Temperature Effects on the Elastic Constants, Stacking Fault Energy and Twinnability of Ni3Si and Ni3Ge: A First-Principles Study

Abstract
:1. Introduction
2. Method of Calculation
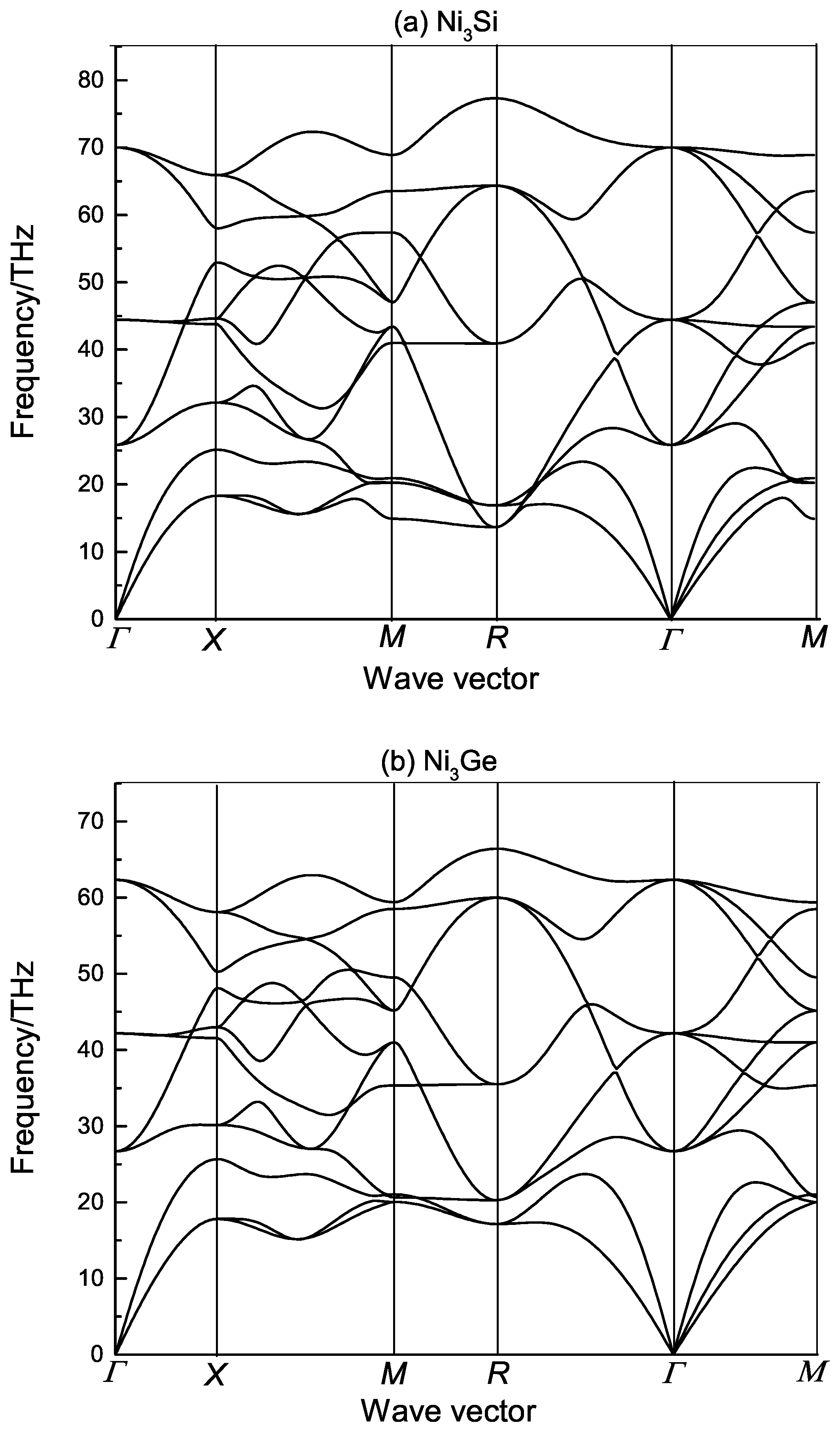
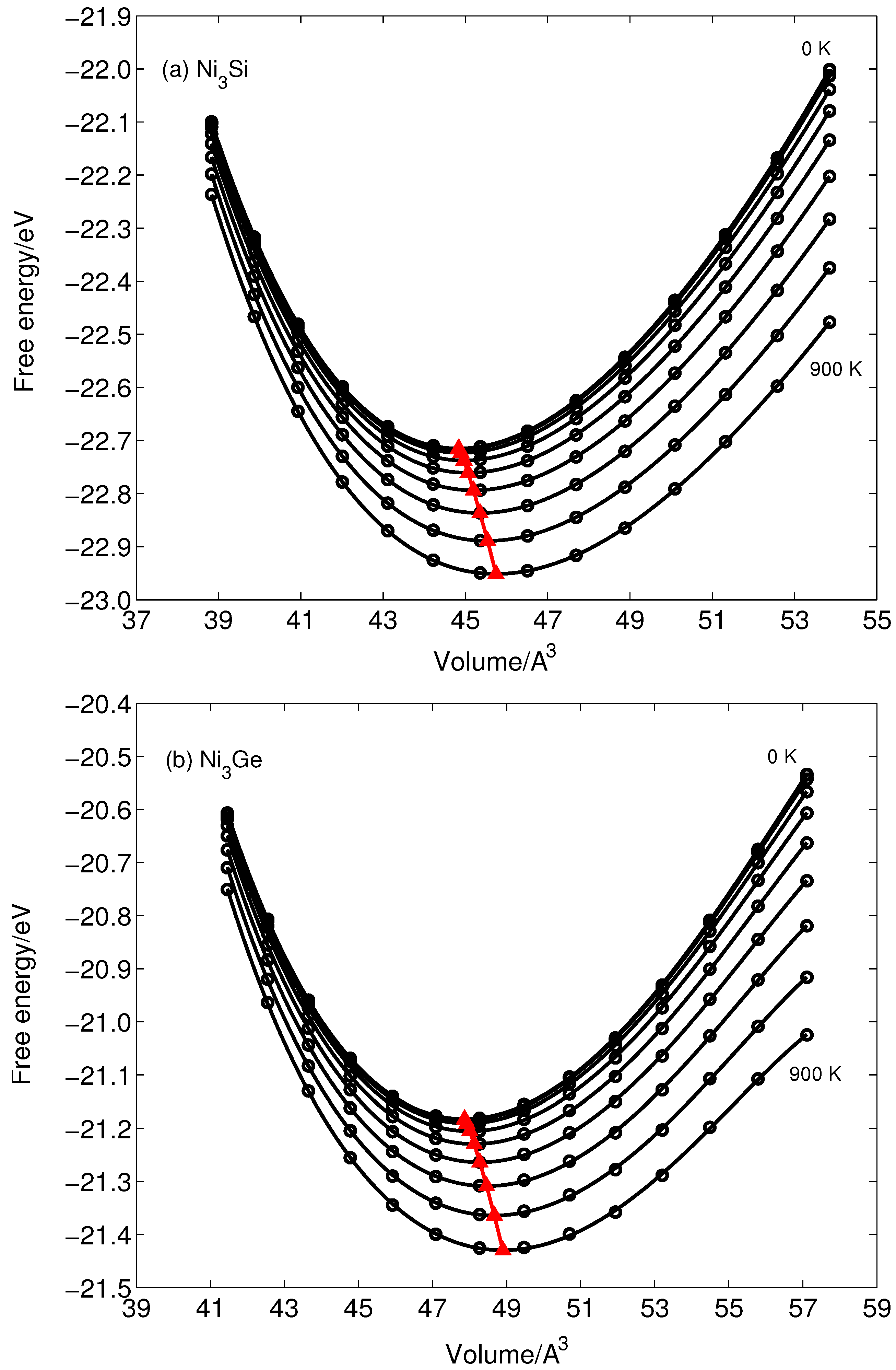
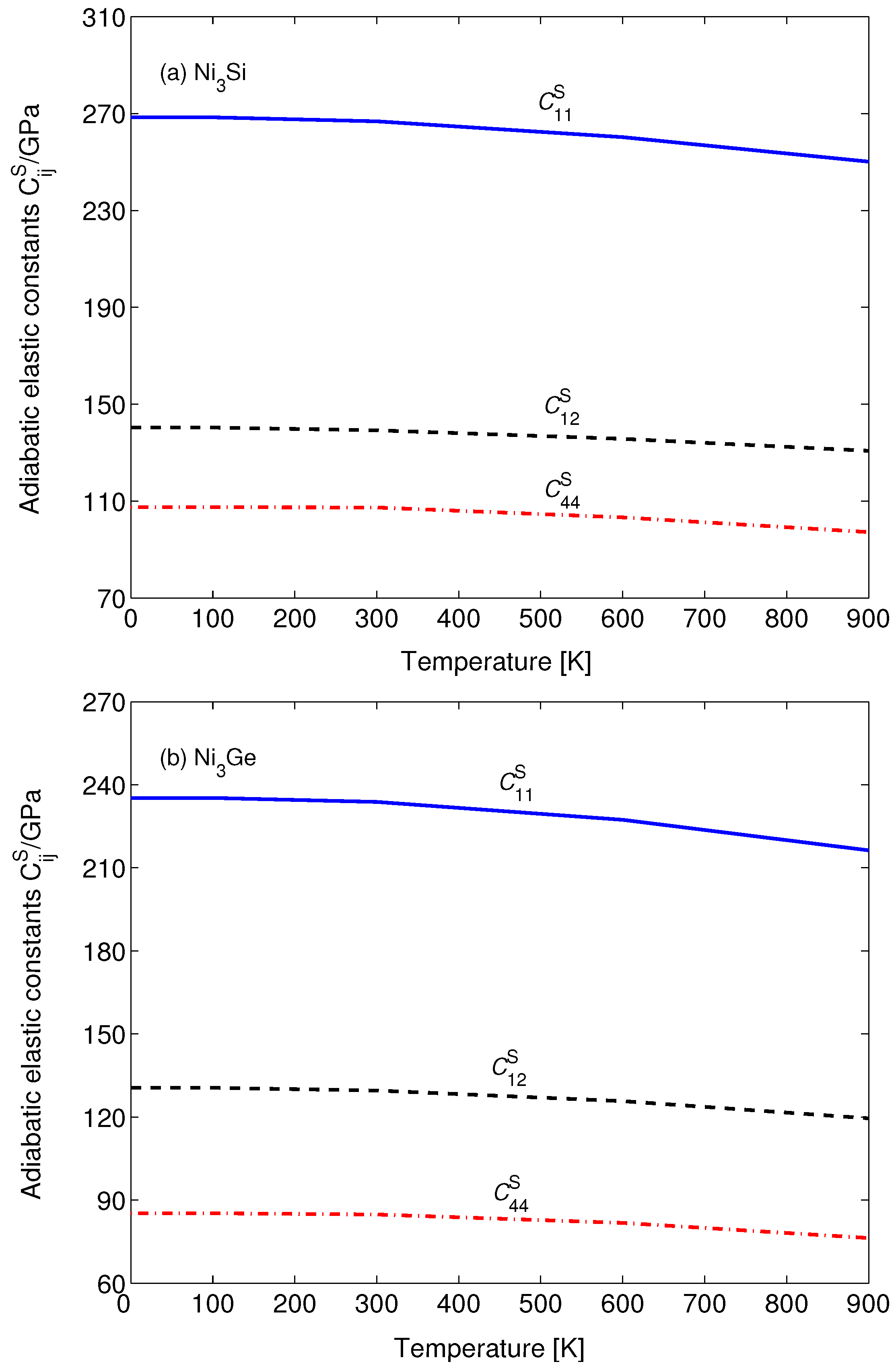
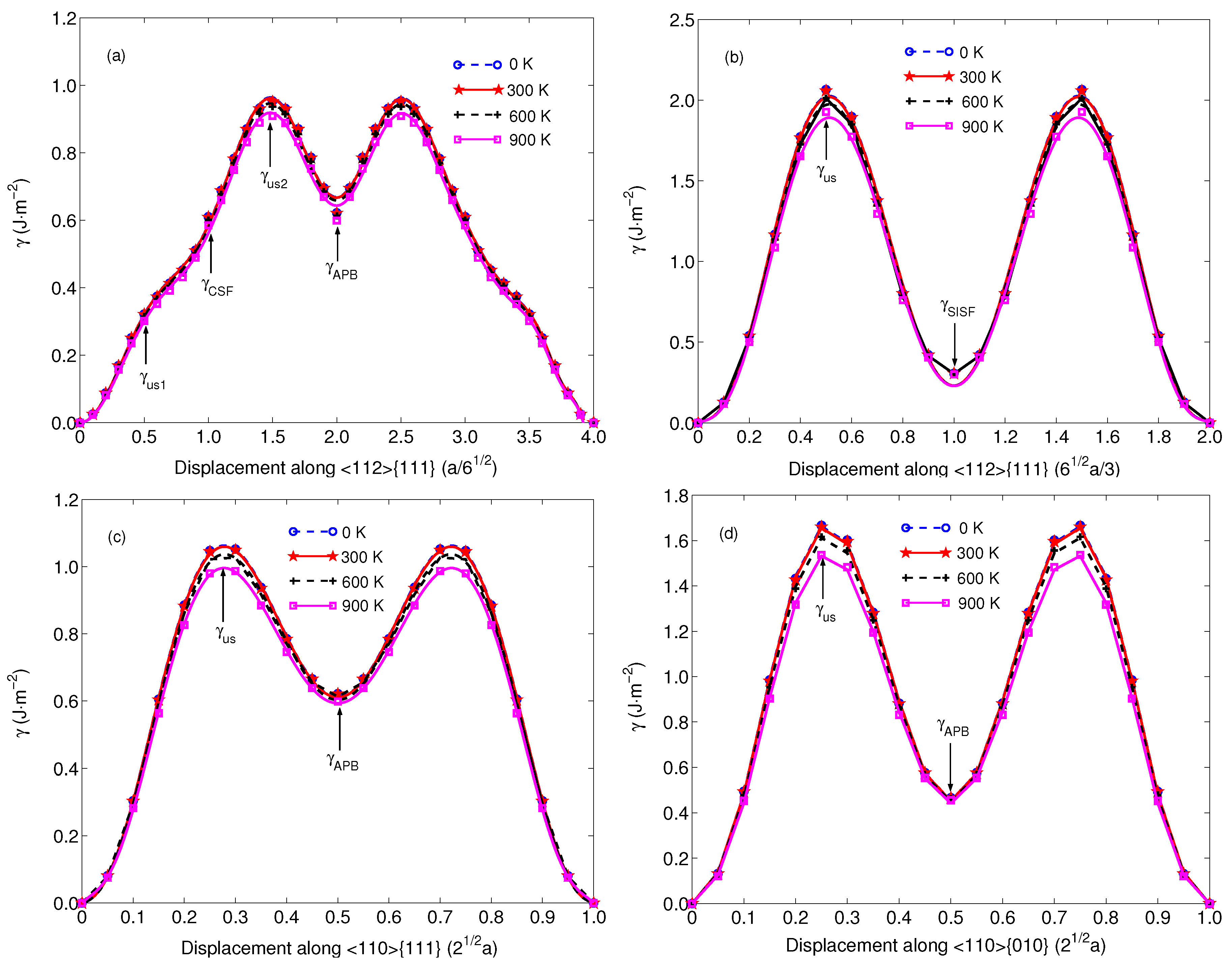
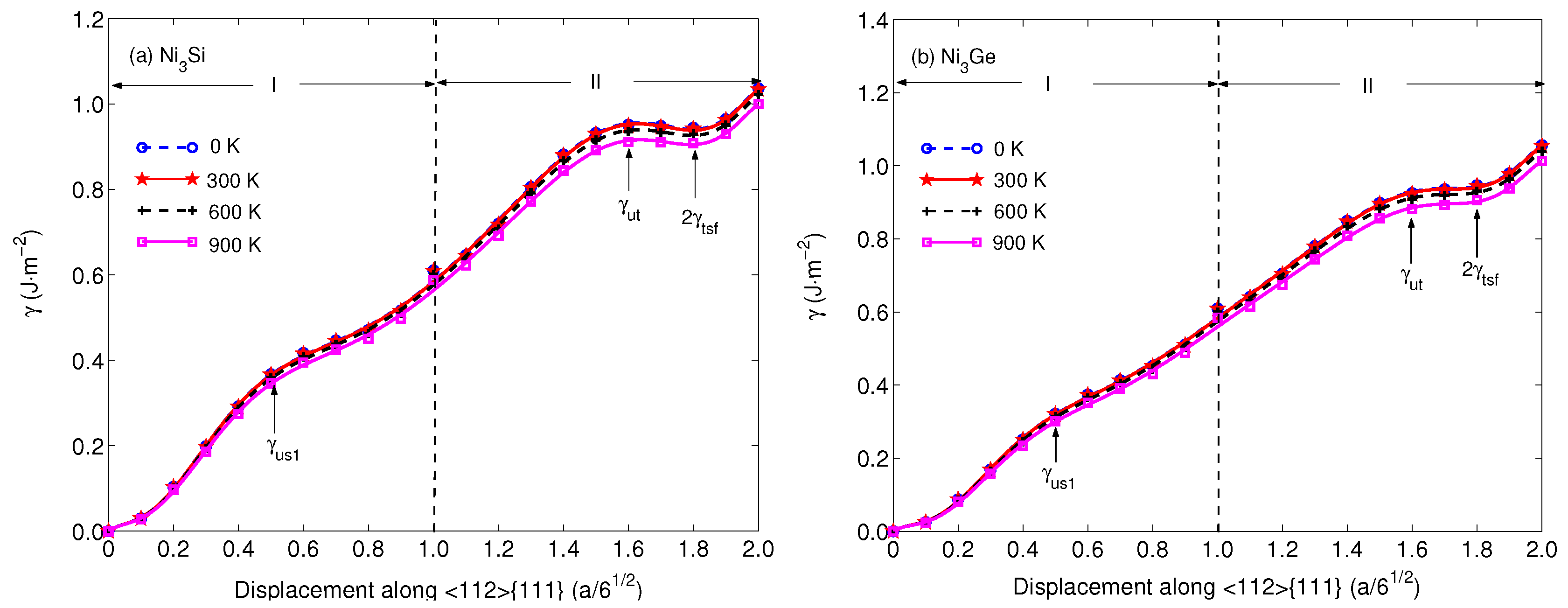
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reed, R.C. The Superalloys: Fundamentals and Applications; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Sims, C.T.; Stoloff, N.S.; Hagel, W.C. Superalloys II; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Ochial, S.; Oya, Y.; Suzuki, T. Alloying behaviour of Ni3Al, Ni3Ga, Ni3Si and Ni3Ge. Acta Metall. 1984, 32, 289–298. [Google Scholar] [CrossRef]
- Yasuda, H.; Takasugi, T.; Koiwa, M. Elasticity of Ni-based L12-type intermetallic compounds. Acta Metall. Mater. 1992, 40, 381–387. [Google Scholar] [CrossRef]
- Iotova, D.; Kioussis, N.; Lim, S.P. Electronic structure and elastic properties of the Ni3X (X = Mn, Al, Ga, Si, Ge) intermetallics. Phys. Rev. B 1996, 54, 14413. [Google Scholar] [CrossRef]
