1. Introduction
Crystallization is the most efficient and economical way of obtaining chemically pure compounds. That is why it is widely used in the pharmaceutical, fertilizer and sugar industries. Single crystal X-ray diffraction, being the most universal, powerful and accurate tool for biological macromolecule structure analysis and protein–substrate interactions, requires relatively large and well-diffracting crystals. However, the major stumbling stone in X-ray diffraction analysis of protein crystals is the lack of a recipe (or definite indications) for growing crystals of newly expressed proteins. The issue is finding conditions that make the homogeneously scattered protein molecules in a solution form stable crystal nuclei; once nucleated, the crystals continue growing spontaneously. Evidently, nucleation is the crucial step that determines the difference between success and failure in protein crystallization trials. Regardless of the numerous auxiliary crystallization tools employed, such as automation and miniaturization of crystallization trials by means of robots, Dynamic Light Scattering, crystallization screening kits, etc., it is researchers’ creativeness and acumen that remain indispensable. Even with state-of-the-art tools, it is exceptionally challenging to probe the nucleation processes in real time. Only most recently, this problem has been addressed successfully by Van Driessche et al. [
1]. Nevertheless, some of the most intimate moments of molecule-by-molecule assembly to form crystal nuclei are still elusive and require further elucidation.
Following the fundamental notion of a kink position (
Halbkristalllage in German) introduced by Kossel [
2] and Stranski [
3], the Bulgarian scientists Stranski and Kaischew [
4,
5,
6] were the first to apply a molecular kinetic (and energetic) approach to crystal nucleation. They proposed the so-called mean work of separation (MWS) method. To calculate the MWS value, all of the different kinds of bonds (between the first, second and third neighboring crystal-building blocks) are counted separately, and each number is multiplied by the corresponding bonding energy. Then, the products are summed up, and the result is divided by the total number of blocks in the corresponding crystal element (crystal face, cluster edge). It has been argued that for vapor phase crystallization MWS value is equal to the chemical potential, taken with a negative sign, plus a substance and temperature dependent constant [
7]. To calculate the work required (energy barrier) for nucleus formation, crystal bond energies are used as well. They are calculated by summing up the number of all intra-crystal bonds, the breaking of which is needed to totally disintegrate a crystal into all its constituting molecules. The same approach is used in the present study. Applying the MWS method to the so-called Kossel-crystal (a crystal build by small cubes held together by equal forces in a cubic primitive crystal lattice) allows the equilibrium crystal shapes to be determined.
A big advantage of the MWS method is its simplicity. It uses the relative bond energies between separate crystal building units instead of the absolute bond energy values, which frequently are unknown. However, the MWS method has two major drawbacks: it is applicable only to the Kossel-crystal model (existing extremely rarely in nature) and considers solely enthalpic effects. Perhaps it is for these very reasons that the MWS method is used rarely nowadays, even though it enables semi-quantitative studies of crystal nucleation and growth.
To find the critical nucleus size, Garcia-Ruiz established the balance between the cohesive energy (ΔG
v), which maintains the integrity of a crystalline cluster, and the sum of destructive energies (ΔG
s), which tend to tear up the crystal, i.e., −ΔG
v + ΔG
s = 0 [
8]. Using a cubic primitive crystal lattice formed by spheres, the author equilibrated the number of bonds shared by the crystal building units with the number of dangling bonds at the crystal surface, pointing toward the solution. This corresponds to the classical nucleation theory where ΔG
v is proportional to the crystal volume while ΔG
s is proportional to its surface, and the critical nucleus size is determined from the compensation of the large surface energy, which is inherent for the undercritical molecule clusters, by the faster volume energy increase resulting from the rising crystal size. Garcia-Ruiz’s intuitive approach accounts for the water molecules acting on the apexes and edges that exist on the polyhedral crystal nuclei but are absent on the droplets. His model shows that at the crystal vertices, water molecules pull protein molecules towards the solution from three (perpendicular) directions, at the crystal edges from two directions, and at the crystal face from one direction only.
