Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes

 ,
,  ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. IL-HCM Preparation
2.2. Protein Crystallization
2.3. X-ray Diffraction Data Collection and Analysis
2.4. Structural Comparisons
3. Results
3.1. Glucose Isomerase Crystallization by Using IL-HCMs
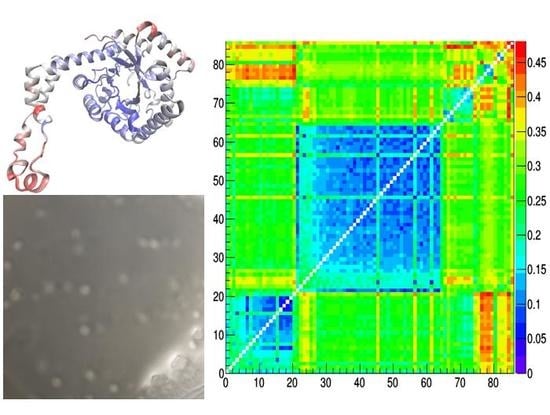
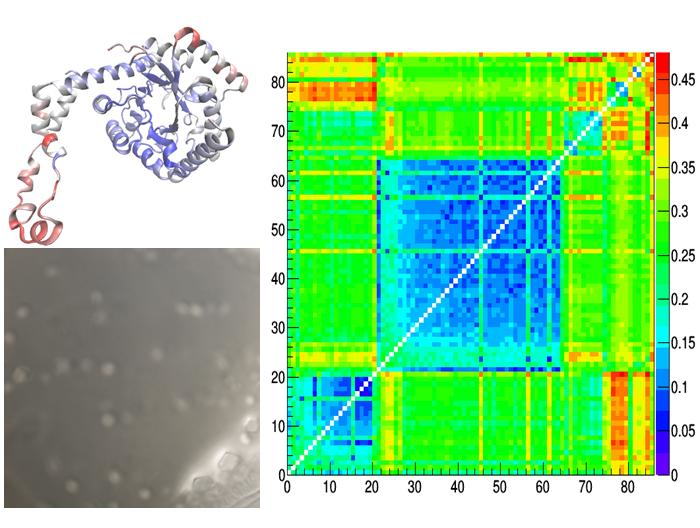
3.2. GI Crystallographic Analysis
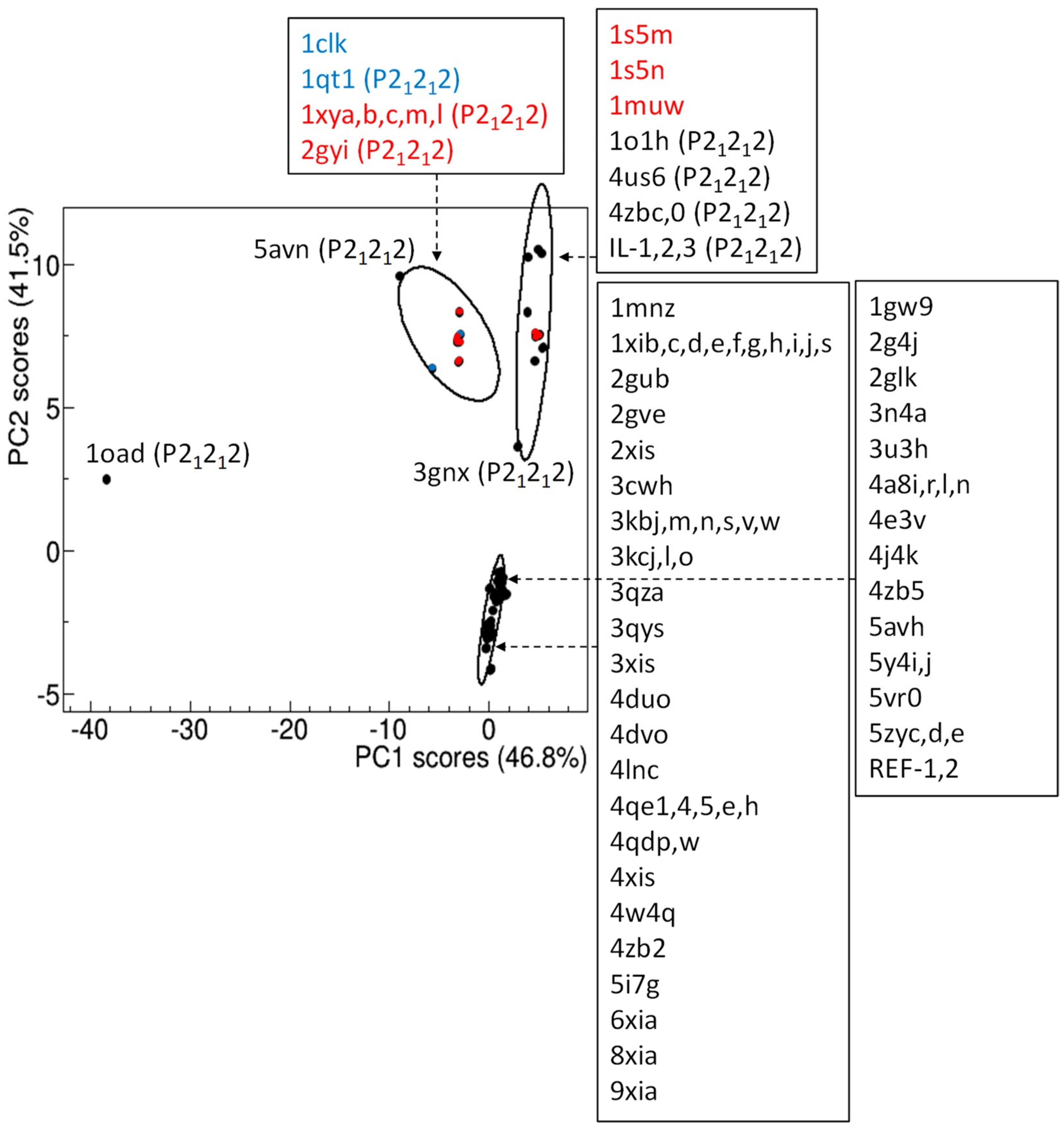
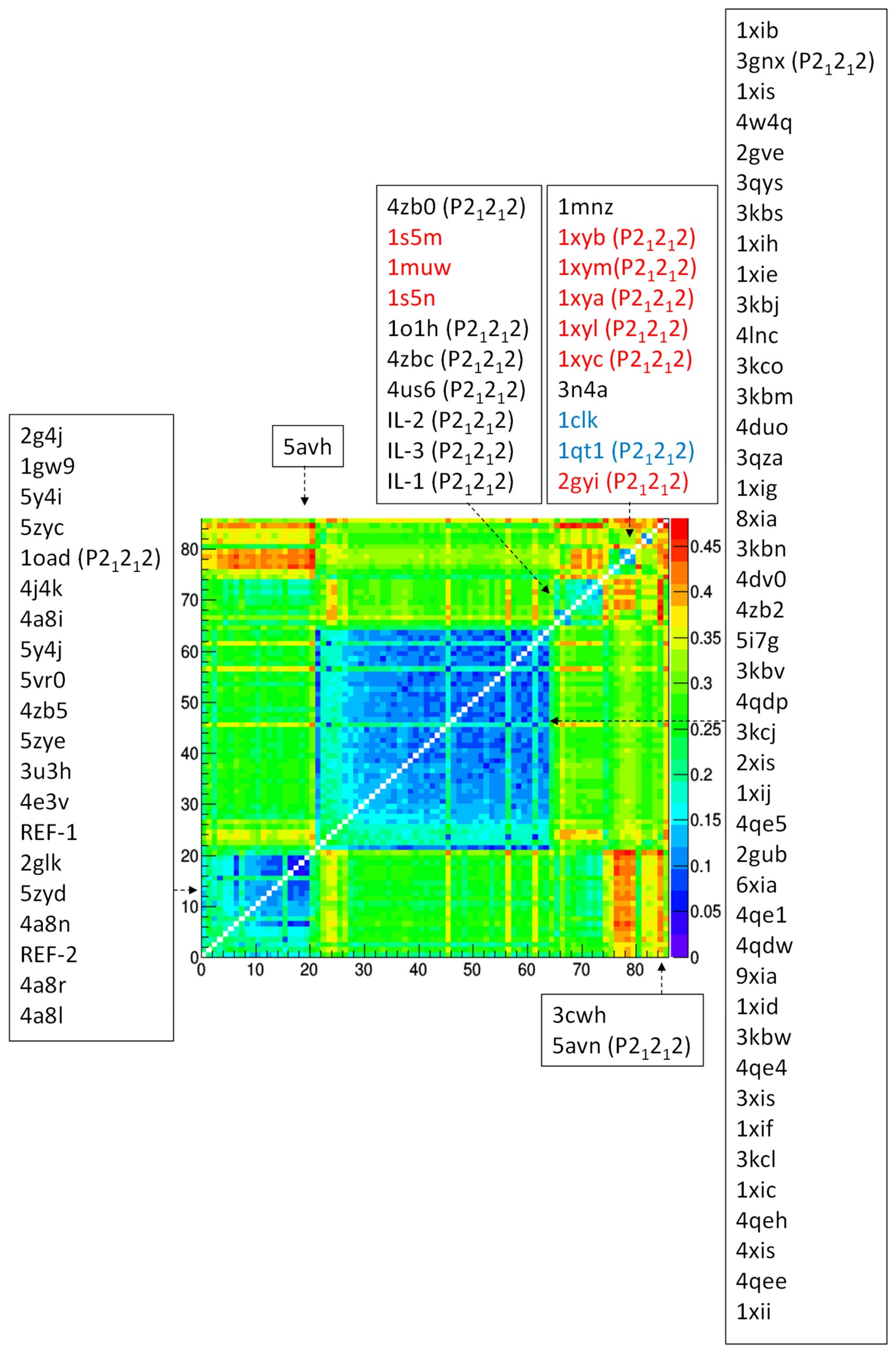
3.3. Comparison of Glucose Isomerase Structural Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pusey, M.L.; Paley, M.S.; Turner, M.B.; Robers, R.D. Protein Crystallization Using Room Temperature Ionic Liquids. Cryst. Growth Des. 2007, 7, 787–793. [Google Scholar] [CrossRef]
- Li, X.; Xu, X.; Dan, Y.; Feng, J.; Ge, L.; Zhang, M. The crystallization of lysozyme in the system of ionic liquid [BMIm][BF4]-water. Cryst. Res. Technol. 2008, 43, 1062–1068. [Google Scholar] [CrossRef]
- Hekmat, D.; Hebel, D.; Joswig, S.; Schmidt, M.; Weuster-Botz, D. Advanced protein crystallization using water-soluble ionic liquids as crystallization additives. Biotechnol Lett. 2007, 29, 1703–1711. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Wang, J. Ionic liquids in the assay of proteins. Anal. Methods 2010, 2, 1222–1226. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, W.; Li, Y.; Zhang, J.; Gu, Q. Korean, A new strategy for protein crystallization: Effect of ionic liquids on lysozyme crystallization and morphology. J. Chem. Eng. 2014, 31, 919–923. [Google Scholar]
- Kowacz, M.; Marchel, M.; Juknaitė, L.; Esperança, J.M.S.S.; Romão, M.J.; Carvalho, A.L.; Rebelo, L.P.N. Ionic-Liquid-Functionalized Mineral Particles for Protein Crystallization. Cryst. Growth Des. 2015, 15, 2994–3003. [Google Scholar] [CrossRef]