- Chen, Q.; Huang, Z.W.; Zhao, Z.D.; Hu, C.K. First-principles study on the structural, elastic, and thermodynamics properties of Ni3X (X: Al, Mo, Ti, Pt, Si, Nb, V, and Zr) intermetallic compounds. Appl. Phys. A 2014, 116, 1161–1172. [Google Scholar] [CrossRef]
- Hou, H.; Wen, Z.Q.; Zhao, Y.H.; Fu, L.; Wang, N.; Han, P.D. First-principles investigations on structural, elastic, thermodynamic and electronic properties of Ni3X (X = Al, Ga and Ge) under pressure. Intermetallics 2014, 44, 110–115. [Google Scholar] [CrossRef]
- Tanaka, K.; Yasuda, H.; Koiwa, M. Temperature Dependence of Elastic Constants of Several Intermetallic Compounds. In Proceedings of the 3rd Japan International SAMPE, Chiba, Japan, 7–10 December 1993. [Google Scholar]
- Prikhodko, S.V.; Isaak, D.G.; Fisher, E.; Starostina, N.V.; Ma, Y.; Ardell, A.J. The elastic constants of FCC Ni-Ga and Ni-Ge alloys up to 1100 K. Scr. Mater. 2006, 54, 1327–1330. [Google Scholar] [CrossRef]
- Hirth, J.P.; Lothe, J. Theory of Dislocations, 2nd ed.; Wiley: New York, NY, USA, 1982. [Google Scholar]
- Schoeck, G. The Peierls model: Progress and limitations. Mater. Sci. Eng. A 2005, 400, 7–17. [Google Scholar] [CrossRef]
- Christian, J.W.; Mahajan, S. Deformation twinning. Prog. Mater. Sci. 1995, 39, 1–157. [Google Scholar] [CrossRef]
