This work includes STM experiments on molecular deuterium interaction with the surface of gold nanoparticles, and quantum chemical simulation of atomic deuterium adsorption onto the gold cluster in contact with two graphene nanocells modeling the edge of the HOPG terrace. In the theoretical portion, we consider the causes of hydrogen dissociative adsorption on the surface of gold nanoparticles and the influence of hydrogen chemisorption on the STM tunneling spectra.
3.1. Experimental Portion
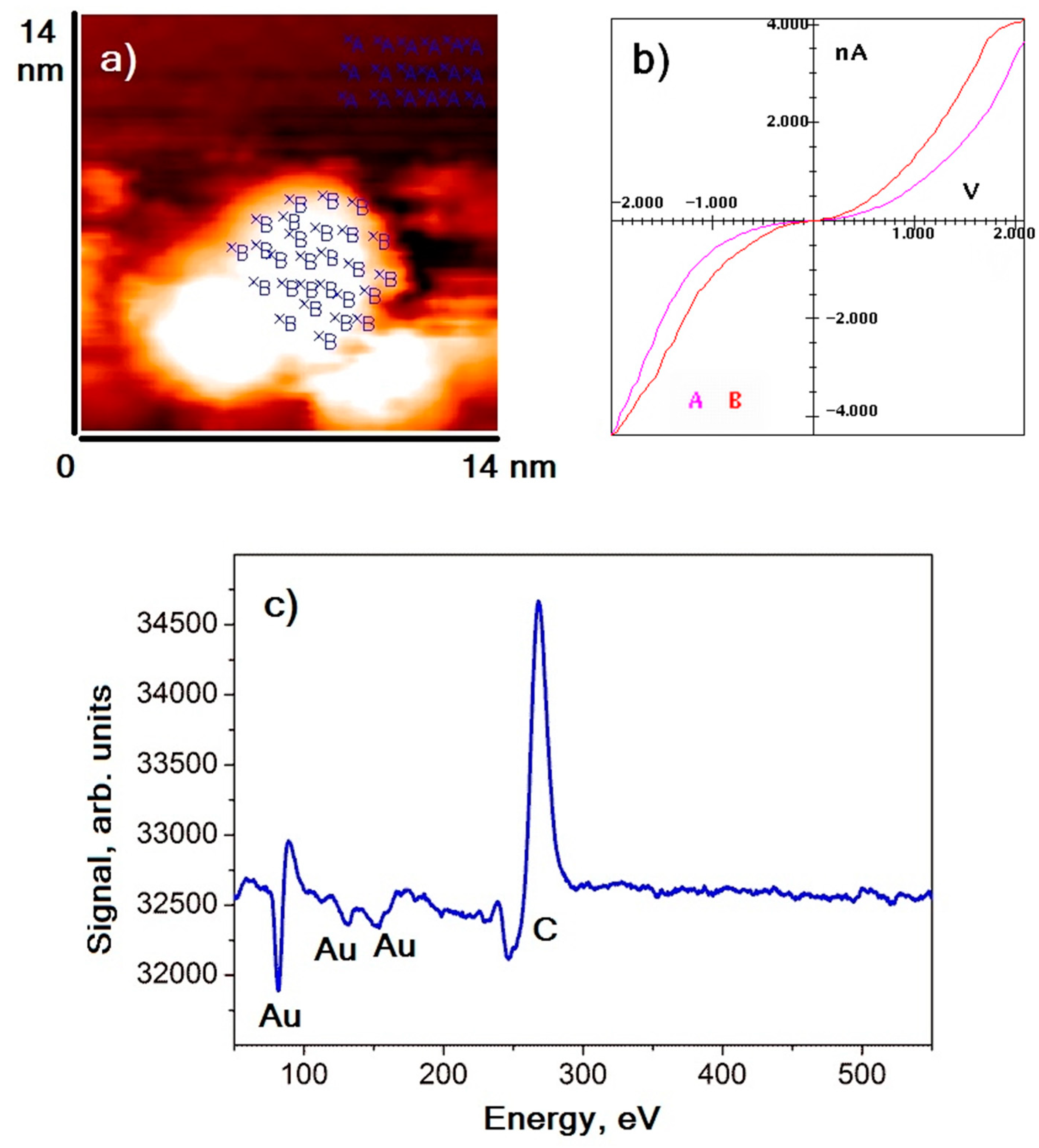
Gold nanoparticles were distributed on the surface of the HOPG in such a way that most of them were arranged on graphite surface defects (terrace edges). Nanoparticles had an oblate spheroidal shape. The maximum of the lateral size distribution was in the range of 4–8 nm, with an average height of approximately 1.5–2 nm. An example image of the HOPG surface area with gold nanoparticles deposited on it is shown in
Figure 1a.
