Block Copolymer Elastomer with Graphite Filler: Effect of Processing Conditions and Silane Coupling Agent on the Composite Properties

 ,
,  and
and 
Abstract
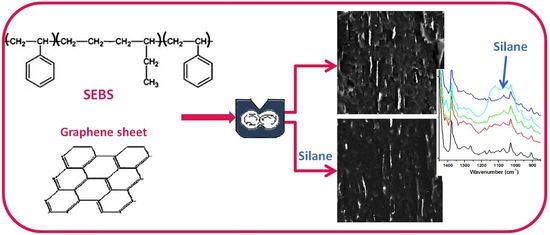
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Composites
2.3. Characterization
3. Results and Discussion
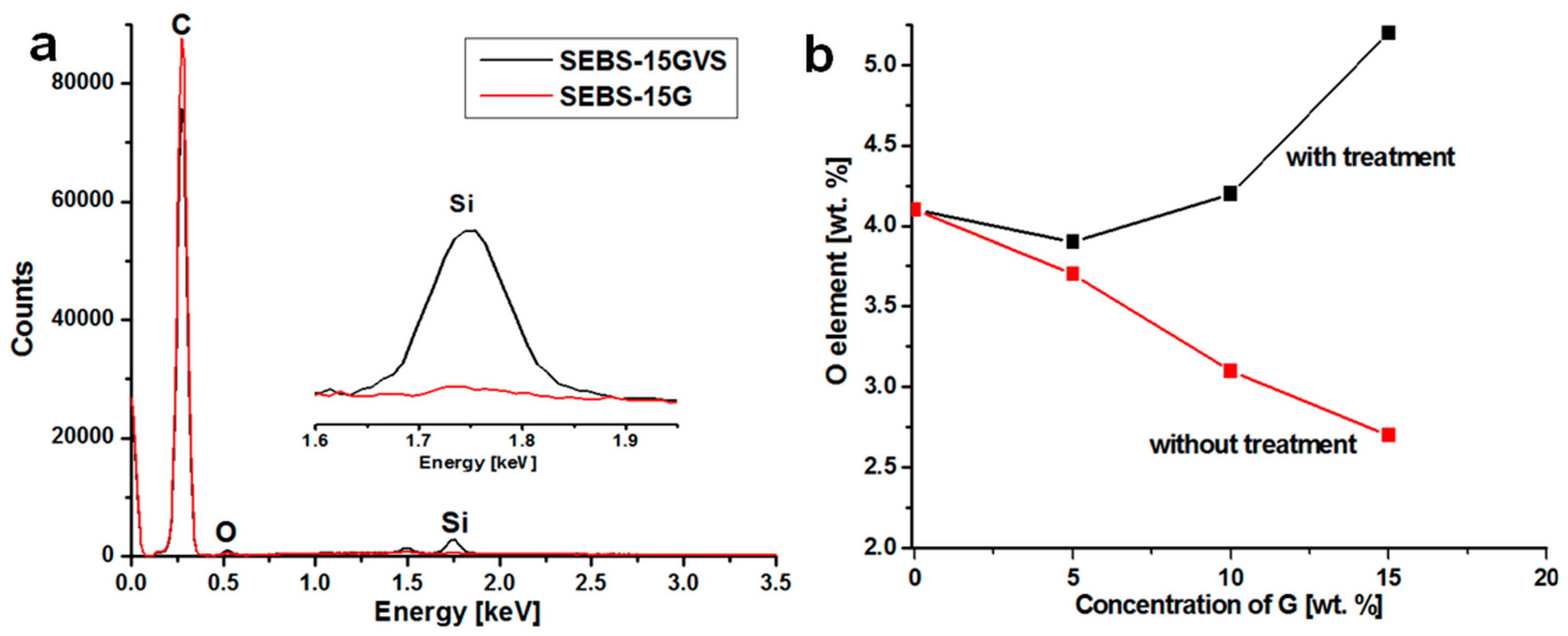
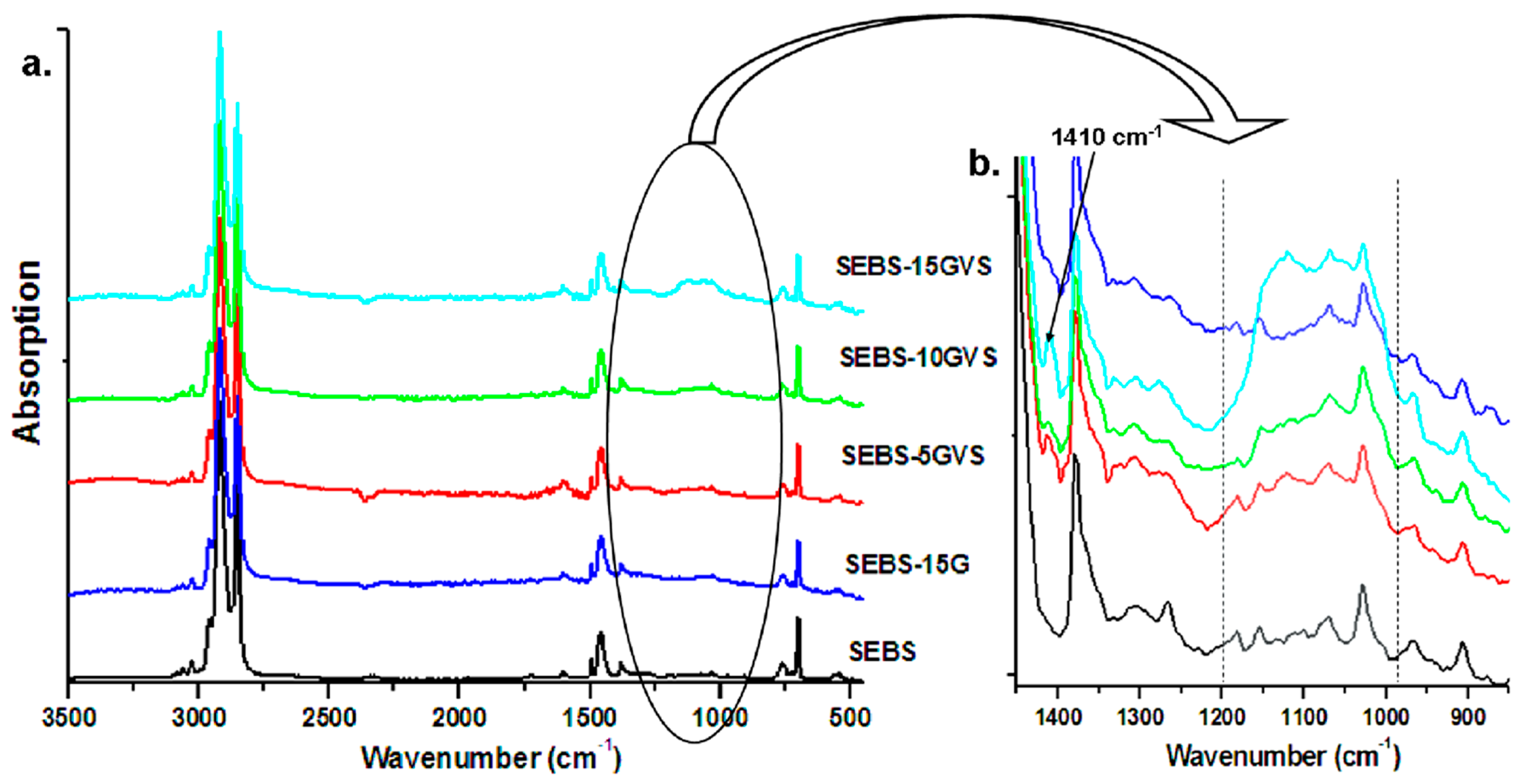
3.1. The Effect of Silane on SEBS-G Composites