- Xie, H.X.; Wang, C.Y.; Yu, T.; Du, J.P. Dislocation formation and twinning from the crack tip in Ni3Al: Molecular dynamics simulations. Chin. Phys. B 2009, 18, 251–258. [Google Scholar]
- van Swygenhoven, H.; Derlet, P.M.; Froseth, A.G. Stacking fault energies and slip in nanocrystalline metals. Nat. Mater. 2004, 3, 399. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, E.B.; Bernstein, N. A first-principles measure for the twinnability of FCC metals. J. Mech. Phys. Solids 2004, 52, 2507–2519. [Google Scholar] [CrossRef]
- Siegel, D.J. Generalized stacking fault energies, ductilities, and twinnabilities of Ni and selected Ni alloys. Appl. Phys. Lett. 2005, 87, 121901. [Google Scholar] [CrossRef]
- Kibey, S.; Liu, J.B.; Johnson, D.D.; Sehitoglu, H. Generalized planar fault energies and twinning in Cu-Al alloys. Appl. Phys. Lett. 2006, 89, 191911. [Google Scholar] [CrossRef]
- Kibey, S.; Liu, J.B.; Johnson, D.D.; Schitoglu, H. Energy pathways and directionality in deformation twinning. Appl. Phys. Lett. 2007, 91, 181916. [Google Scholar] [CrossRef]
- Muzyk, M.; Pakiela, Z.; Kurzydlowski, K.J. Ab initio calculations of the generalized stacking fault energy in aluminium alloys. Scr. Mater. 2011, 64, 916–918. [Google Scholar] [CrossRef]