Figure 1b,c demonstrate the averaged VAC of nanoparticles (dots B) and graphite (dots A) and the Auger electrons spectrum of the sample, respectively. The results of spectroscopic measurements revealed that VACs of nanoparticles and graphite were similar—they possessed an S-shape VAC similar to cases of tunnel contact formed by conductors. It could also be noted that the absolute value of the tunneling current measured on the nanoparticle in the entire voltage range was slightly higher than that of the tunneling current measured on graphite. That is, the local conductivity of nanoparticles was slightly higher than the conductivity of graphite. Such behavior occurred because the density of states in the vicinity of the Fermi level of metal nanoparticles was higher than that of the HOPG (semimetal). The absence of residual impurities on gold was confirmed by the elemental analysis of the sample surface: Auger spectra show maxima at energies of 272 eV and 69, 141, and 160 eV, corresponding to carbon and gold, respectively. Noticeable signals of other elements are not detected.
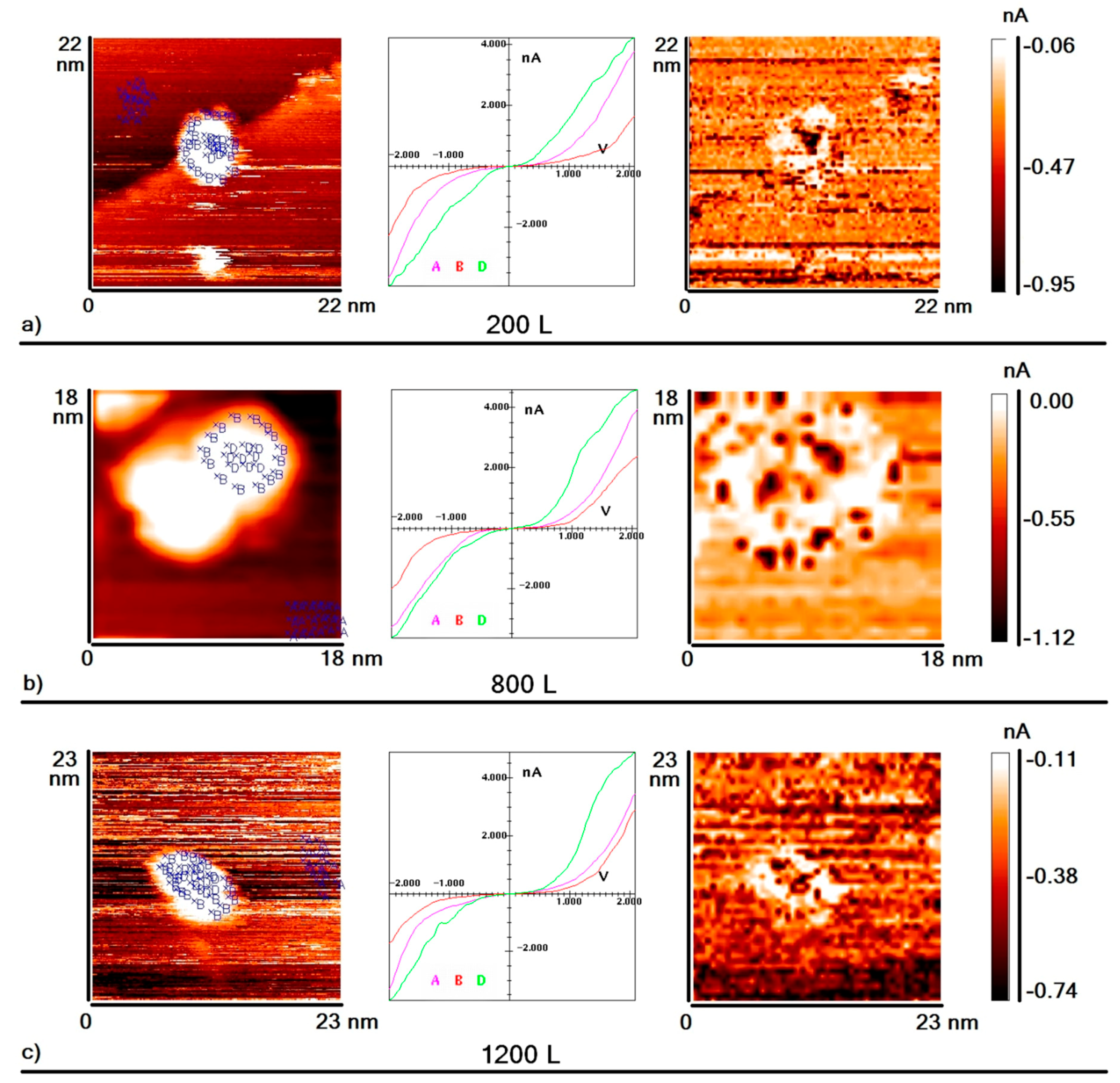
The exposure of the sample to deuterium resulted in a significant perturbation of the local electron density of nanoparticles. The change in the electronic structure of nanoparticles as a result of increased exposure to D
2 is shown in
Figure 2a–f. Each part in this figure consists of three elements:
(1) The first element is a topography image of the HOPG surface area with nanoparticles deposited on it. Blue crosses indicate points where VAC was measured;
(2) The second element shows the averaged VAC curves corresponding to the points indicated on the topography image (curve A: area of graphite without nanoparticles; curve B: peripheral area of nanoparticles; curve D: central area of nanoparticles);
(3) The last element is a “current” image of the same surface area, which is a map of the surface distribution of tunneling current values at a certain voltage value (darker points correspond to higher values of tunneling current). This type of image informs the local electron density of the sample surface in an explicit form.
