Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Quantum Chemical Calculation
2.2. Crystal Preparation
2.3. Measurement
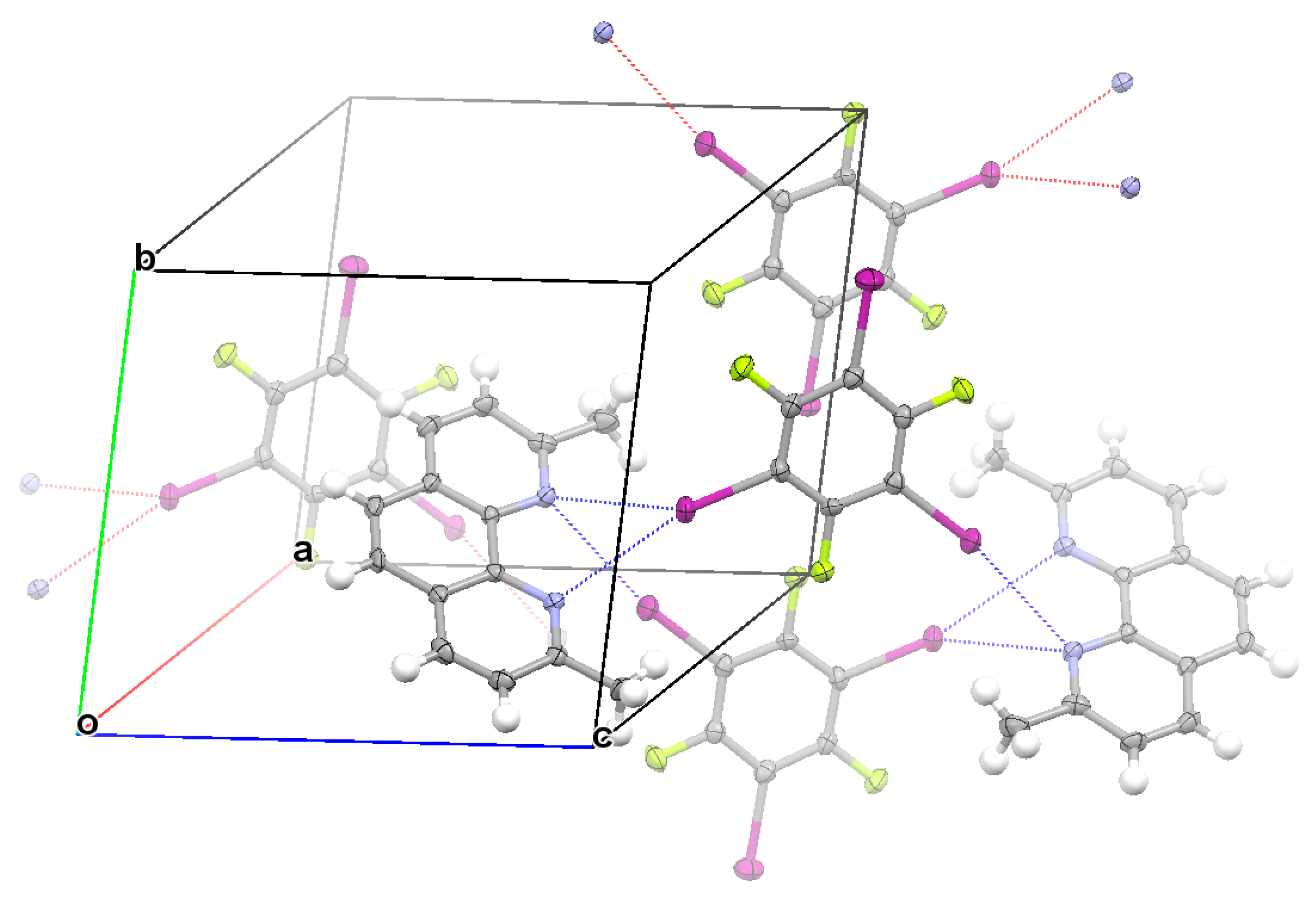
3. Results and Discussion
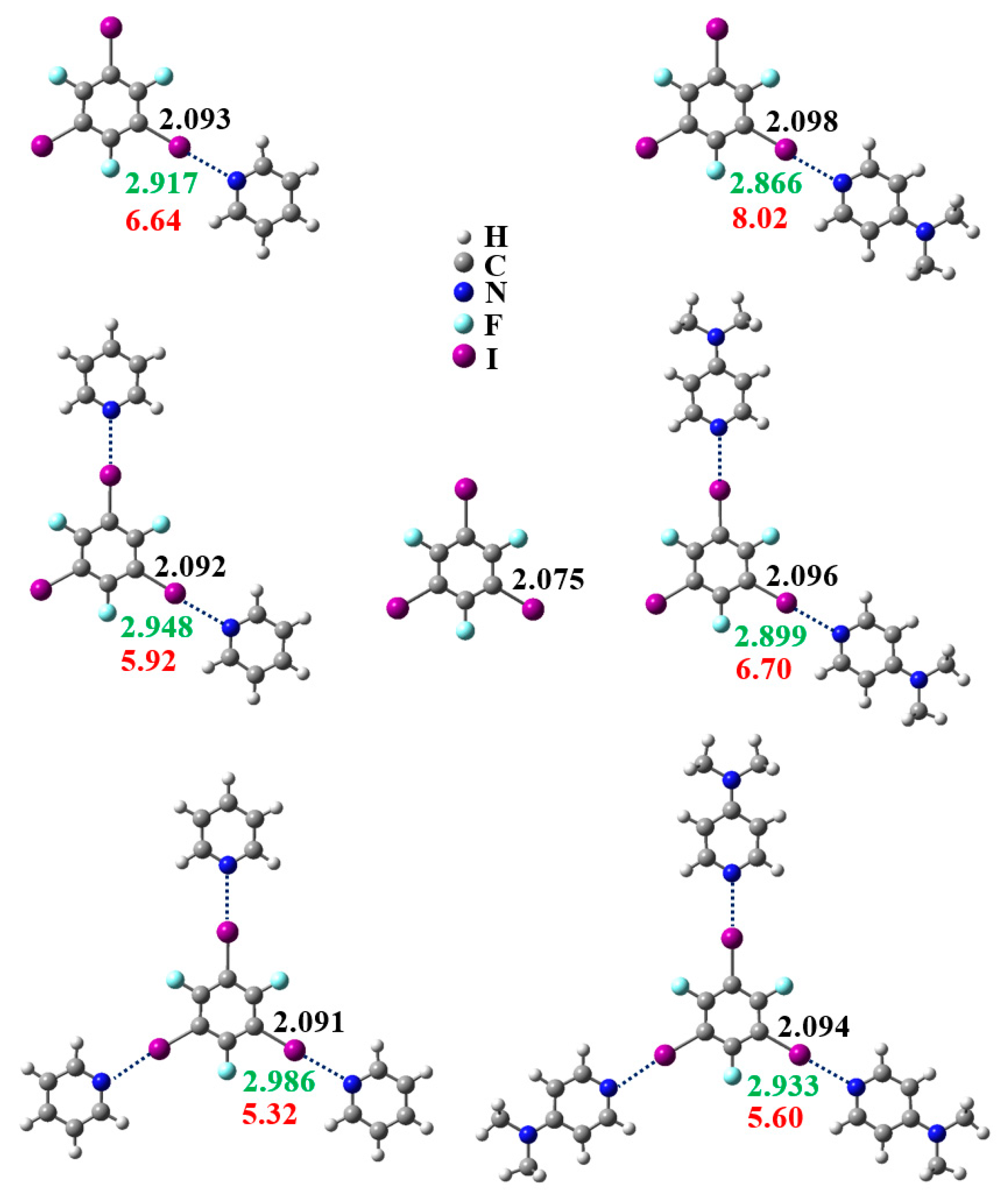
3.1. Anticooperativity of the Halogen Bonds
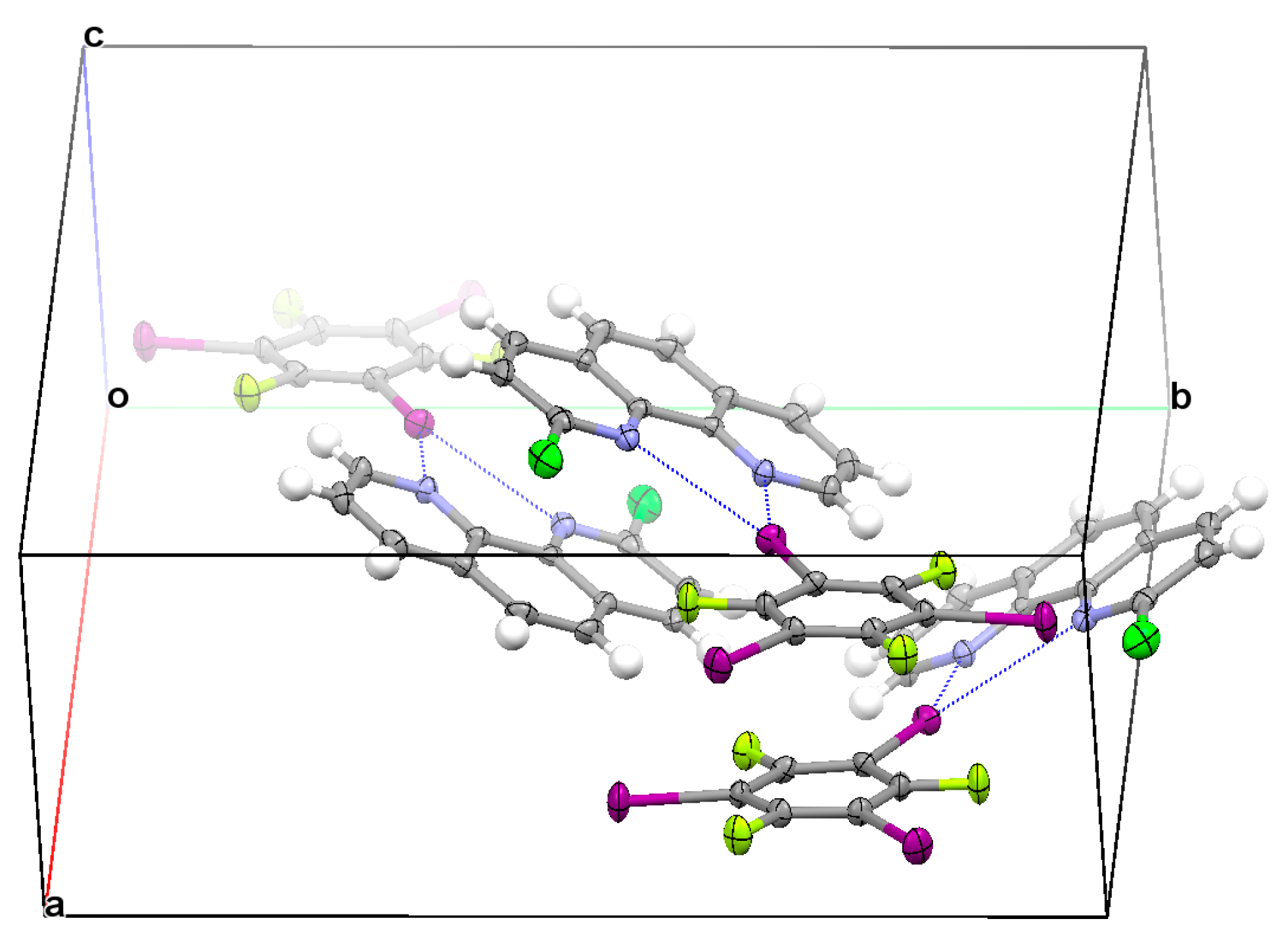
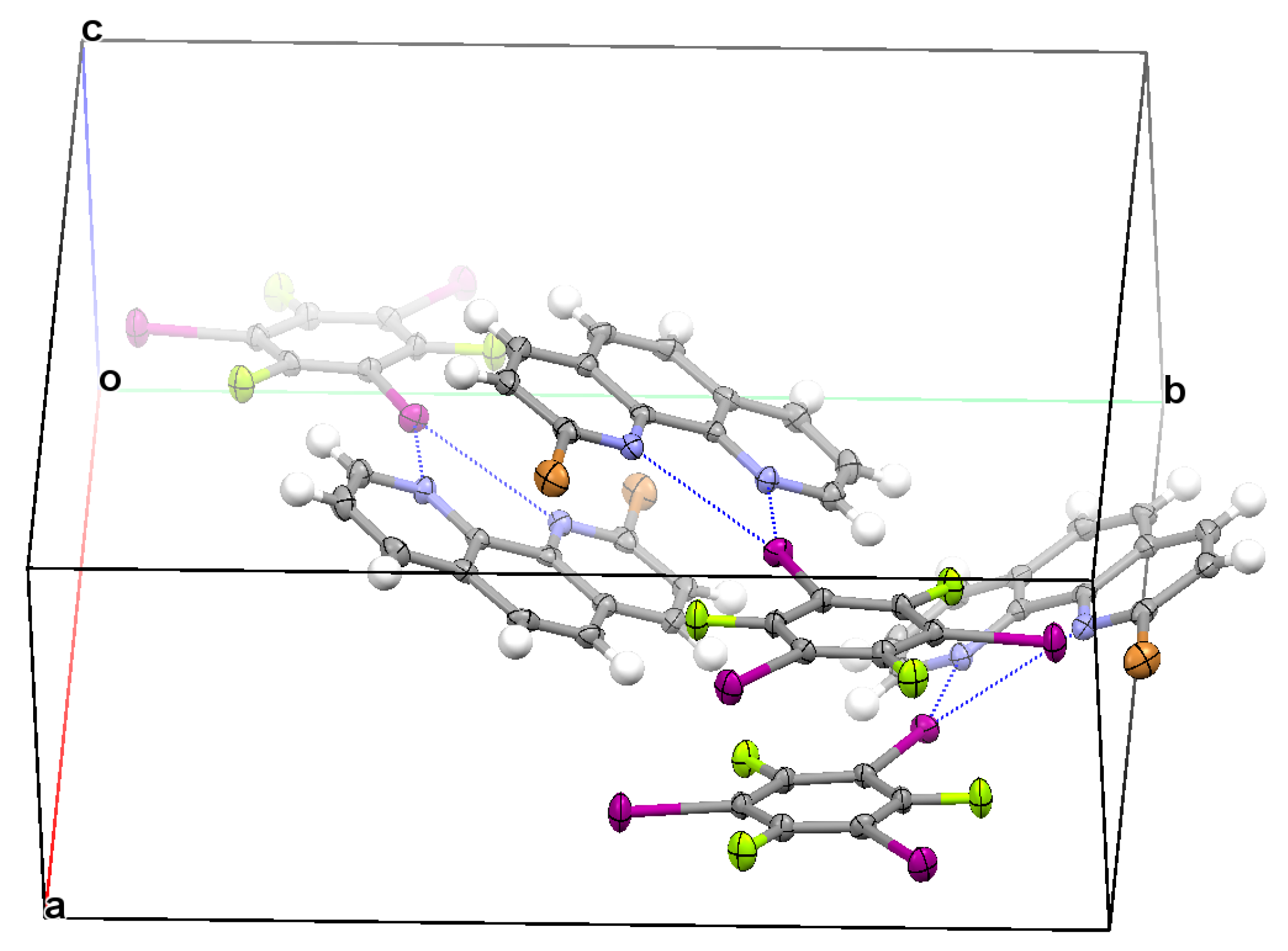
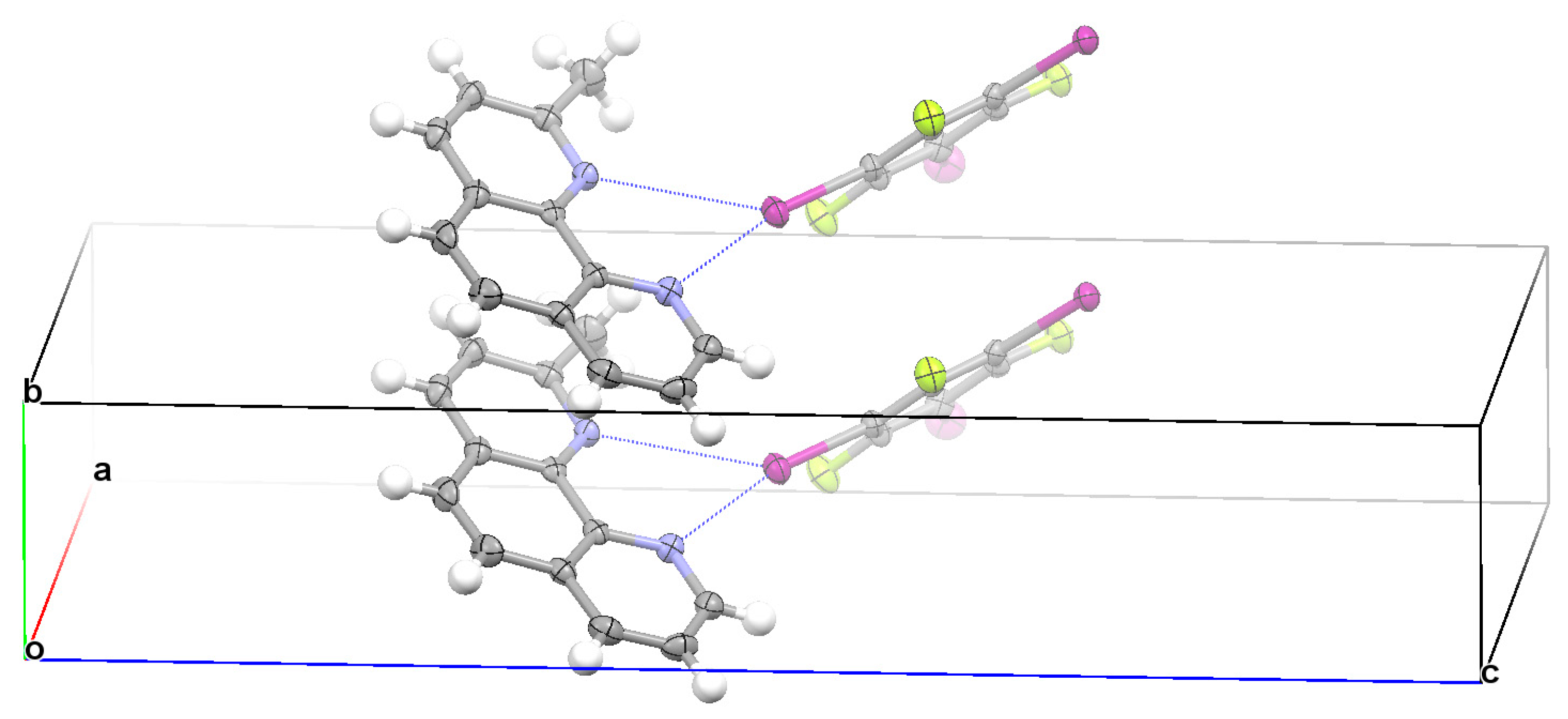
3.2. Structural Competition between C−I⋯N Halogen Bond and π⋯π Stacking Interaction in the Crystal Structures
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Amico, V.; Meille, S.V.; Corradi, E.; Messina, M.T.; Resnati, G. Perfluorocarbon-Hydrocarbon Self-Assembling. 1D Infinite Chain Formation Driven by Nitrogen⋯Iodine Interactions. J. Am. Chem. Soc. 1998, 120, 8261–8262. [Google Scholar] [CrossRef]
- Farina, A.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G.; Vecchio, G. Resolution of Racemic 1,2-Dibromohexafluoropropane through Halogen-Bonded Supramolecular Helices. Angew. Chem. Int. Ed. 1999, 38, 2433–2436. [Google Scholar] [CrossRef]