- Li, B.Q.; Sui, M.L.; Mao, S.X. Twinnability predication for fcc metals. J. Mater. Sci. Technol. 2011, 27, 97–100. [Google Scholar] [CrossRef]
- Wen, Y.F.; Sun, J. Generalized planar fault energies and mechanical twinning in gamma TiAl alloys. Scr. Mater. 2013, 68, 759–762. [Google Scholar] [CrossRef]
- Shang, S.L.; Wang, W.Y.; Zhou, B.C.; Wang, Y.; Darling, K.A.; Kecskes, L.J.; Mathaudhu, S.N.; Liu, Z.K. Generalized stacking fault energy, ideal strength and twinnability of dilute Mg-based alloys: A first-principles study of shear deformation. Acta Mater. 2014, 67, 168–180. [Google Scholar] [CrossRef]
- Wang, J.; Sehitoglu, H.; Maier, H.J. Dislocation slip stress prediction in shape memory alloys. Int. J. Plasticity 2014, 54, 247–266. [Google Scholar] [CrossRef]
- Cai, T.; Zhang, Z.J.; Zhang, P.; Yang, J.B.; Zhang, Z.F. Competition between slip and twinning in face-centered cubic metals. J. Appl. Phys. 2014, 116, 163512. [Google Scholar] [CrossRef]
- Fu, C.L.; Ye, Y.Y.; Yoo, M.H. Theoretical investigation of the elastic constants and shear fault energies of Ni3Si. Philos. Mag. Lett. 1993, 67, 179–185. [Google Scholar] [CrossRef]