As seen from the above data, after the exposure of the sample to deuterium equal to 200 L, an area appeared on the nanoparticles’ periphery in which the measured value of the tunneling current was lower than one measured on the graphite (hereafter this area is called the “area of reduced conductivity”). However, the value of the tunneling current measured in the center of nanoparticle was still bigger than the one measured on the graphite (
Figure 2a). That is, after exposure to deuterium, we could observe two different areas on the surface of the gold nanoparticle—the central area with high conductivity, and the peripheral one with low conductivity. In addition, the VAC measured at several individual points on the periphery of the nanoparticle had a zero-current section. Before the exposition of the sample to deuterium, the existence of such curves was not observed.
As we can see, further increasing the sample’s exposure to deuterium up to 800 L resulted in significant changes: the width of the reduced conductivity region at the periphery of the particle increased, as did the number of points where such VACs containing zero-current sections were detected (
Figure 2b). The central part of the nanoparticles remained an area of high conductivity. With an increase in exposure to deuterium up to 1200 L, some points in the central region of nanoparticles started to demonstrate VAC with a zero-current section. However, the average conductivity of the nanoparticle central region was still slightly higher than the conductivity of graphite (
Figure 2c). With further exposure to deuterium up to 1600 L, the width of the high conductivity area decreased, and the number of points with zero-current section within the VAC increased even further (
Figure 2d). Finally, when the exposure reached approximately 2000 L, it became impossible to identify areas with high and low conductivity on the surface of the nanoparticle. The fraction of points with VAC containing a zero-current section was about one-third of the total number of points where VAC measurements were carried out. So, after the exposure to 2000 L, the surface of nanoparticles became homogeneous, and VACs with and without zero-current sections became almost randomly distributed over the entire surface of the nanoparticle. Formal averaging of VACs measured at the center of the particle and at its periphery demonstrates that conductivity of both the central and peripheral regions of the particle was lower than that of the graphite (
Figure 2e). Note that this effect was observed both for isolated nanoparticles and for nanoparticles agglomerated in clusters. A further increase in exposure up to 3000 L did not lead to noticeable changes in the form of VAC curves measured on the surfaces of nanoparticles (
Figure 2f).
In our previous work [
22], it was shown that adsorption of atomic hydrogen on the surface of free gold nanoclusters results in a decrease in the density of states in the vicinity of the Fermi level, leading to a decrease in the conductivity of the gold nanocluster. Taking into account the results of this work and the experimental data described above, one can conclude that dissociative adsorption of deuterium on the surface of gold nanoparticles takes place and that the filling of nanoparticles with adsorbate begins mainly with the gold–graphite interface, followed by the filling of the rest of the nanoparticle surface. It should be noted that the time of transition of the system to the final (stationary) state is very long (up to 25–30 h). This long transition time indicates the presence of significant activation barriers both for the acts of surface migration of deuterium atoms and for the processes of structural rearrangement of nanoparticles.
3.2. Theoretical Portion and Quantum Chemical Simulation
In this section we consider why gold nanoparticles, unlike monocrystals, are able to adsorb hydrogen, and why hydrogen adsorption significantly affects the tunnel spectra of STM. Our considerations are built on the basis of model concepts that allow an analytical solution.
