Decomposition Characterizations of Methane Hydrate Confined inside Nanoscale Pores of Silica Gel below 273.15 K



Abstract
:1. Introduction
2. Experimental
2.1. Materials and Experimental Apparatus
2.2. Experimental Procedures
2.2.1. Pore Water Confined Inside the Silica Gel Pores
2.2.2. Raman Spectra of Pore Water
2.2.3. Hydrate Formation
2.2.4. Micro Decomposition of Hydrate
2.2.5. Macro Decomposition of Hydrate
3. Results and Discussion
3.1. Raman Spectra of Pore Water
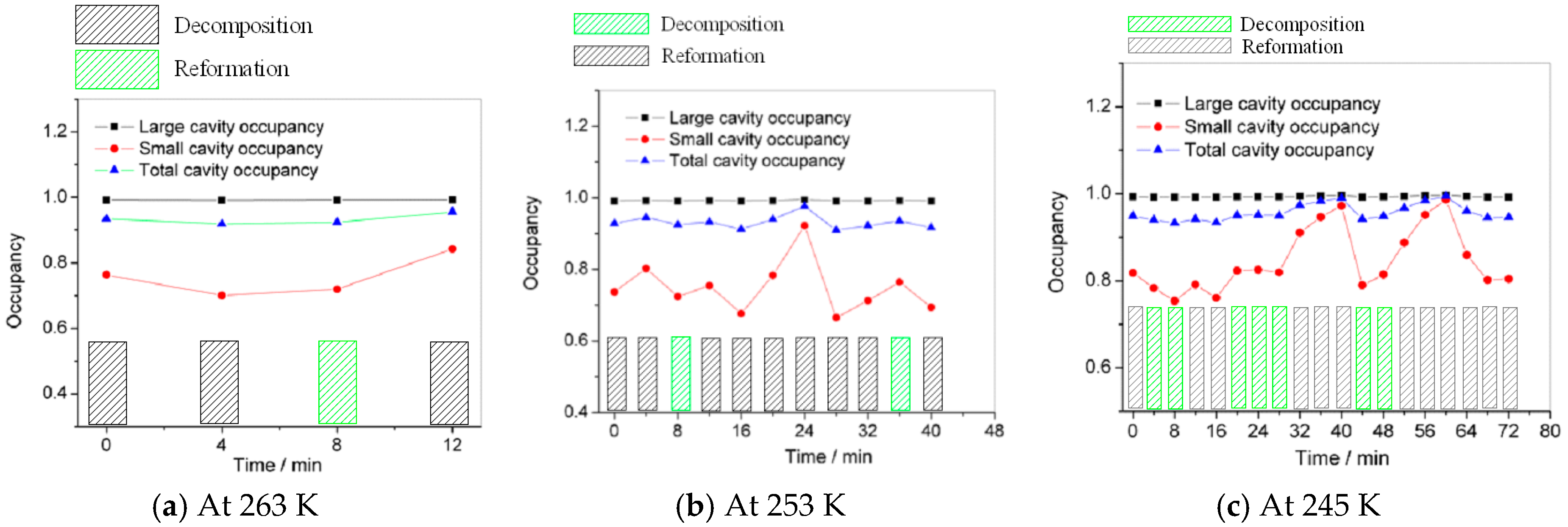
3.2. Micro Decomposition of Hydrate
3.3. Macro Decomposition of Hydrate
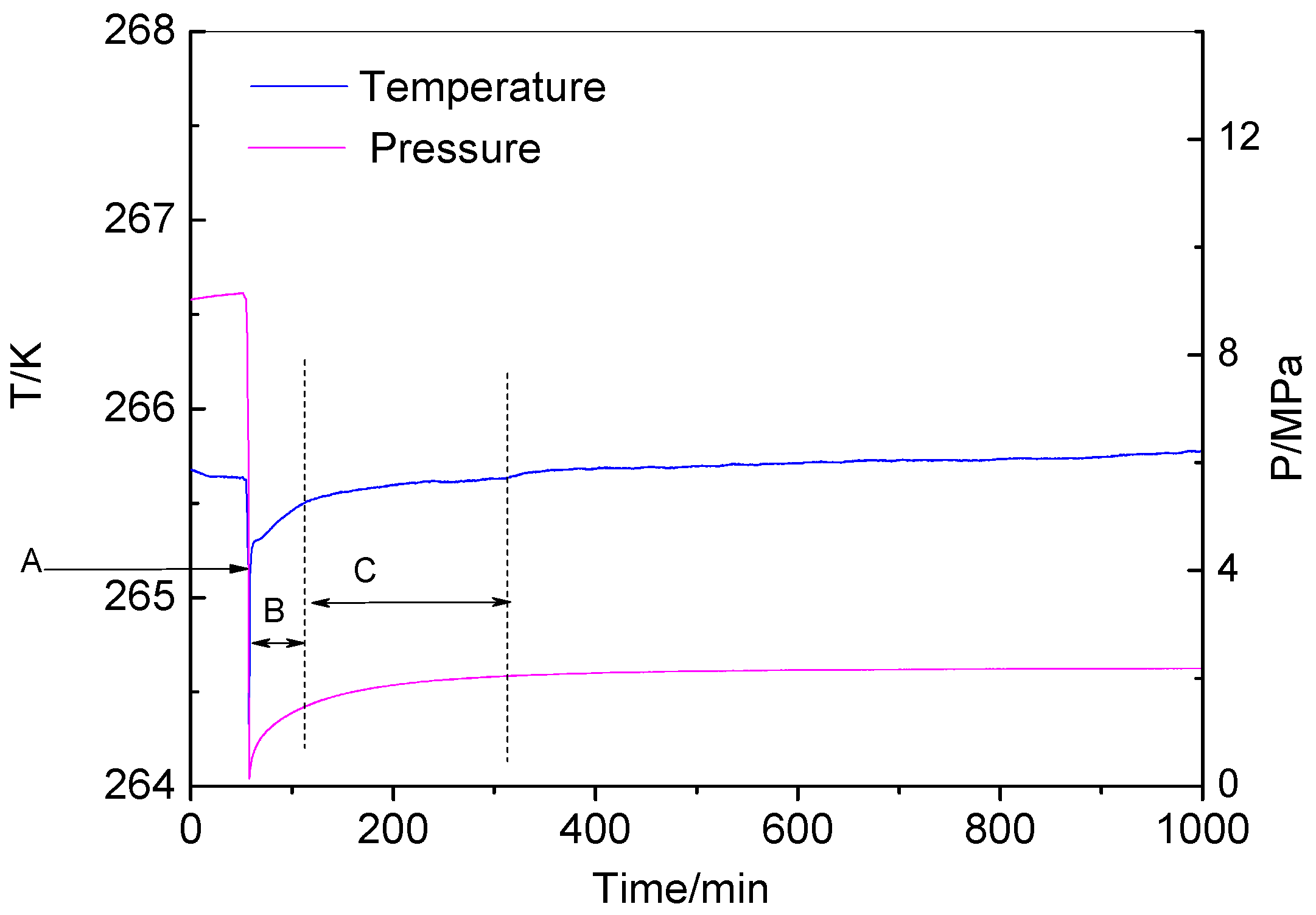
3.3.1. Decomposition from High Rre-Decomposition Pressure
3.3.2. Decomposition from Low Pre-Decomposition Pressure
3.4. Mechanism of Hydrate Decomposition
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Makogon, Y.F.; Trebin, F.A.; Trofimuk, A.A.; Tsarev, V.P.; Cherskiy, N.V. Detection of a Pool of Natural Gas in a Solid (Hydrated Gas) State. Doklady Akademii Nauk SSSR 1971, 196, 203–206. [Google Scholar]
- Bily, C.; Dick, J.W.L. Naturally Occurring Gas hydrates in the Mackenzie Delta, NWT. Bull. Can. Pet. Geol. 1974, 22, 320–352. [Google Scholar]
- Yefremova, A.G.; Zhizchenko, B.P. Occurrence of Crystal Hydrates of Gases in the Sediments of Modern Marine Basins. Doklady Akademii Nauk SSSR 1974, 214, 1179–1181. [Google Scholar]
- Wu, N.Y.; Zhang, H.Q.; Yang, S.X.; Liang, J.Q.; Wang, H.B. Preliminary Discussion on Natural Gas Hydrate (NGH) Reservoir System of Shenhu Area, North Slope of South China Sea. Nat. Gas Ind. 2007, 27, 1. [Google Scholar]
- Makogon, Y.F.; Holditch, S.A.; Makogon, T.Y. Natural-gas Hydrates—A Potential Energy Source for the 21st Century. J. Pet. Sci. Eng. 2007, 56, 14–31. [Google Scholar] [CrossRef]
- Sultan, N.; Cochonat, P.; Foucher, P.J. Effect of Gas Hydrates Melting on Seafloor Slope Instability. Mar. Geol. 2004, 213, 379–401. [Google Scholar] [CrossRef]
- Park, T.; Kyung, D.; Lee, W. Effect of Organic Matter on CO2 Hydrate Phase Equilibrium in Phyllosilicate Suspensions. Environ. Sci. Technol. 2014, 48, 6597–6603. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.Y.; Germanovich, L.N. Excess Pore Pressure Resulting from Methane Hydrate Dissociation in Marine Sediments: A Theoretical Approach. J. Geophys. Res. Solid Earth 2006, 111, 1978–2012. [Google Scholar] [CrossRef]
- Kwon, T.H.; Cho, G.C.; Santamarina, J.C. Gas Hydrate Dissociation in Sediments: Pressure-temperature Evolution. Geochem. Geophys. Geosyst. 2008, 9, Q03019. [Google Scholar] [CrossRef]