- Mryasov, O.N.; Gornostyrev, Y.N.; van Schilfgaarde, M.; Freeman, A.J. Superdislocation core structure in L1 NiAl, NiGe and FeGe: Peierls-Nabarro analysis starting from ab-initio GSF energetics calculations. Acta Mater. 2002, 50, 4545–4554. [Google Scholar] [CrossRef]
- Brugger, K. Thermodynamic definition of higher order elastic coefficients. Phys. Rev. 1964, 133, 1611–1612. [Google Scholar] [CrossRef]
- Vinet, P.; Rose, J.H.; Ferrante, J.; Smith, J.R. Universal features of the equation of state of solids. J. Phys. Condens. Matter 1989, 1, 1941–1963. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.J.; Zhang, H.; Manga, V.R.; Shang, S.L.; Chen, L.Q.; Liu, Z.K. A first-principles approach to finite temperature elastic constants. J. Phys. Condens. Matter 2010, 22, 225404. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.L.; Zhang, H.; Wang, Y.; Liu, Z.K. Temperature-dependent elastic stiffness constants of α- and θ- Al2O3 from first-principles calculations. J. Phys. Condens. Matter 2010, 22, 375403. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.K.; Zhang, H.; Ganeshan, S.; Wang, Y.; Mathaudhu, S.N. Computational modeling of effects of alloying elements on elastic coefficients. Scr. Mater. 2010, 63, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 3115. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Wang, R.; Wang, S.F.; Wu, X.Z.; Liu, A.P. First-principles phonon calculations of thermodynamic properties for ductile rare-earth intermetallic compounds. Intermetallics 2011, 19, 1599–1604. [Google Scholar] [CrossRef]
- Liu, L.L.; Wang, R.; Wu, X.Z.; Gan, L.Y.; Wei, Q.Y. Temperature effects on the generalized planar fault energies and twinnbilities of Al, Ni and Cu: First principles calculations. Comput. Matter. Sci. 2014, 88, 124–130. [Google Scholar] [CrossRef]
- Gülsern, O.; Cohen, R.E. High pressure thermoelasticity of body-centered cubic tantalum. Phys. Rev. B 2002, 65, 064103. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals; Clarendon Press: Oxford, UK, 1964. [Google Scholar]
- Paidar, V.; Pope, D.P.; Vitek, V. A theory of the anomalous yield behavior in L12 ordered alloys. Acta Metall. 1984, 32, 435–438. [Google Scholar] [CrossRef]
- Rice, J.R. Dislocation nucleation from a crack tip: An analysis based on the Peierls concept. J. Mech. Phys. Solids 1992, 40, 239–271. [Google Scholar] [CrossRef]
- Wang, T.C.; Wang, K.R.; Zhang, Y.W. A unified model for dislocation nucleation, dislocation emission and dislocation free zone. Int. J. Fract. 1996, 78, 227–239. [Google Scholar] [CrossRef]