Hydrogen adsorption on the surface of metals, and gold in particular, has been studied many times. According to the Newns-Anderson model, one can describe the surface as a set of single-electron states with energy
εk, and the adsorbate interacting with it as a single valence state with energy
εa. Therefore, when the adsorbate approaches the surface, the two sets of states will relate through the interaction matrix with elements
Vak, and the adsorbate density of states will be
where
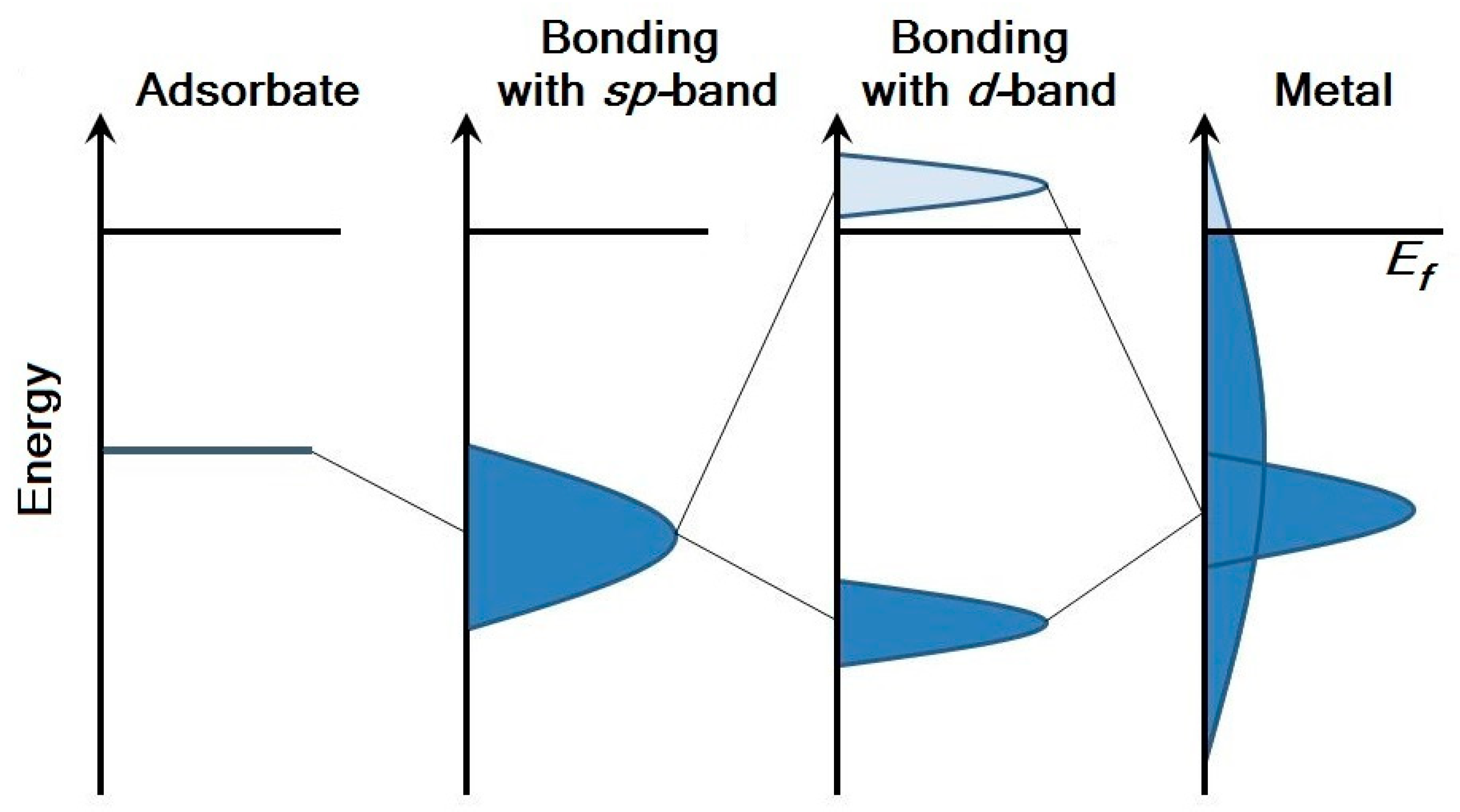
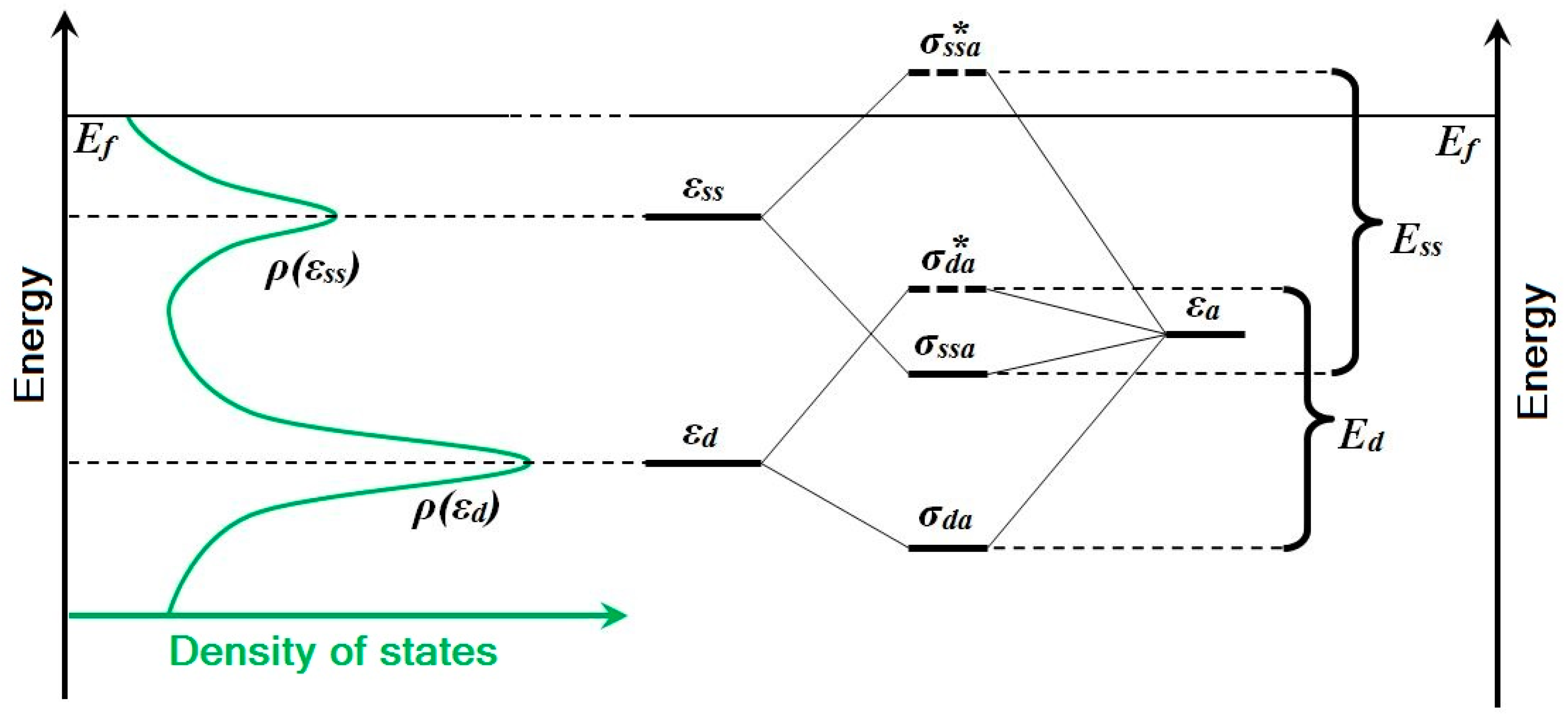
As can be seen, Δ(ε) is a local projection of the density of states of the metal near the adsorbate. If we consider the valence shells formed by the sp- and d-states in the case of noble and transition metals — the most interesting case for catalysis — then we can simplify the dependence Δ(ε) with the next assumption—Δ(ε) does not depend on energy for the sp-band, and the width of Δ(ε) is small for the d-band.
Thus, two stages can be distinguished in the process of atom/molecule adsorption on the metal surface: the interaction of the adsorbate with the
sp-states of the metal in the weak-chemisorption approximation, and the interaction of the adsorbate with the
d-states of the metal in the strong-chemisorption approximation. Weak chemisorption leads to broadening of the density of adsorbate states. Bonding and antibonding states are formed below and above the
d-band and the level of adsorbate as a result of strong chemisorption (
Figure 3).
The interactions of the adsorbate with the
sp-band (with the surface states) differ little for various noble and transition metals. Therefore, the metal
d-band—mainly the location of its center—plays the key role in determining the adsorption properties. For example, for Pt-group transition metals, the
d-band is not completely occupied and is located in the vicinity of the Fermi level; therefore, in the case of adsorption, the anti-bonding state will be above the Fermi level, providing remarkable chemisorption properties of the surface. However, the
d-band of noble metals (copper, silver and gold) is below the Fermi level and is completely occupied. Therefore, in the case of adsorption, the antibonding state will be below the Fermi level too, and the surface will be more inert (
Figure 4a). In addition, within the subgroup of noble metals, Pauli’s repulsion is maximal for gold, which is the most inert and the “noblest of metals” due to this fact [
28]. The binding energy of the hydrogen atom on the surface of a gold monocrystal is less than half of the dissociation energy of a hydrogen molecule. As a result, the layer of adsorbed H atoms is unstable.