- Jain, K.; Juanes, R. Preferential Mode of Gas Invasion in Sediments: Grain-scale Mechanistic Model of Coupled Multiphase Fluid Flow and Sediment Mechanics. J. Geophys. Res. 2009, 114, B08101. [Google Scholar] [CrossRef]
- Holtzman, R.; Juanes, R. Thermodynamic and Hydrodynamic Constraints on Overpressure Caused by Hydrate Dissociation: A Pore-Scale Model. Geophys. Res. Lett. 2011, 38, L14308. [Google Scholar] [CrossRef]
- Jang, J.; Santamarina, J.C. Recoverable Gas from Hydrate-bearing Sediments: Pore Network Model Simulation and Macroscale Analyses. J. Geophys. Res. Solid Earth 2011, 116, 1978–2012. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Ioannis, N.; Lichtner, P.C. Gas Saturation Resulting from Methane Hydrate Dissociation in a Porous Medium: Comparison between Analytical and Pore-network Results. J. Phys. Chem. C 2013, 117, 11104–11116. [Google Scholar] [CrossRef]
- Wang, D.G.; Wang, C.C.; Li, C.F.; Liu, C.L.; Lu, H.L.; Wu, N.Y.; Hu, G.W.; Liu, L.L.; Meng, Q.G. Effect of Gas Hydrate Formation and Decomposition on Flow Properties of Fine-grained Quartz Sand Sediments Using X-ray CT Based Pore Network Model Simulation. Fuel 2018, 226, 516–526. [Google Scholar] [CrossRef]
- Chen, X.Y.; Espinoza, N.D. Surface Area Controls Gas Hydrate Dissociation Kinetics in Porous Media. Fuel 2018, 234, 358–363. [Google Scholar] [CrossRef]
- Zhan, L.; Wang, Y.; Li, X.S. Experimental Study on Characteristics of Methane Hydrate Formation and Dissociation in Porous Medium with Different Particle Sizes Using Depressurization. Fuel 2018, 230, 37–44. [Google Scholar] [CrossRef]
- Zhang, J.H.; Zhao, C.; Xiong, Z.S. Meso-level Simulation of Gas Hydrate Dissociation in Low-permeability Sediments. Theor. Appl. Mech. Lett. 2014, 4, 062002. [Google Scholar] [CrossRef]
- Wang, X.; Dong, B.; Li, W.Z.; Yu, M.H.; Song, Y.C. Microscale Effects on Methane Hydrate Dissociation at Low Temperature in the Micro Porous Media Channels by Depressurization. Int. J. Heat. Mass. Trans. 2018, 122, 1182–1197. [Google Scholar] [CrossRef]
- Wang, P.F.; Yang, M.J.; Chen, B.B.; Zhao, Y.C.; Zhao, J.F.; Song, Y.C. Methane Hydrate Reformation in Porous Media with Methane Migration. Chem. Eng. Sci. 2017, 168, 344–351. [Google Scholar] [CrossRef]
- Uchida, T.; Takeya, S.; Chuvilin, E.M.; Ohmura, R.; Nagao, J.; Yakushev, V.S.; Istomin, V.A.; Minagawa, H.; Ebinuma, T.; Narita, H. Decomposition of Methane Hydrates in Sand, Sandstone, Clays, and Glass beads. J. Geophys. Res. Solid Earth 2004, 109, B05206. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Takeya, S.; Nagao, J.; Narita, H. Effects of Pore Sizes on Dissociation Temperatures and Pressures of Methane, Carbon Dioxide, and Propane Hydrates in Porous Media. J. Phys. Chem. B 2002, 106, 820–826. [Google Scholar] [CrossRef]