Recently, Garcia-Ruiz’s brilliant idea was been further elaborated [
9]. When the system is undersaturated, i.e., crystallization is impossible, the protein ‘affinity’ to water molecules prevails over the crystallization propensity. Therefore, to evoke crystallization, it is necessary to impose supersaturation and the higher the latter, the more thermodynamically stable the crystal is, with respect to the solution. Thus, it is feasible to assume that the imposed supersaturation decreases the protein-to-water affinity, i.e., supersaturation diminishes the destructive energy (
ψd) per bond. This means that the tendency to tear up the crystal depends on the degree of supersaturation in contrast to the cohesive energy per bond in the crystal lattice (
ψb) which is supersaturation independent. This means that any supersaturation increase will lead to an increase in
ψb/
ψd ratio. On this basis, the critical nucleus size dependence on supersaturation has been determined (from the balance between the sum of all intra-crystal bonds and the sum of surface destructive energies) for the Kossel-crystal model [
9].
The objective of the present work is to shed additional light on the thermodynamic and molecular aspects of the homogeneous nucleation of non-Kossel crystals, such as nucleation of protein crystals in solution. To determine the supersaturation dependent critical nucleus size, the crystal bond energy and the sum of supersaturation dependent surface destructive energies are equilibrated (referred to as EBDE) and compared to the MWS method. In view of the nature of lattice binding forces among huge biomolecules, only the first nearest neighbor interactions are considered. For simplicity purposes, equal bond energy interactions throughout the whole crystal are assumed. The consequences of the highly anisotropic and multivalent interactions of proteins during crystal nucleation were considered elsewhere [