However, experimentation on hydrogen interaction with gold nanoparticles given in this paper and other works [
20,
29,
30,
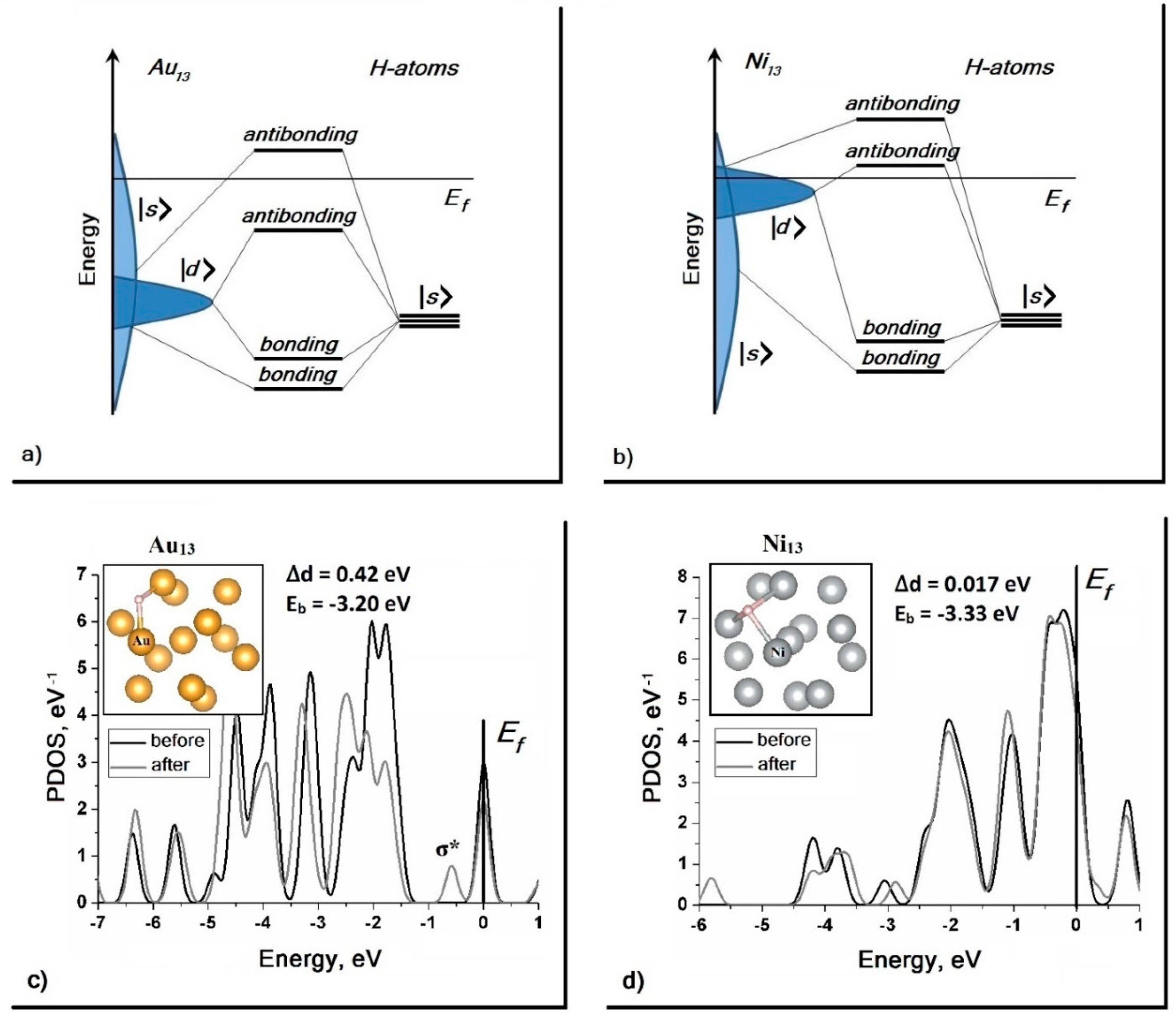
31] demonstrates the existence of a fairly stable form of hydrogen adsorption. How can these results be explained? PDOS calculations for clusters of noble Au
13 and transitional Ni
13 have shown that the conclusions drawn from a simple model of surface adsorption are also valid in cases of limited systems. The
d-band of the Au
13 cluster is located below the Fermi level, and after adsorption of hydrogen its center is shifted down, and the antibonding state
σ* is formed (
Figure 4c). In the case of Ni
13 cluster — cluster of transition metal — the
d-band is in the vicinity of the Fermi level, and the
d-band shift is much smaller; therefore, no formation of the occupied antibonding state
σ* takes place (
Figure 4d).