- Schroder, C. Proteins in ionic liquids: current status of experiments and simulations. Top. Curr. Chem (Z) 2017, 375–425. [Google Scholar]
- Judge, R.A.; Takahashi, S.; Longenecker, K.L.; Fry, E.H.; Abad-Zapatero, C.; Chiu, M.L. The Effect of Ionic Liquids on Protein Crystallization and X-ray Diffraction Resolution. Cryst. Growth Des. 2009, 9, 3463–3469. [Google Scholar] [CrossRef]
- Marr, P.C.; Marr, A.C. Ionic liquid gel materials: Applications in green and sustainable chemistry. Green Chem. 2016, 18, 105–128. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Li, J.; Li, Z.; He, P.; Liu, H.; Li, J. Highly active horseradish peroxidase immobilized in 1-butyl-3-methylimidazolium tetrafluoroborate room-temperature ionic liquid based sol–gel host materials. Chem. Comm. 2005, 13, 1778–1780. [Google Scholar] [CrossRef] [PubMed]
- Di Profio, G.; Polino, M.; Nicoletta, F.P.; Belviso, B.D.; Caliandro, R.; Fontananova, E.; De Filpo, G.; Curcio, E.; Drioli, E. Tailored Hydrogel Membranes for Efficient Protein Crystallization. Adv. Func. Mat. 2014, 24, 1582–1590. [Google Scholar] [CrossRef]
- Drioli, E.; Di Profio, G.; Curcio, E. Membrane-assisted crystallization technology. In Advances in Chemical and Process Engineering; Imperial College Press: London, UK, 2015; Volume 2. [Google Scholar]
- Mirabelli, V.; Salehi, S.M.; Angiolillo, L.; Belviso, B.D.; Conte, A.; Del Nobile, M.A.; Di Profio, G.; Caliandro, R. Enzyme Crystals and Hydrogel Composite Membranes as New Active Food Packaging Material. Glob. Chall. 2018, 2, 1700089. [Google Scholar] [CrossRef] [Green Version]