- Wang, W.; Wong, N.B.; Zheng, W.; Tian, A. Theoretical Study on the Blueshifting Halogen Bond. J. Phys. Chem. A 2004, 108, 1799–1805. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Investigations into the Nature of Halogen Bonding Including Symmetry Adapted Perturbation Theory Analyses. J. Chem. Theory Comput. 2008, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Supramolecular Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C. The halogen bond: an interim perspective. Phys. Chem. Chem. Phys. 2010, 12, 7736–7747. [Google Scholar] [CrossRef] [PubMed]
- Bertani, R.; Sgarbossa, P.; Venzo, A.; Lelj, F.; Amati, M.; Resnati, G.; Pilati, T.; Metrangolo, P.; Terraneo, G. Halogen Bonding in Metal–Organic–Supramolecular Networks. Coord. Chem. Rev. 2010, 254, 677–695. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Sansotera, M.; Terraneo, G. Halogen Bonding: A General Route in Anion Recognition and Coordination. Chem. Soc. Rev. 2010, 39, 3772–3783. [Google Scholar] [CrossRef]
- Wang, W. Halogen Bond Involving Hypervalent Halogen: CSD Search and Theoretical Study. J. Phys. Chem. A 2011, 115, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Pennington, W.T.; Resnati, G.; Taylor, M.S. Halogen Bonding: From Self-Assembly to Materials and Biomolecules. CrystEngComm 2013, 15, 3057. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming Interactions from the Electrophilic Site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Giese, M.; Albrecht, M.; Rissanen, K. Anion−π Interactions with Fluoroarenes. Chem. Rev. 2015, 115, 8867–8895. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Resnati, G.; Pennington, W.T. The Halogen Bond: A New Avenue in Recognition and Self-Assembly. New J. Chem. 2018, 42, 10461–10462. [Google Scholar] [CrossRef]
- Ding, X.H.; Ou, C.J.; Wang, S.; Xie, L.H.; Lin, J.Y.; Wang, J.P.; Huang, W. Co-Crystallization of 1,3,5-Trifluoro-2,4,6-triiodobenzene (1,3,5-TFTIB) with a Variety of Lewis Bases through Halogen-Bonding Interactions. CrystEngComm 2017, 19, 5504–5521. [Google Scholar] [CrossRef]
- Lucassen, A.C.B.; Karton, A.; Leitus, G.; Shimon, L.J.W.; Martin, J.M.L.; van der Boom, M.E. Co-Crystallization of Sym-Triiodo-Trifluorobenzene with Bipyridyl Donors: Consistent Formation of Two Instead of Anticipated Three N⋯I Halogen Bonds. Cryst. Growth Des. 2007, 7, 386–392. [Google Scholar] [CrossRef]