However, in the case of gold nanoparticles in comparison with the bulk, the contribution of surface states to the whole binding energy
Eads increases with size reduction of nanoparticle—the well-known “size effect”:
where
ρ(εd) and
ρ(εss) are the peak amplitudes of the density of
d-states and surface states, and
Ed and
Ess are corresponding interaction energies. The
ρ(εd)/ρ(εss) ratio is negligible for the surface of bulk gold, but it increases rapidly with nanoparticle size reduction. Also note that
Ess >
Ed. These changes are due to the location of the “band” of the surface-states. For noble metals, the peak
ρ(εss) is ~0.5 eV lower than the Fermi level. In terms of two-state formalism, this means that the antibonding state of an Au-H “surface molecule” is unoccupied and does not compensate for the bonding-state contribution to the binding energy, in contrast with interaction just with the
d-band states. This concept is shown schematically in
Figure 5. Thus, it is due to the interaction with surface states of gold nanoparticles that the binding energy of the H adatom increases and provides existence of stable adsorption forms observed in experiments.
We now consider how the conductivity of STM tunneling contact is affected by hydrogen adsorption on the surface of gold nanoparticles. At
kT >>
eU, where
U is bias voltage, the Fermi distribution
can be given as a Heaviside function and the tunneling current can be written as
where
ρtip(ε) is density of states of the probe,
ρk(ε) is local (projected on the k
th atom) density of states of the sample, and
Mk is the matrix element of wave function overlaps for the probe and the k
th atom of the sample. To more clearly establish the dependence we are interested in, we assume that
ρtip(ε) has no singularities in the vicinity of the Fermi level as it is usually observed in experiments.
ρk(ε) and
Mk can be approximated by values averaged over the region of tunneling contact:
The tunneling current is now determined only by the density of states of the sample:
At low-bias voltages, the decisive contribution to binding energy is made by the interaction of the H-adatom state with surface states of the gold nanoparticle. Note that the peak of surface states decreases as hydrogen atoms fill the nanoparticle surface—adsorption is known to influence the distribution of surface state densities [
32]. Therefore, it is due to the reduction of density of states in the vicinity of the Fermi level that we can detect the acts of hydrogen (deuterium) interaction (dissociative adsorption) with gold nanoparticles in STM-STS experiments.
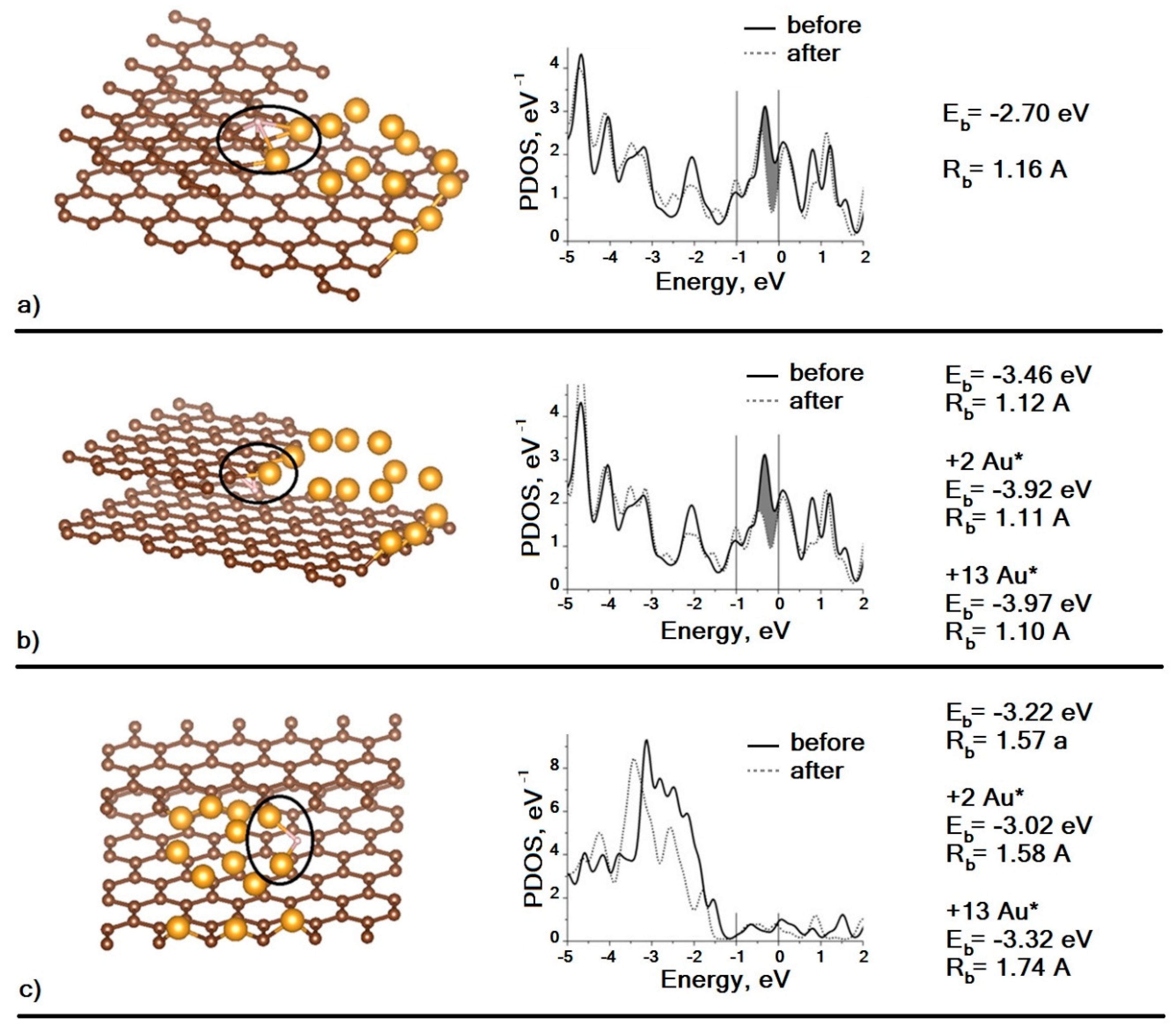
The density of states of individual atoms can also be calculated using DFT modeling and compared with the VAC measured in STM experiments, and we have performed this for the abovementioned supercluster Au
13C