- Salehi, S.M.; Santagada, R.; De Pietra, S.; Fontananova, E.; Curcio, E.; Di Profio, G. Ionic Liquid Hydrogel Composite Membranes (IL-HCMs). ChemEngineering 2019, 3, 47. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gillialand, G.; Bhat, T.N.; Weissing, H.; Shindyalov, P.E.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. XDS. Acta Cryst. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mazzone, A.; Siliqi, D. Molecular replacement: The probabilistic approach of the program REMO09 and its applications. Acta Cryst. A 2009, 65, 512–527. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst. D 2008, 64, 61–69. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Bricogne, G.; Blanc, E.; Brandl, M.; Flensburg, C.; Keller, P.; Paciorek, W.; Roversi, P.; Sharff, A.; Smart, O.S.; Vonrhein, C.; et al. BUSTER version 2.X; Global Phasing Ltd.: Cambridge, UK, 2017. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Cryst. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Cryst. D 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Caliandro, R.; Belviso, B.D. RootProf: Software for multivariate analysis of unidimensional profiles. J. Appl. Cryst. 2014, 47, 1087–1096. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst. D 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulte, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sugiyama, S.; Maruyama, M.; Sazaki, G.; Hirose, M.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; More, Y.; Matsumura, H. Growth of Protein Crystals in Hydrogels Prevents Osmotic Shock. J. Am. Chem. Soc. 2012, 134, 5786–5789. [Google Scholar] [CrossRef]
- Salehi, S.M.; Manjua, A.C.; Belviso, B.D.; Portugal, C.A.M.; Coelhoso, I.M.; Mirabelli, V.; Fontananova, E.; Caliandro, R.; Crespo, J.G.; Curcio, E.; et al. Hydrogel Composite Membranes Incorporating Iron Oxide Nanoparticles as Topographical Designers for Controlled Heteronucleation of Proteins. Cryst. Growth Des. 2018, 18, 3317–3327. [Google Scholar] [CrossRef]