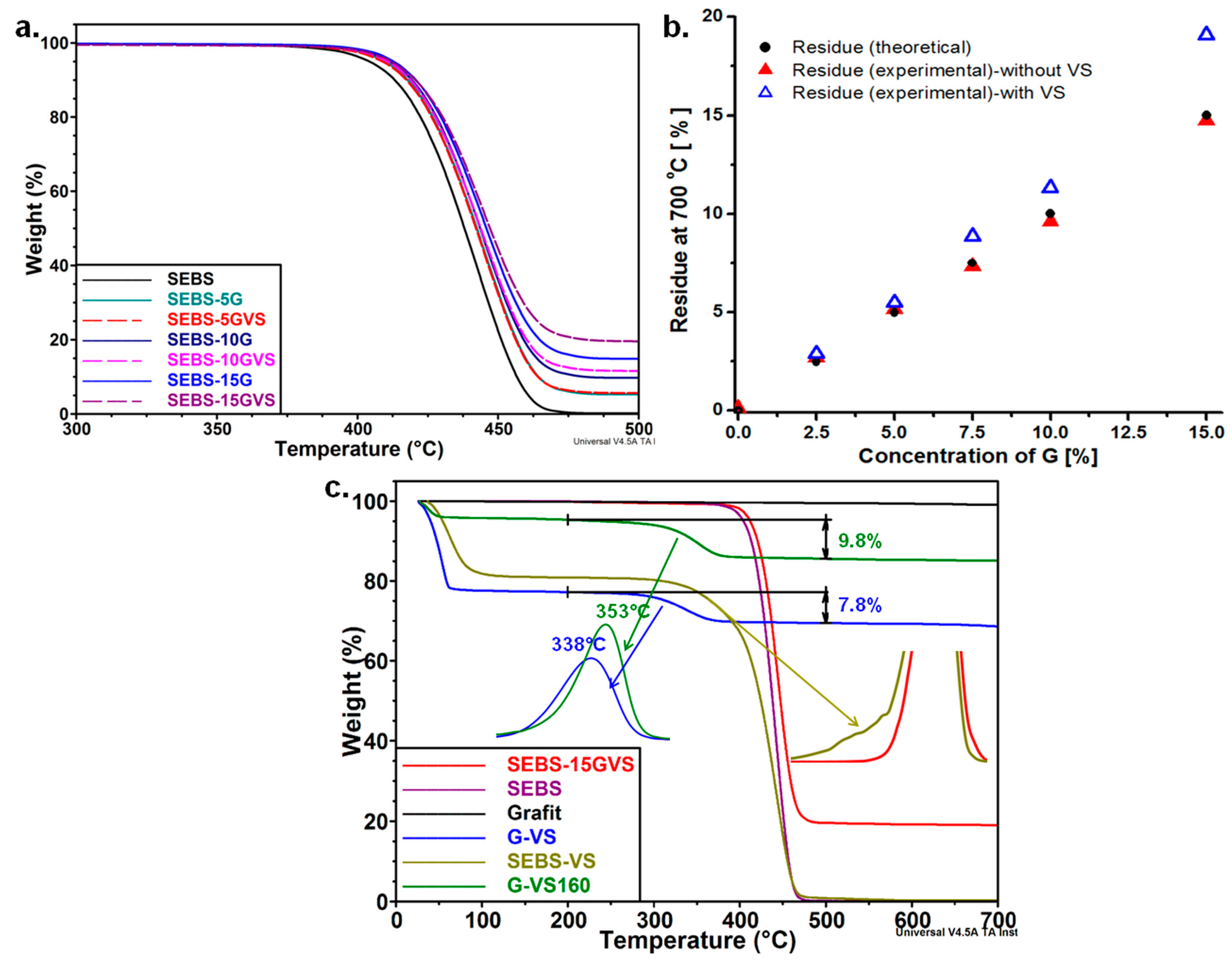
3.2. The Effect of G and VS on the Thermal Behavior of Composites