- Vartanian, M.; Lucassen, A.C.B.; Shimon, L.J.W.; van der Boom, M.E. Cocrystallization of a Tripyridyl Donor with Perfluorinated Iodobenzene Derivatives: Formation of Different N⋯I Halogen Bonds Determining Network vs Plain Packing Crystals. Cryst. Growth Des. 2008, 8, 786–790. [Google Scholar] [CrossRef]
- Roper, L.C.; Präsang, C.; Kozhevnikov, V.N.; Whitwood, A.C.; Karadakov, P.B.; Bruce, D.W. Experimental and Theoretical Study of Halogen-Bonded Complexes of DMAP with Di- and Triiodofluorobenzenes. A Complex with a Very Short N⋯I Halogen Bond. Cryst. Growth Des. 2010, 10, 3710–3720. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J. Practical Crystal Engineering Using Halogen Bonding: A Hierarchy Based on Calculated Molecular Electrostatic Potential Surfaces. J. Mol. Struct. 2014, 1072, 20–27. [Google Scholar] [CrossRef]
- Hidalgo, P.I.; Leal, S.; Jiménez, C.A.; Vöhringer-Martinez, E.; Herrera, B.; Pasán, J.; Ruiz-Pérez, C.; Bruce, D.W. Extending the Halogen-Bonded Supramolecular Synthon Concept to 1,3,4-Oxadiazole Derivatives. CrystEngComm 2016, 18, 42–47. [Google Scholar] [CrossRef]