- Clarke, M.A.; Pooladi-Darvish, M.; Bishnoi, P.R. A Method to Predict Equilibrium Conditions of Gas Hydrate Formation in Porous Media. Ind. Eng. Chem. Res. 1999, 38, 2485–2490. [Google Scholar] [CrossRef]
- Seshadri, K.; Wilder, J.W.; Smith, D.H. Measurements of Equilibrium Pressures and Temperatures for Propane Hydrate in Silica Gels with Different Pore-size Distributions. J. Phys. Chem. B 2001, 105, 2627–2631. [Google Scholar] [CrossRef]
- Wilder, J.W.; Seshadri, K.; Smith, D.H. Resolving Apparent Contradictions in Equilibrium Measurements for Clathrate Hydrates in Porous Media. J. Phys. Chem. B 2001, 105, 9970–9972. [Google Scholar] [CrossRef]
- Klauda, J.B.; Sandler, S.I. Modeling Gas Hydrate Phase Equilibria in Laboratory and Natural Porous Media. Ind. Eng. Chem. Res. 2001, 40, 4197–4208. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, H.; Uchida, T. Methane and Carbon Dioxide Hydrate Phase Behavior in Small Porous Silica Gels: Three-Phase Equilibrium Determination and Thermodynamic Modeling. Langmuir 2002, 18, 9164–9170. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, H. Hydrate Phase Equilibria of the Ternary CH4 + NaCl + Water, CO2 + NaCl + Water and CH4 + CO2 + Water Mixtures in Silica Gel Pores. J. Phys. Chem. B 2003, 107, 889–894. [Google Scholar] [CrossRef]
- Anderson, R.; Llamedo, M.; Tohidi, B.; Burgass, R.W. Experimental Measurement of Methane and Carbon Dioxide Clathrate Hydrate Equilibria in Mesoporous Silica. J. Phys. Chem. B 2003, 107, 3507–3514. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, S.; Cha, I.; Lee, J.D.; Lee, H. Phase Equilibria and Thermodynamic Modeling of Ethane and Propane Hydrates in Porous Silica Gels. J. Phys. Chem. B 2009, 113, 5487–5492. [Google Scholar] [CrossRef]
- Lee, S.; Seo, Y. Experimental Measurement and Thermodynamic Modeling of the Mixed CH4 + C3H8 Clathrate Hydrate Equilibria in Silica Gel Pores: Effects of Pore Size and Salinity. Langmuir 2010, 26, 9742–9748. [Google Scholar] [CrossRef]
- Liu, X.; Flemings, P.B. Capillary Effects on Hydrate Stability in Marine Sediments. J. Geophys. Res. 2011, 116, B07102. [Google Scholar] [CrossRef]
- Anderson, R.; Tohidi, B.; Webber, J.B.W. Gas Hydrate Growth and Dissociation in Narrow Pore Networks: Capillary Inhibition and Hysteresis Phenomena. Geol. Soc. Spec. Publ. 2009, 319, 145–159. [Google Scholar] [CrossRef]