- Lorber, B.; Sauter, C.; Ng, J.D.; Zhu, D.W.; Giegé, R.; Vidal, O.; Robert, M.C.; Capelle, B. Characterization of protein and virus crystals by quasi-planar wave X-ray topography: A comparison between crystals grown in solution and in agarose gel. J. Cryst. Growth 1999, 204, 357–368. [Google Scholar] [CrossRef]
- Lorber, B.; Sauter, C.; Robert, M.C.; Capelle, B.; Giegé, R. Crystallization within agarose gel in microgravity improves the quality of thaumatin crystals. Acta Cryst. D 1999, 55, 1491–1494. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, J.M.; Gavira, J.A.; Otálora, F.; Guash, A.; Coll, M. Reinforced protein crystals. Mater. Res. Bull. 1998, 33, 1593–1598. [Google Scholar] [Green Version]
- Vuolanto, A.; Uotila, S.; Leisola, M.; Visuri, K. Solubility and crystallization of xylose isomerase from Streptomyces rubiginosus. J. Cryst. Growth. 2003, 257, 403–411. [Google Scholar] [CrossRef]
- Gillespie, C.M.; Asthagiti, D.; Lenhoff, A.M. Polymorphic Protein Crystal Growth: Influence of Hydration and Ions in Glucose Isomerase. Cryst. Growth Des. 2014, 14, 46–57. [Google Scholar] [CrossRef]
- Farber, G.K.; Petsko, G.A.; Ringe, D. The 3.0 Å crystal structure of xylose isomerase from Streptomyces olivochromogenes. Protein Eng. 1987, 1, 459–466. [Google Scholar] [CrossRef]
- Conejero-Muriel, M.; Gavira, J.A.; Pineda-Molina, E.; Belsom, A.; Bradley, M.; Moral, M.; Garcia-Lopez Durán, J.D.D.; Luque Gonzȧlez, A.; Diaz-Mochȯn, J.J.; Contreras-Montoya, R.; et al. Influence of the chirality of short peptide supramolecular hydrogels in protein crystallogenesis. Chem. Comm. 2015, 51, 3862–3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobley, C.M; Sandy, J.; Sanchez-Weatherby, J.; Mazzorana, M.; Krojer, T.; Nowak, R.P.; Sorensen, T.L. A