- Yoo, M.H.; Fu, C.L.; Horton, J.A. Crack-tip dislocations and fracture behavior in Ni3Al and Ni3Si. Mater. Sci. Eng. A 1994, 176, 431–437. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Ref. | a | |||
|---|---|---|---|---|---|
| NiSi | This work | 3.553 | 268.5 | 140.4 | 107.5 |
| [6] | 3.517 | 317 | 162 | 129 | |
| NiGe | This work | 3.629 | 235.3 | 130.6 | 85.2 |
| [7] | 3.584 | 268.9 | 148.4 | 103.4 | |
| [4] | 263.0 | 143.0 | 103.0 |
| NiSi | (K) | 0 | 300 | 600 | 900 |
| 0.367 | 0.366 | 0.359 | 0.346 | ||
| 0.990 | 0.988 | 0.973 | 0.948 | ||
| 1.193 | 1.189 | 1.164 | 1.122 | ||
| 0.610 | 0.609 | 0.602 | 0.588 | ||
| 0.308 | 0.307 | 0.304 | 0.298 | ||
| 0.951 | 0.949 | 0.935 | 0.911 | ||
| 2 | 0.944 | 0.942 | 0.929 | 0.908 | |
| 1.984 | 1.975 | 1.925 | 1.840 | ||
| 0.599 | 0.598 | 0.592 | 0.580 | ||
| 0.477 | 0.476 | 0.471 | 0.463 | ||
| 0.584 | 0.583 | 0.576 | 0.565 | ||
| 0.550 | 0.549 | 0.547 | 0.541 | ||
| NiGe | (K) | 0 | 300 | 600 | 900 |
| 0.321 | 0.320 | 0.314 | 0.301 | ||
| 0.953 | 0.951 | 0.936 | 0.908 | ||
| 1.047 | 1.044 | 1.021 | 0.980 | ||
| 0.609 | 0.608 | 0.600 | 0.584 | ||
| 0.306 | 0.305 | 0.303 | 0.298 | ||
| 0.925 | 0.923 | 0.908 | 0.882 | ||
| 2 | 0.947 | 0.944 | 0.931 | 0.906 | |
| 1.667 | 1.661 | 1.617 | 1.537 | ||