10]. However, specifics of protein and small molecule crystal nucleation are kept in mind [
11].
3. MWS Method Application to Closest-Packed 3D Crystals; Comparison of EBDE and MWS
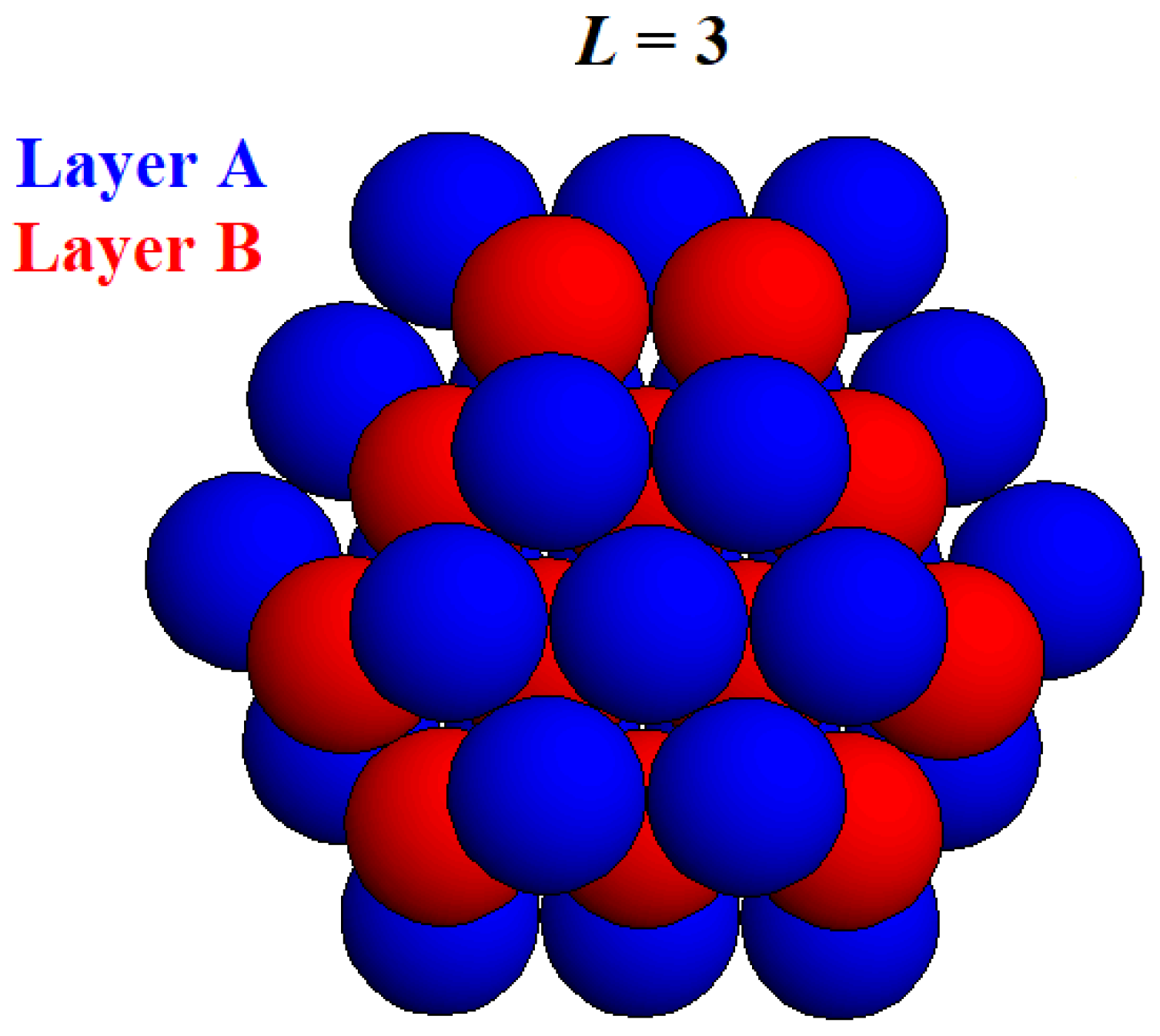
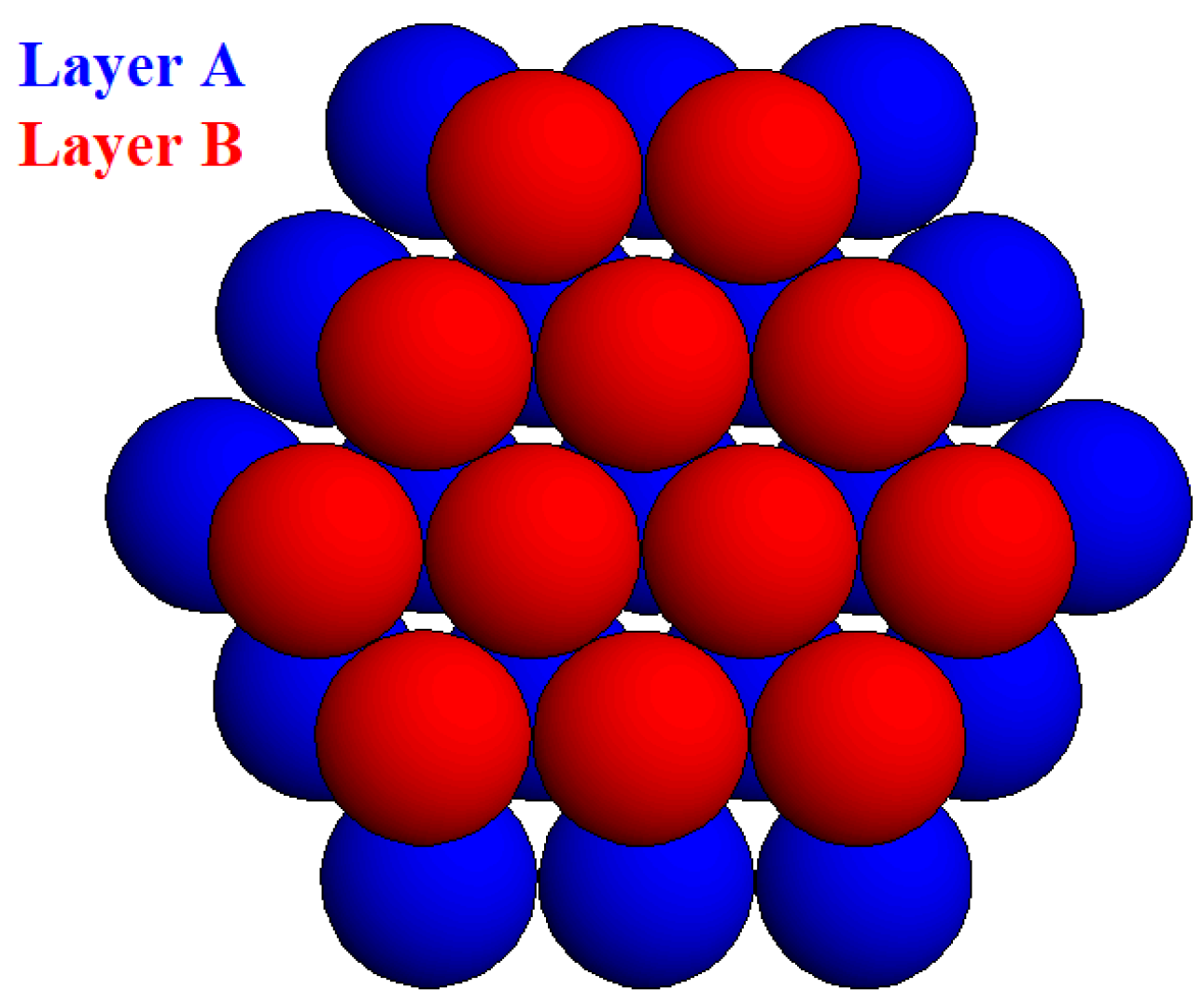
Crystallographic computer programs provide crystal lattice images and numerical data, in contrast to the classical MWS method which uses analytical expressions. Simple models of modestly sized 3D crystals with complete shapes were used to calculate the MWSs of faces forming the habitus of non-Kossel crystal nuclei, the reason being that incomplete crystalline clusters would have sites for subsequent attachment and formation of minimum surface free energy clusters. Considered herein are the closest-packed (HCP and FCC) crystals. To prepare such models, closest-packed monomolecular layers were stacked consecutively onto both sides of a basic A-layer. Three-layered (
Figure 7), five-layered (
Figure 5), etc. homologous HCP crystal nuclei with
L = 3, 4, etc. were created. (It is worth noting that heaping 7 and more close-packed layers need larger foundations, i.e. larger
L values. As already mentioned however, a nucleus size limit is inevitable.) In contrast, to calculate the MWS of a {0001} crystal face, a whole upper mono-layer was stripped-off from the rest of the crystal. Then the layer disintegrated completely into its constituent molecules. The results made it obvious that MWS value calculations introduced uncertainty, caused by the alternatively stacked hexagonal and ditrigonal layers in the HCP crystals. This is why two different MWS values (but not one like by the Kossel-crystal) were calculated for the closest-packed surface layers. Oddly enough, depending on the type of surface layer to be disintegrated, the crystal stood in equilibrium with two different supersaturations.