generic protocol for protein crystal dehydration using the HC1b humidity controller. Acta Cryst. D 2016, 72, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Lavie, A.; Allen, K.N.; Petsko, G.A.; Ringe, D. X-ray Crystallographic Structures of D-Xylose Isomerase-Substrate Complexes Position the Substrate and Provide Evidence for Metal Movement during Catalysis. Biochem. 1994, 33, 5469–5480. [Google Scholar] [CrossRef]
- Nordwald, E.M.; Plaks, J.G.; Snell, J.R.; Scousa, M.C.; Kaar, J.L. Crystallographic Investigation of Imidazolium Ionic Liquid Effects on Enzyme Structure. ChembioChem 2015, 16, 2456–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ji, Y.; Wang, J. Improvement on the crystallization of lysozyme in the presence of hydrophilic ionic liquid. J. Analyst. 2010, 135, 2241–2248. [Google Scholar] [CrossRef]
- Kowacs, M.; Mukhopadhyay, A.; Carvalho, A.L.; Esperança, J.M.S.S.; Romão, M.J.; Rebelo, L.P.N. Hofmeister effects of ionic liquids in protein crystallization: Direct and water-mediated interactions. CrystEngComm 2012, 14, 4912–4921. [Google Scholar] [CrossRef]
- Zhu, X.; Teng, M.; Niu, L.; Xu, C.; Wang, Y. Structure of xylose isomerase from Streptomyces diastaticus No. 7 strain M1033 at 1.85 Å resolution. Acta Cryst D 2000, 56, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Ramagopal, U.A.; Dauter, M.; Dauter, Z. SAD manganese in two crystal forms of glucose isomerase. Acta Cryst. D 2003, 59, 868–875. [Google Scholar] [CrossRef]
- Nowak, E.; Pajikar, S.; Tucker, P.A. Structure of Glucose Isomerase Derivatized with Kr. 2002. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
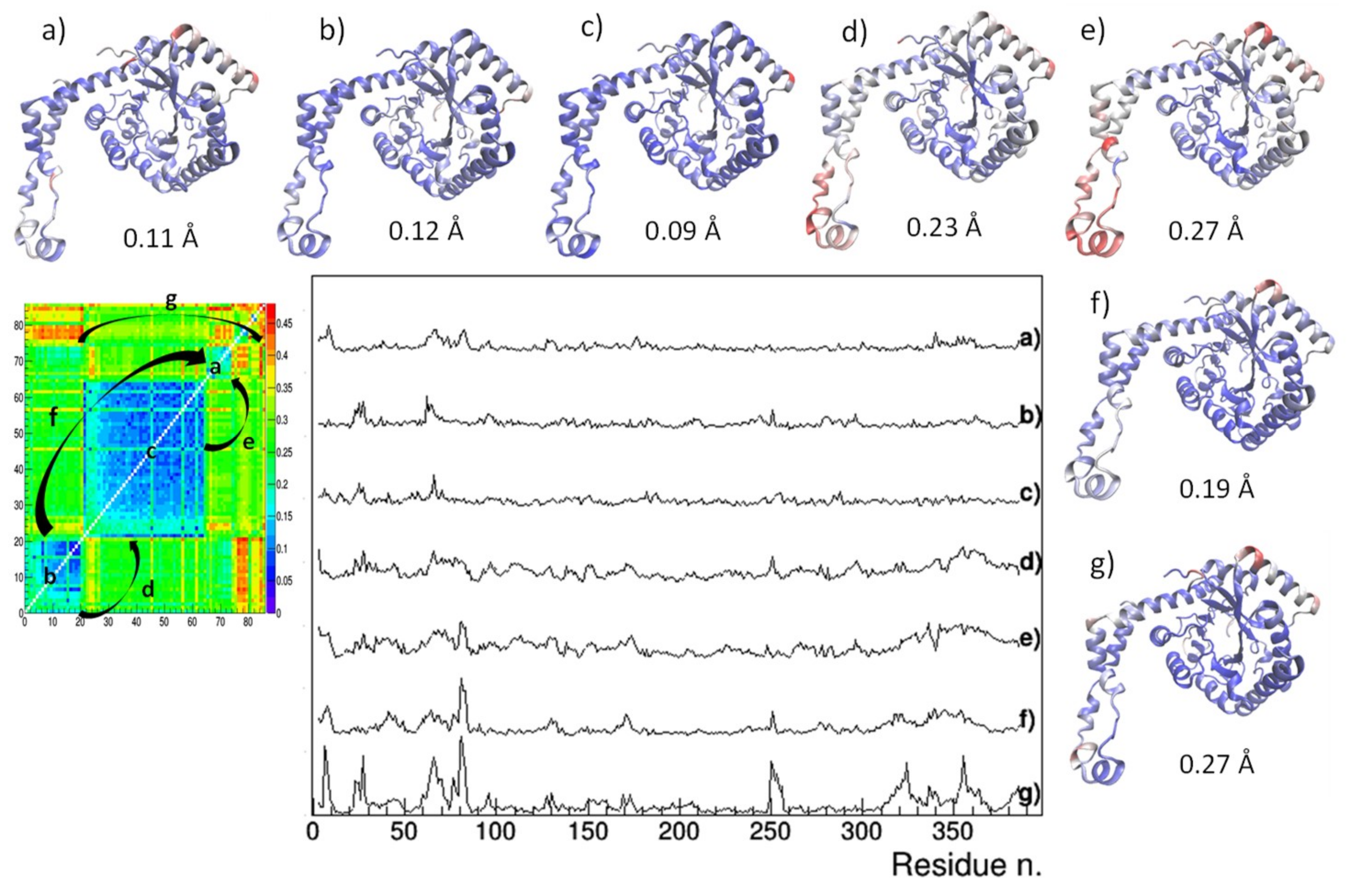{kind=link}
| IL-1 | IL-2 | IL-3 | REF-1 | REF-2 | |
|---|---|---|---|---|---|