- Triguero, S.; Llusar, R.; Polo, V.; Formigué, M. Halogen Bonding Interactions of sym-Triiodotrifluorobenzene with Halide Anions: A Combined Structural and Theoretical Study. Cryst. Growth Des. 2008, 8, 2241–2247. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Mutual Induced Coordination in Halogen-Bonded Anionic Assemblies with (6,3) Cation-Templated Topologies. Chem. Commun. 2008, 1635–1637. [Google Scholar] [CrossRef]
- Cauliez, P.; Polo, V.; Roisnel, T.; Llusar, R.; Formigué, M. The Thiocyanate Anion as a Polydentate Halogen Bond Acceptor. CrystEngComm 2010, 12, 558–566. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. The π⋯π Stacking Interactions between Homogeneous Dimers of C6FxI(6-x) (x =0, 1, 2, 3, 4 and 5): A Comparative Study with the Halogen Bond. J. Phys. Chem. A 2012, 116, 12486–12491. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Unexpected Strong Stacking Interactions between the Homogeneous Dimers of C6FxI(6-x) (x =0, 1, 2, 3, 4 and 5). Comput. Theor. Chem. 2013, 1023, 88–94. [Google Scholar] [CrossRef]
- Ji, B.; Wang, W.; Deng, D.; Zhang, Y.; Cao, L.; Zhou, L.; Ruan, C.; Li, T. Structural Competition between π⋯π Interactions and Halogen Bonds: A Crystallographic Study. CrystEngComm 2013, 15, 769–774. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Reed, A.E.; Curitss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1998, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cocrystal | I | II | III | IV |
|---|---|---|---|---|
| CCDC No. | 1891221 | 1891219 | 1891220 | 1891222 |
| Formula | C18H7ClF3I3N2 | C18H7BrF3I3N2 | C19H10F3I3N2 | C20H12F3I3N2 |
| Formula weight | 724.41 | 768.87 | 703.99 | 718.02 |
| Crystal size/mm3 | 0.24 × 0.15 × 0.017 | 0.30 × 0.27 × 0.26 | 0.27 × 0.26 × 0.19 | 0.31 × 0.28 × 0.24 |
| Crystal system | monoclinic | monoclinic | monoclinic | triclinic |
| Space group | P21/c | P21/c | P21/n | P-1 |
| a/Å | 14.2733(5) | 14.2515(4) | 17.9454(9) | 9.6529(7) |
| b/Å | 18.2696(6) | 18.2923(6) | 4.4461(3) | 9.6760(5) |
| c/Å | 7.6371(4) | 7.6939(3) | 25.0595(14) | 11.8940(7) |
| α/° | 90 | 90 | 90 | 81.258(5) |
| β/° | 91.662(3) | 90.725(3) | 93.433(5) | 88.575(5) |
| γ/° | 90 | 90 | 90 | 72.537(6) |
| Volume/Å3 | 1990.69(13) | 2005.57(12) | 1995.81(19) | 1047.15(12) |
| Z | 4 | 4 | 4 | 2 |
| ρcalc/g cm–3 | 2.417 | 2.546 | 2.343 | 2.277 |