Well-known crystallography equations were applied for the MWS value calculations. Since each molecule in the closest-packed surface layer (regardless whether hexagonal or ditrigonal) is related to three molecules beneath it, the work (energy) needed for striping-off one such layer is always three times the number (z) of molecules in the layer. By denoting the number of molecules in the edge of a hexagonal layer by
λ, we obtain
which gives z = 7, 19, 37, 61, 91, 127 ... for
λ = 2, 3, 4, 5, 6, 7 ... respectively.
In addition, for the number (Z′) of molecules in the ditrigonal layer, which is situated onto a hexagonal layer:
which gives Z′ = 3, 12, 27, 48, … for
λ = 2, 3, 4, 5 ... respectively.
A complete disintegration of the hexagonal and ditrigonal layers into their constituent molecules requires different amounts of work (ΔG
v2) which, in turn, implies two different MWS values. Though disintegration can be done in various ways, the result must be one and the same. Here, the crystallographic formula concerning the number of bonds in the hexagonal crystal layer is used:
Thus, the MWS value (W) for HCP crystals with hexagonal {0001} surface layers is
Neglecting (for L→∞) all numbers smaller than the quadratic terms in Equation (11) gives W→6ψb, i.e., the bonding energy of the molecule in a kink position.
The formula for ΔG
v2 of ditrigonal layers is
and the MWS value (W) for HCP crystals with ditrigonal {0001} surface lattice planes is
Here, again, all numbers smaller than the quadratic terms in Equation (13) can be neglected for L→∞, and again, W→6ψb.