- Bangs, N.L.B.; Musgrave, R.J.; Tréhu, A.M. Upward Shifts in the Southern Hydrate Ridge Gas Hydrate Stability Zone Following Postglacial Warming, Offshore Oregon. J. Geophys. Res. 2005, 110, B03102. [Google Scholar] [CrossRef]
- Liu, X.; Flemings, P.B. Passing Gas through the Hydrate Stability Zone at Southern Hydrate Ridge, Offshore Oregon. Earth Planet. Sci. Lett. 2006, 241, 211–226. [Google Scholar] [CrossRef]
- Daigle, H.; Dugan, B. Pore Size Controls on the Base of the Methane Hydrate Stability Zone in the Kumano Basin, Offshore Japan. Geophys. Res. Lett. 2014, 41, 8021–8028. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Ishizaki, T. Dissociation Condition Measurements of Methane Hydrate in Confined Small Pores of Porous Glass. J. Phys. Chem. B 1999, 103, 3659–3662. [Google Scholar] [CrossRef]
- Park, T.; Lee, J.Y.; Kwon, T.-H. Effect of Pore Size Distribution on Dissociation Temperature Depression and Phase Boundary Shift of Gas Hydrate in Various Fine-Grained Sediments. Energy Fuels 2018, 32, 5321–5330. [Google Scholar] [CrossRef]
- Handa, Y.P.; Stupin, D. Thermodynamic Properties and Dissociation Characteristics of Methane and Propane Hydrates in 70-Å-Radius Silica Gel Pores. J. Phys. Chem. 1992, 96, 8599–8603. [Google Scholar] [CrossRef]
- Watanabe, K.; Mizoguchi, M. Amount of Unfrozen Water in Frozen Porous Media Saturated with Solution. Cold Reg. Sci. Technol. 2002, 34, 103–110. [Google Scholar] [CrossRef]
- Istomin, V.; Chuvilin, E.; Bukhanov, B.; Uchida, T. Pore Water Content in Equilibrium with Ice or Gas Hydrate in Sediments. Cold Reg. Sci. Technol. 2017, 137, 60–67. [Google Scholar] [CrossRef]
- Morishige, K.; Iwasaki, H. X-ray Study of Freezing and Melting of Water Confined within SBA-15. Langmuir 2003, 19, 2808–2811. [Google Scholar] [CrossRef]
- Morishige, K. Influence of Pore Wall Hydrophobicity on Freezing and Melting of Confined Water. J. Phys. Chem. C 2018, 122, 5013–5019. [Google Scholar] [CrossRef]
- Handa, Y.P.; Zakrzewski, M.; Fairbridge, C. Effect of Restricted Geometries on the Structure and Thermodynamic Properties of Ice. J. Phys. Chem. 1992, 96, 8594–8599. [Google Scholar] [CrossRef]
- Sun, Q. Local Statistical Interpretation for Water Structure. Chem. Phys. Lett. 2013, 568–569, 90–94. [Google Scholar] [CrossRef]
- Baschenko, S.M.; Marchenko, L.S. On Raman Spectra of Water, Its Structure and Dependence on Temperature. Semicond. Phys. Quan. Elec. Optoelec. 2011, 14, 77–79. [Google Scholar] [CrossRef]