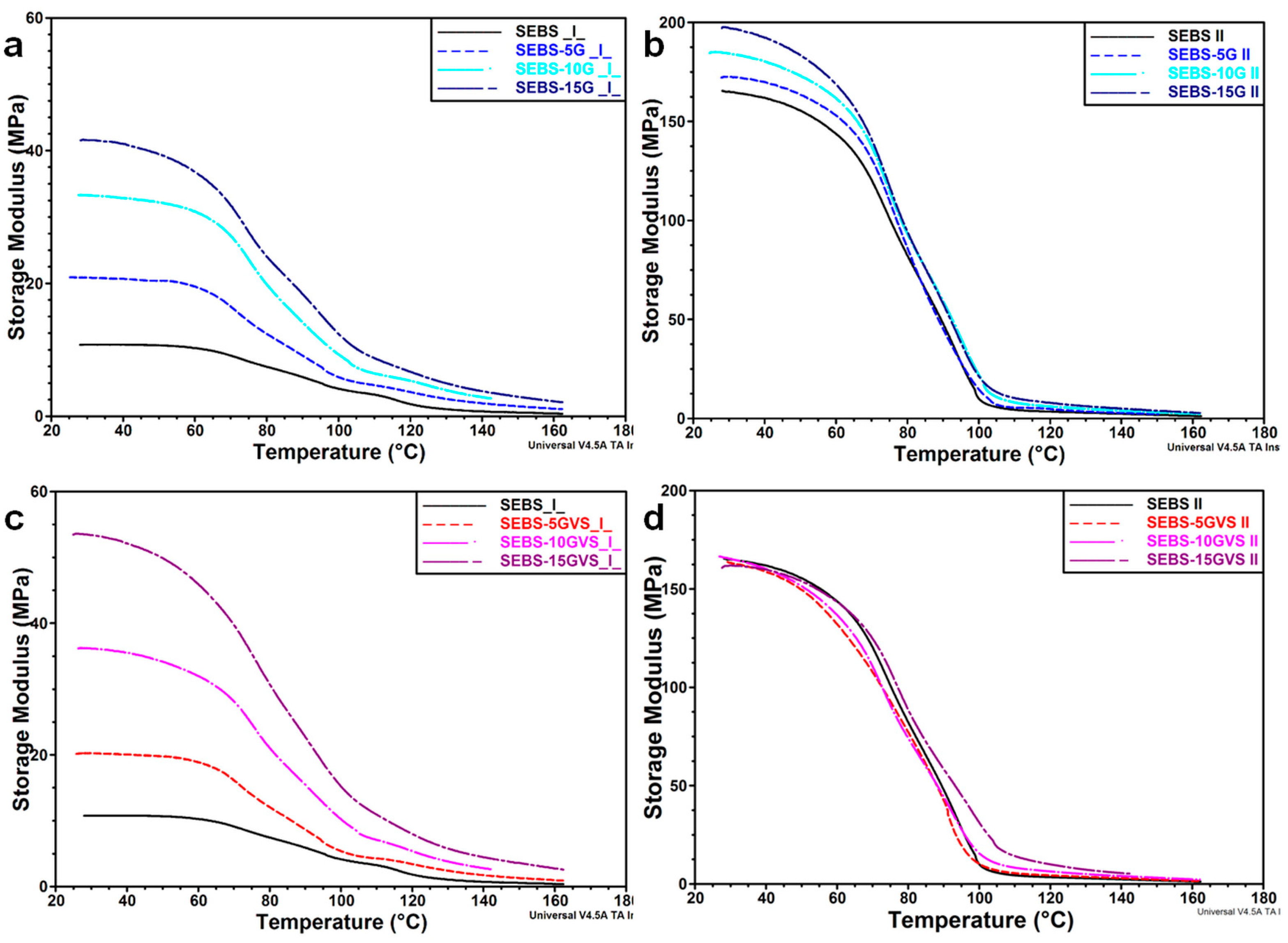
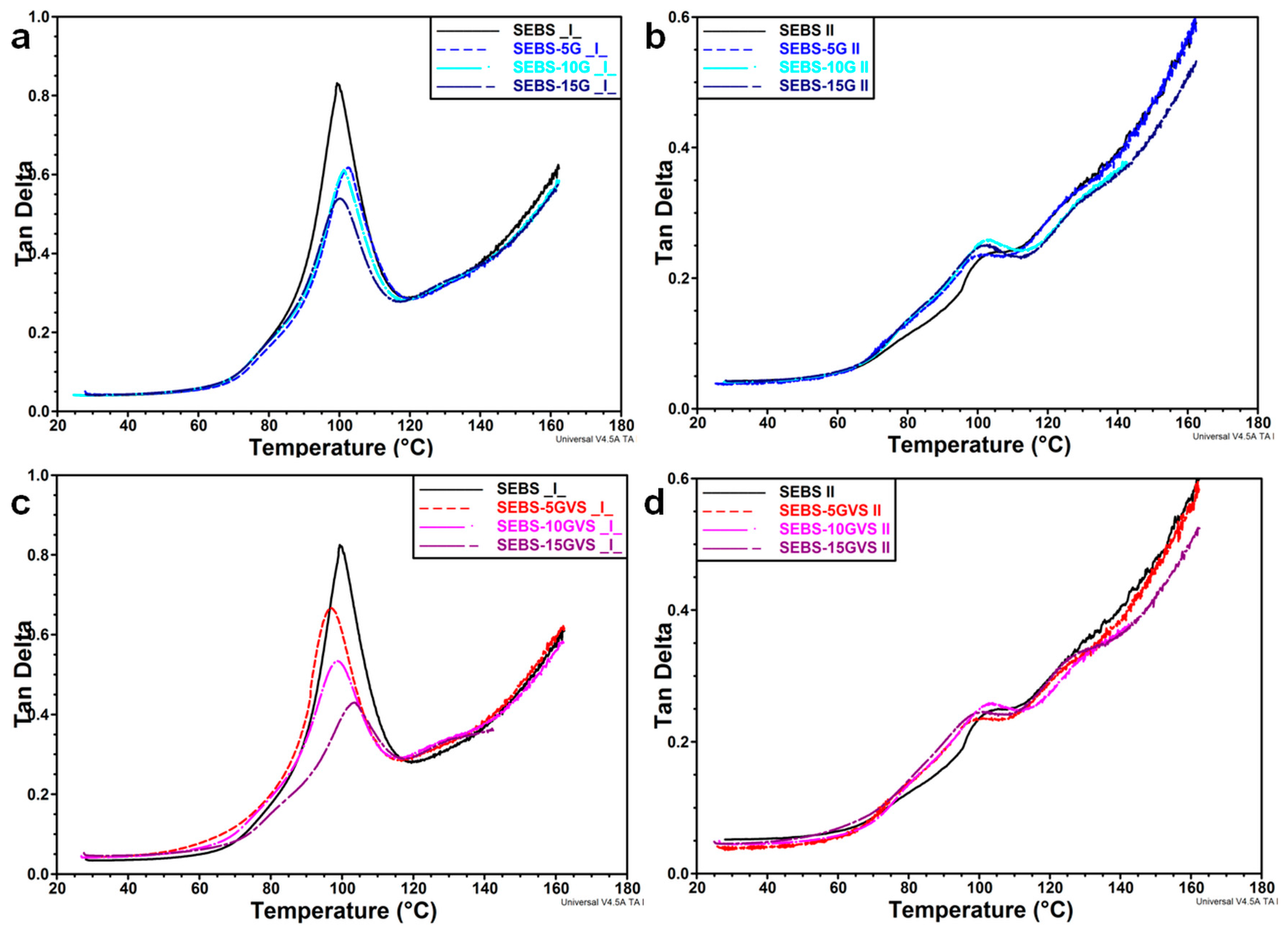