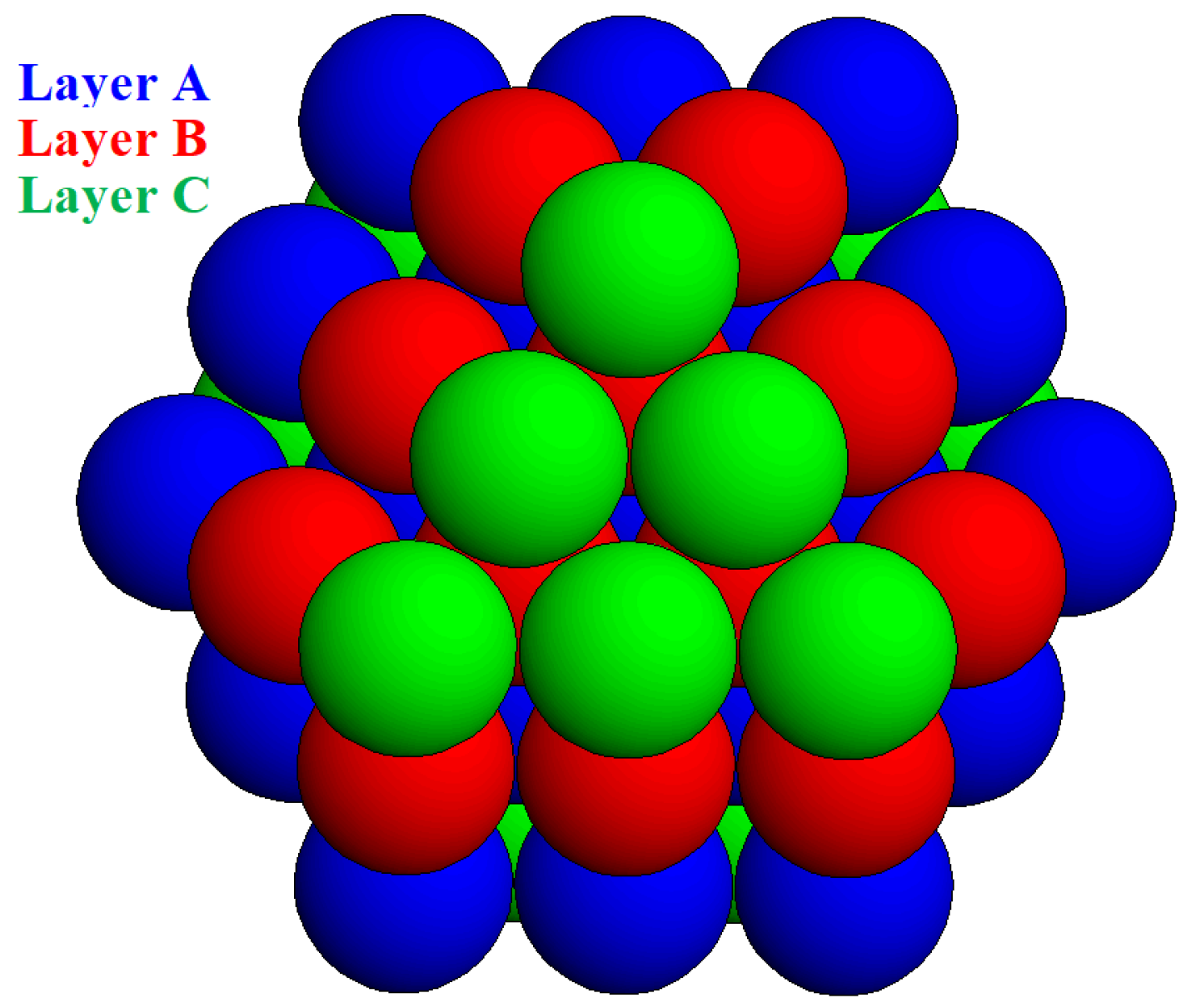
Relevant to the application of the MWS method to FCC crystals is the cubo-octahedron crystal shown in
Figure 8 (which is composed by five layers, BCABC, and has eight equal octahedral faces of triangular shape, all of them containing six molecules). The EBDE method shows that this crystal can be a critical nucleus under supersaturation, which is determined from the ratio
ψb/
ψd ≈ 0.42. It is almost equal to the
ψb/
ψd ratio for the HCP crystal shown in
Figure 5 (
ψb/
ψd ≈ 0.43, see
Table 2). Note that the FCC crystal is comprised of 55 molecules, while the HCP crystal has two additional molecules.
By applying the MWS method, we see that the stripping-off of the whole upper triangular layer {111} (containing green balls,
Figure 8) requires work (energy) amounting to 18
ψb, while each layer here has a bond energy of 9
ψb. This gives a MWS value of 4.5
ψb. The cubic faces {100} are built from nine molecules of bond energy 12
ψb, and the bond energy of the front cubic face in
Figure 8 to the layer beneath it is 32
ψb. This gives a MSW value ≈ 4.9
ψb. The different MWSs of the {111} and {100} faces indicate that, according to the MSW method, this crystal does not have an equilibrium shape.
The difficulties encountered with the highly symmetrical HPC and FCC crystals indicate that the MWS method has low applicability to less-symmetrical crystals. This notwithstanding, historically, the classical MWS method has played an important role in the crystal growth theory, regardless of being demonstrated merely on the Kossel-crystal model [
24]. Markedly, present-day computer programs are capable of elucidating at least one of the basic notions used in the classical MWS method, namely the meaning of an ‘infinitely’ large crystal. The latter is used as a benchmark in Stranski-Kaischew’s molecular kinetic theory.