138 which is a model of a gold nanoparticle at the edge of the graphite terrace. Summation over a set of projectors simulates a situation typical for STM experiments on metal clusters when the curvature radius of the probe is much less than the curvature radius of the cluster and when the tunneling current is formed by several atoms. In these cases, matrix elements of the wave function of the probe and the cluster atoms are assumed to be the same for simplicity. The results are shown in
Figure 6.
As can be seen, in the case of D atom adsorption over the Au–C interface, the density of surface states decreases (
Figure 6a). In the case of adsorption under the interface, a decrease in the density of surface states is also observed, and the binding energy of the adatom with the supercluster is higher than in the previous case (
Figure 6b). For adsorption under the interface, the effect of the Au atoms’ mobility on the value of the binding energy with the D atom was also investigated. As shown by calculations, if we allow the two nearest Au atoms to move, the binding energy will increase by 0.46 eV (+2Au*), and if we remove the “freezing” from degrees of freedom for all Au atoms, the binding energy will increase by only 0.05 eV (+13Au*). Thus, it is possible to describe the interaction of D with a supercluster as local, since the perturbation of the atomic and electronic structure occurs on the atoms closest to D. In
Figure 6c, one can see that the adsorption of the D atom at the top of the Au
13 cluster does not lead to any noticeable decrease in the density of surface states. The binding energy of the D atom with the supercluster is calculated for three similar cases. Note that the difference in the adsorption energy at the interface and at the top is small (i.e., the surface migration of D adatoms is constrained because of a significant activation barrier). Existence of this barrier was confirmed experimentally in our previous work [
20], where we described strong bonding between hydrogen and gold nanoparticles on the surface of graphite and the long transition time (up to 25–30 h) of the system to the final (stationary) state. Of course, the real values of binding energy are likely to be significantly lower than the calculated ones, which is at least partly due to the absence of the correction of basis set superposition error, but such comparison of calculated values is valid.
Thus, the results of quantum chemical calculations correlate with results of the above experiment, which together demonstrate that at small exposures of HOPG-supported gold nanoparticles to deuterium, adsorption occurs predominantly on the periphery of gold nanoparticles close to the gold–carbon interface. In addition, our calculations prove the conclusions of [
20,
21], where hydrogen adsorption on HOPG-supported gold nanoparticles was shown to decrease local conductivity of tunnel nanocontact.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