- Webber, J.B.W.; Dore, J.C.; Strange, J.H.; Anderson, R.; Tohidi, B. Plastic Ice in Confined Geometry: The Evidence from Neutron Diffraction and NMR Relaxation. J. Phys. Condens. Matter 2007, 19, 415117. [Google Scholar] [CrossRef]
- Sum, A.K.; Burruss, R.C.; Sloan, E.D. Measurement of Clathrate Hydrates via Raman Spectroscopy. J. Phys. Chem. B 1997, 101, 7371–7377. [Google Scholar] [CrossRef]
- Liu, C.L.; Meng, Q.G.; He, X.L.; Li, C.F.; Ye, Y.G.; Zhang, G.X.; Liang, J.Q. Characterization of Natural Gas Hydrate Recovered from Pearl River Mouth Basin in South China Sea. Mar. Petrol. Geol. 2015, 61, 14–21. [Google Scholar] [CrossRef]
- Kuhs, W.F.; Genov, G.; Staykova, D.K.; Hansen, T. Ice Perfection and Onset of Anomalous Preservation of Gas Hydrates. Phys. Chem. Chem. Phys. 2004, 6, 4917–4920. [Google Scholar] [CrossRef]
- Takeya, S.; Shimada, W.; Kamata, Y.; Ebinuma, T.; Uchida, T.; Nagao, J.; Narita, H. In Situ X-ray Diffraction Measurements of the Self-preservation Effect of CH4 Hydrate. J. Phys. Chem. A 2001, 105, 9756–9759. [Google Scholar] [CrossRef]
- Stern, L.A.; Circone, S.; Kirby, S.H.; Durham, W.B. Temperature, Pressure, and Compositional Effects on Anomalous or “Self” Preservation of Gas Hydrates. Can. J. Phys. 2003, 81, 271–283. [Google Scholar] [CrossRef]
- English, N.J.; Phelan, G.M. Molecular Dynamics Study of Thermal-driven Methane Hydrate Dissociation. J. Chem. Phys. 2009, 131, 074704. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Baghel, V.S.; Roy, S.; Kumar, R. Molecular Dynamics Study of Model SI Clathrate Hydrates: Effect of Guest Size and Guest–Water Interaction on Decomposition Kinetics. Phys. Chem. Chem. Phys. 2015, 17, 9509–9518. [Google Scholar] [CrossRef] [PubMed]
- Van der Waals, J.H.; Platteeuw, J.C. Clathrate Solutions. Adv. Chem. Phys. 1959, 2, 1–57. [Google Scholar]
- Susilo, R.; Ripmeester, J.A.; Englezos, P. Characterization of Gas Hydrates with PXRD, DSC, NMR, and Raman Spectroscopy. Chem. Eng. Sci. 2007, 62, 3930–3939. [Google Scholar] [CrossRef]
- Subramanian, S.; Sloan, E.D. Trends in Vibrational Frequencies of Guests Trapped in Clathrate Hydrate Cages. J. Phys. Chem. B 2002, 106, 4348–4355. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Diamantonis, N.I.; Economou, I.G.; Papadimitriou, N.I.; Stubos, A.K. Influence of Combining Rules on the Cavity Occupancy of Clathrate Hydrates Using van der Waals–Platteeuw-Theory-Based Modelling. Chem. Eng. Res. Des. 2014, 92, 2992–3007. [Google Scholar] [CrossRef]
- Goussard, J.O.; Roulet, B. Free Expansion for Real Gases. Am. J. Phys. 1993, 61, 845–848. [Google Scholar] [CrossRef]