| Resolution (Å) | 1.58 (1.62–1.58) | 1.77 (1.87–1.77) | 1.98 (2.01–1.98) | 1.19 (1.21–1.19) | 1.19 (1.22–1.19) |
| Space group | P21212 | P21212 | P21212 | I222 | I222 |
| Crystal Cell (Å) a | 85.90 | 81.62 | 81.75 | 92.52 | 92.52 |
| b | 92.60 | 94.92 | 93.35 | 99.12 | 98.17 |
| c | 99.11 | 97.47 | 97.85 | 102.48 | 101.85 |
| Rmerge (%) | 9.9 (66.4) | 9.1 (54.8) | 22.0 (122) | 11.7 (48.0) | 4.2 (16.6) |
| I/σ | 10.2 (3.4) | 9.2 (2.1) | 3.1 (0.9) | 5.3 (1.0) | 13.8 (2.6) |
| Completeness (%) | 95.6 (97.4) | 97.9 (99.6) | 99.7 (96.9) | 75.8 (2.9) | 76.7 (4.9) |
| Multiplicity | 4.2 (4.1) | 4.0 (4.2) | 3.9 (4.1) | 3.4 (1.0) | 3.4 (1.0) |
| Mosaicity (°) | 0.16 | 0.32 | 0.59 | 0.14 | 0.05 |
| B Wilson (Å2) | 9.60 | 19.17 | 22.12 | 9.65 | 8.59 |
| Rwork | 0.167 | 0.170 | 0.220 | 0.171 | 0.107 |
| Rfree | 0.194 | 0.205 | 0.271 | 0.188 | 0.129 |
| Number of water molecules | 929 | 902 | 491 | 584 | 538 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belviso, B.D.; Caliandro, R.; Salehi, S.M.; Di Profio, G.; Caliandro, R. Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes. Crystals 2019, 9, 253. https://doi.org/10.3390/cryst9050253
Belviso BD, Caliandro R, Salehi SM, Di Profio G, Caliandro R. Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes. Crystals. 2019; 9(5):253. https://doi.org/10.3390/cryst9050253
Chicago/Turabian StyleBelviso, Benny Danilo, Rosanna Caliandro, Shabnam Majidi Salehi, Gianluca Di Profio, and Rocco Caliandro. 2019. "Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes" Crystals 9, no. 5: 253. https://doi.org/10.3390/cryst9050253
APA StyleBelviso, B. D., Caliandro, R., Salehi, S. M., Di Profio, G., & Caliandro, R. (2019). Protein Crystallization in Ionic-Liquid Hydrogel Composite Membranes. Crystals, 9(5), 253. https://doi.org/10.3390/cryst9050253