| T/K | 287.12(10) | 293(2) | 289.78(10) | 293(2) |
| 2θ Range for data collection/° | 6.52–50.992 | 6.92–51.00 | 6.92–51.00 | 6.612–50.994 |
| Reflections collected | 10704 | 11373 | 10763 | 12066 |
| No. unique data [R(int)] | 3622 [0.0282] | 3729 [0.0273] | 3689 [0.0384] | 3892 [0.0379] |
| Final R (I > 2σ(I)) | 0.0319 | 0.0302 | 0.0392 | 0.0508 |
| Final wR2 (all data) | 0.0611 | 0.0567 | 0.0877 | 0.1550 |
| Goodness-of-fit | 1.065 | 1.039 | 1.113 | 1.072 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, J.-G.; Wang, W. Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study. Crystals 2019, 9, 140. https://doi.org/10.3390/cryst9030140
Zhang Y, Wang J-G, Wang W. Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study. Crystals. 2019; 9(3):140. https://doi.org/10.3390/cryst9030140
Chicago/Turabian StyleZhang, Yu, Jian-Ge Wang, and Weizhou Wang. 2019. "Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study" Crystals 9, no. 3: 140. https://doi.org/10.3390/cryst9030140
APA StyleZhang, Y., Wang, J. -G., & Wang, W. (2019). Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study. Crystals, 9(3), 140. https://doi.org/10.3390/cryst9030140