- Wang, J.J.; Dong, M.Z.; Yang, Z.H.; Gong, H.J.; Li, Y.J. Investigation of Methane Desorption and its Effect on the Gas Production Process from Shale: Experimental and Mathematical Study. Energy Fuels 2017, 31, 205–216. [Google Scholar] [CrossRef]
- Jung, J.W.; Santamarina, J.C. Hydrate Formation and Growth in Pores. J. Cryst. Growth 2012, 345, 61–68. [Google Scholar] [CrossRef]
- Aladko, E.Y.; Dyadin, Y.A.; Fenelonov, V.B.; Larionov, E.G.; Mel’gunov, M.S.; Manakov, A.Y.; Nesterov, A.N.; Zhurko, F.V. Dissociation Conditions of Methane Hydrate in Mesoporous Silica Gels in Wide Ranges of Pressure and Water Content. J. Phys. Chem. B 2004, 108, 16540–16547. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Working Parameters | Setting Conditions |
|---|---|
| Laser source | 532 nm |
| Laser power | 80 mW |
| Objective lens magnification | ×1–100 |
| Grating scale | 600 Grooves/mm |
| Raman shift range | 500–4000 cm−1 |
| Integration time | 1–10 s |
| Temperature (K) | DDAA | DA | Reference |
|---|---|---|---|
| 293 | 3220 | 3430 | Water [44] |
| 276 | 3223 | 3434 | Water [45] |
| 293 | 3231 | 3441 | Water [45] |
| 298 | 3227 | 3430 | Water [This work] |
| 293 | 3087 | 3288 | Pore water [This work] |
| 223 | 3063 | 3267 | Pore water [This work] |
| Items | Decomposition Temperature K | Pre-Decomposition Pressure MPa | Stage B Time Consumption min | Stage C Time Consumption min | Total Time Consumption min |
|---|---|---|---|---|---|
| Run 1 | 265.64 | 9.11 | 56 | 149 | 205 |
| Run 2 | 265.76 | 2.41 | 5 | 210 | 215 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, L.; Zhou, X.; Chen, P.; Zang, X.; Liang, D.; Guan, J. Decomposition Characterizations of Methane Hydrate Confined inside Nanoscale Pores of Silica Gel below 273.15 K. Crystals 2019, 9, 200. https://doi.org/10.3390/cryst9040200
Wan L, Zhou X, Chen P, Zang X, Liang D, Guan J. Decomposition Characterizations of Methane Hydrate Confined inside Nanoscale Pores of Silica Gel below 273.15 K. Crystals. 2019; 9(4):200. https://doi.org/10.3390/cryst9040200
Chicago/Turabian StyleWan, Lihua, Xuebing Zhou, Peili Chen, Xiaoya Zang, Deqing Liang, and Jinan Guan. 2019. "Decomposition Characterizations of Methane Hydrate Confined inside Nanoscale Pores of Silica Gel below 273.15 K" Crystals 9, no. 4: 200. https://doi.org/10.3390/cryst9040200
APA StyleWan, L., Zhou, X., Chen, P., Zang, X., Liang, D., & Guan, J. (2019). Decomposition Characterizations of Methane Hydrate Confined inside Nanoscale Pores of Silica Gel below 273.15 K. Crystals, 9(4), 200. https://doi.org/10.3390/cryst9040200