| 0.622 | 0.620 | 0.613 | 0.598 | ||
| 0.466 | 0.465 | 0.462 | 0.456 | ||
| 0.604 | 0.603 | 0.594 | 0.581 | ||
| 0.500 | 0.499 | 0.498 | 0.492 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Chen, L.; Jiang, Y.; He, C.; Xu, G.; Wen, Y. Temperature Effects on the Elastic Constants, Stacking Fault Energy and Twinnability of Ni3Si and Ni3Ge: A First-Principles Study. Crystals 2018, 8, 364. https://doi.org/10.3390/cryst8090364
Liu L, Chen L, Jiang Y, He C, Xu G, Wen Y. Temperature Effects on the Elastic Constants, Stacking Fault Energy and Twinnability of Ni3Si and Ni3Ge: A First-Principles Study. Crystals. 2018; 8(9):364. https://doi.org/10.3390/cryst8090364
Chicago/Turabian StyleLiu, Lili, Liwan Chen, Youchang Jiang, Chenglin He, Gang Xu, and Yufeng Wen. 2018. "Temperature Effects on the Elastic Constants, Stacking Fault Energy and Twinnability of Ni3Si and Ni3Ge: A First-Principles Study" Crystals 8, no. 9: 364. https://doi.org/10.3390/cryst8090364
APA StyleLiu, L., Chen, L., Jiang, Y., He, C., Xu, G., & Wen, Y. (2018). Temperature Effects on the Elastic Constants, Stacking Fault Energy and Twinnability of Ni3Si and Ni3Ge: A First-Principles Study. Crystals, 8(9), 364. https://doi.org/10.3390/cryst8090364