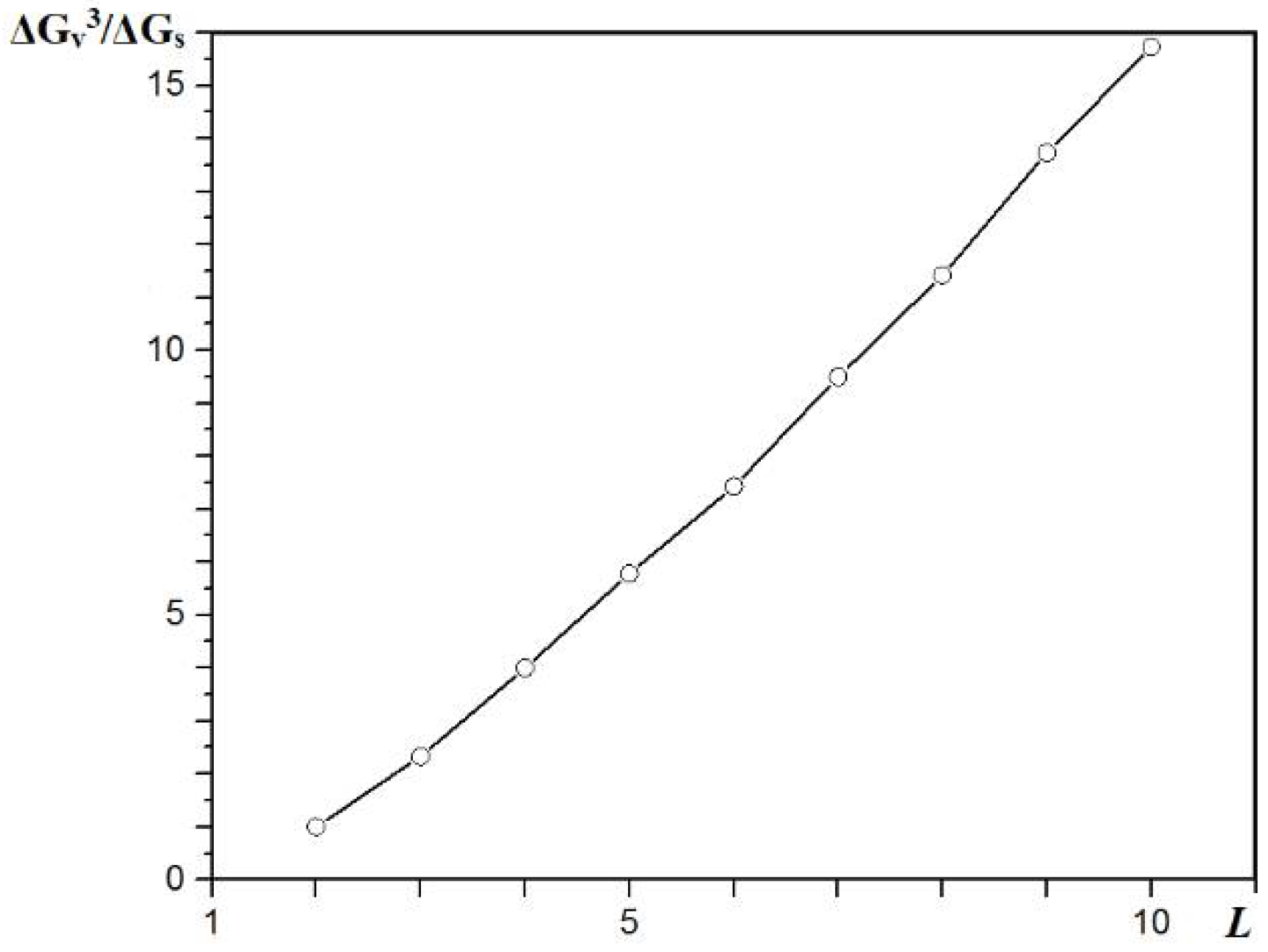
Figure 9 shows how the ‘infinitely’ large crystal size is approached asymptotically. Here, the N
b/N
a ratio (where N
b is the total number of bonds in the crystal and N
a is the number of molecules in the truncated HCP bipyramid) is plotted versus the crystal edge length,
L. It is well-known that the N
b/N
a for a 12-coordinated molecule is six, and the bond energy of a molecule in the kink position is 6
ψb.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}