-TTF)2Cu2(CN)3: Comprehensive explanation of the electrodynamic response in a spin-liquid compound. Phys. Rev. B 2016, 93, 081201(R). [Google Scholar] [CrossRef]
- Alemany, P.; Pouget, J.-P.; Canadell, E. Structural and electronic control of the metal to insulator transition and local orderings in the θ-(BEDT-TTF)2X organic conductors. J. Phys. Condens. Matter 2015, 27, 465702. [Google Scholar] [CrossRef] [PubMed]
- Tomić, S.; Dressel, M. Ferroelectricity in molecular solids: A review of electrodynamic properties. Rep. Prog. Phys. 2015, 78, 096501. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Itoh, H.; Naka, M.; Saito, S.; Hosako, I.; Yoneyama, N.; Ishihara, S.; Sasaki, T.; Iwai, S. Collective Excitation of an Electric Dipole on a Molecular Dimer in an Organic Dimer-Mott Insulator. Phys. Rev. Lett. 2013, 110, 106401. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, H.; Kishine, J.-I.; Ogata, M. Energy Landscape of Charge Excitations in the Boundary Region between Dimer–Mott and Charge Ordered States in Molecular Solids. J. Phys. Soc. Jpn. 2017, 86, 123706. [Google Scholar] [CrossRef]
- Foury-Leylekian, P.; Ilakovac, V.; Fertey, P. Experimental Report n°: 20160753; Cristal Beamline Experiment; Synchrotron Soleil: Saint-Aubin, France, 2017. [Google Scholar]
- The 2c superstructure which develops below 200K in the Br salt should suppress the inter-donor inversion symmetry; See Nogami, Y.; Pouget, J.P.; Ito, H.; Ishiguro, T.; Saito, G. Superlattice structural transition in the organic superconductor χ-(BEDT-TTF)2Cu [N(CN)2]Br. Solid State Commun. 1994, 89, 113–116. [Google Scholar] [CrossRef]
- The site symmetry breaking leads to a broad distribution of electronic density detected by 13C NMR; De Soto, S.M.; Slichter, C.P.; Kini, A.M.; Wang, H.H.; Geiser, U.; Williams, J.M. 13C NMR studies of the normal and superconducting states of the organic superconductor κ-(ET)2Cu[N(CN)2]Br. Phys. Rev. B 1995, 52, 10364–10368. [Google Scholar] [CrossRef]
- Wzietek, P.; Mayaffre, H.; Jérome, D.; Brazovskii, S. NMR in the 2D Organic Superconductors. J. Phys. I 1996, 6, 2011–2041. [Google Scholar] [CrossRef]
- Shimizu, A.; Hiramatsu, T.; Maesato, M.; Otsuka, A.; Yamochi, H.; Ono, A.; Itoh, M.; Yoshida, M.; Takigawa, M.; Yoshida, Y.; et al. Pressure-Tuned Exchange Coupling of a Quantum Spin Liquid in the Molecular Triangular Lattice κ-(ET)2Ag2(CN)3. Phys. Rev. Lett. 2016, 117, 107203. [Google Scholar] [CrossRef] [PubMed]
- Pinterić, M.; Lazić, P.; Pustogow, A.; Ivek, T.; Kuveždić, M.; Milat, O.; Gumhalter, B.; Basletić, M.; Čulo, M.; Korin-Hamzić, B.; et al. Anion effects on electronic structure and electrodynamic properties of the Mott insulator κ-(BEDT-TTF)2Ag2(CN)3. Phys. Rev. B 2016, 94, 161105(R). [Google Scholar] [CrossRef]
- Nakamura, Y.; Hiramatsu, T.; Yoshida, Y.; Saito, G.; Kishida, H. Optical Properties of a Quantum Spin Liquid Candidate Material, κ-(BEDT-TTF)2Ag2(CN)3. J. Phys. Soc. Jpn. 2017, 86, 014710. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature | 300 K |
|---|---|
| d1(Å) | 3.866(20) |
| d2(Å) | 3.872(20) |
| C1-C2 Dimer 1 (Å) | 1.369(20) |
| C1-C2 Dimer 2 (Å) | 1.372(20) |
| 2δ (structural correlation) [52] | ±0.06 |
| 2δ (DFT) | ±0.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foury-Leylekian, P.; Ilakovac, V.; Balédent, V.; Fertey, P.; Arakcheeva, A.; Milat, O.; Petermann, D.; Guillier, G.; Miyagawa, K.; Kanoda, K.; et al. (BEDT-TTF)2Cu2(CN)3 Spin Liquid: Beyond the Average Structure. Crystals 2018, 8, 158. https://doi.org/10.3390/cryst8040158
Foury-Leylekian P, Ilakovac V, Balédent V, Fertey P, Arakcheeva A, Milat O, Petermann D, Guillier G, Miyagawa K, Kanoda K, et al. (BEDT-TTF)2Cu2(CN)3 Spin Liquid: Beyond the Average Structure. Crystals. 2018; 8(4):158. https://doi.org/10.3390/cryst8040158
Chicago/Turabian StyleFoury-Leylekian, Pascale, Vita Ilakovac, Victor Balédent, Pierre Fertey, Alla Arakcheeva, Ognjen Milat, Denis Petermann, Gilles Guillier, Kazuya Miyagawa, Kazushi Kanoda, and et al. 2018. "(BEDT-TTF)2Cu2(CN)3 Spin Liquid: Beyond the Average Structure" Crystals 8, no. 4: 158. https://doi.org/10.3390/cryst8040158
APA StyleFoury-Leylekian, P., Ilakovac, V., Balédent, V., Fertey, P., Arakcheeva, A., Milat, O., Petermann, D., Guillier, G., Miyagawa, K., Kanoda, K., Alemany, P., Canadell, E., Tomic, S., & Pouget, J. -P. (2018). (BEDT-TTF)2Cu2(CN)3 Spin Liquid: Beyond the Average Structure. Crystals, 8(4), 158. https://doi.org/10.3390/cryst8040158