3.3. Mechanical Behavior by DMA Analysis
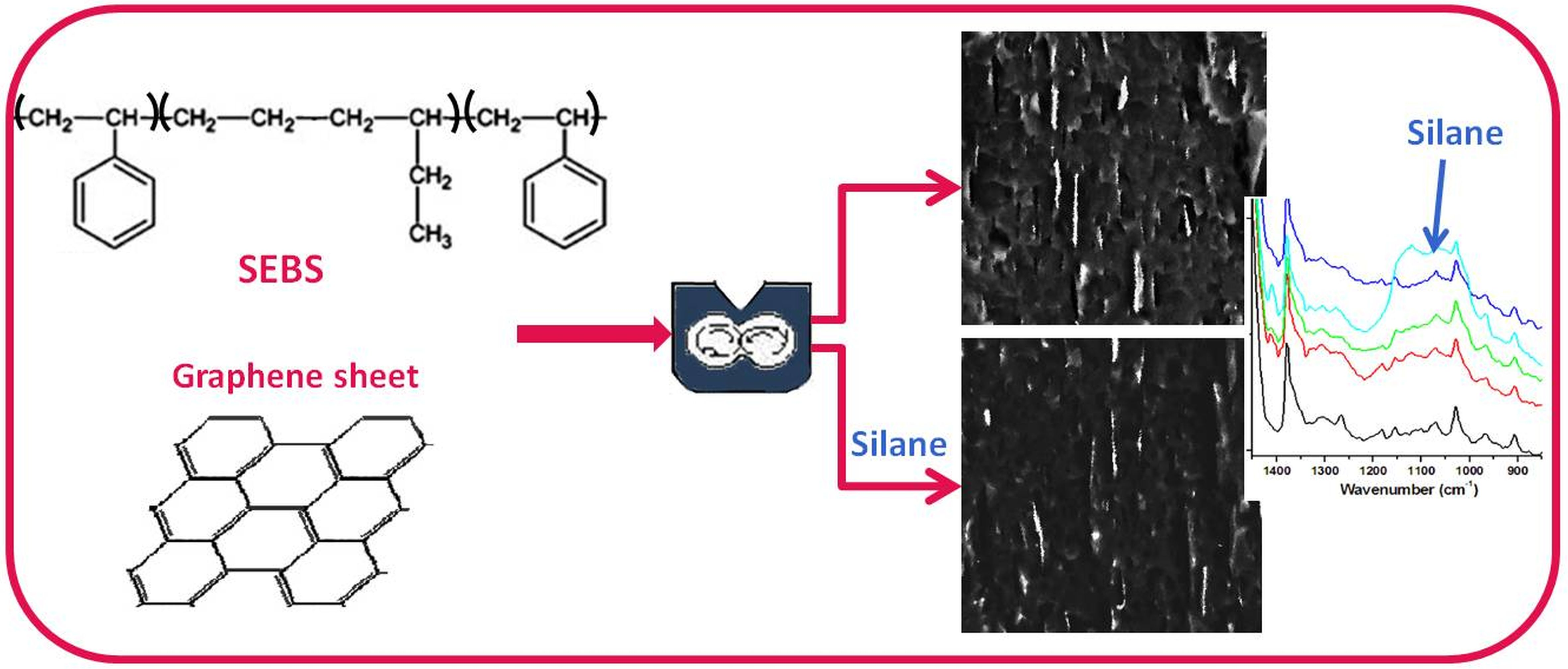
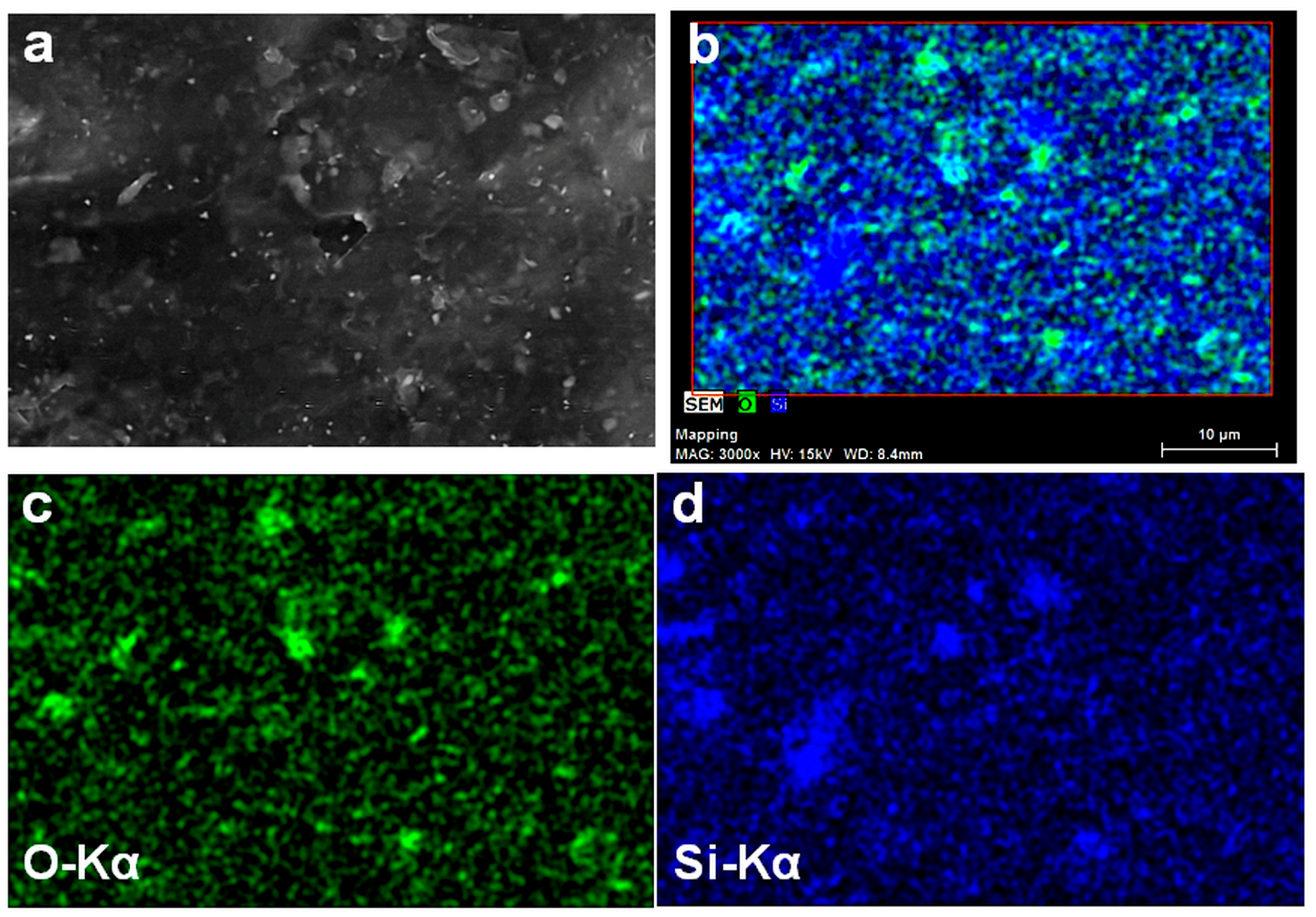
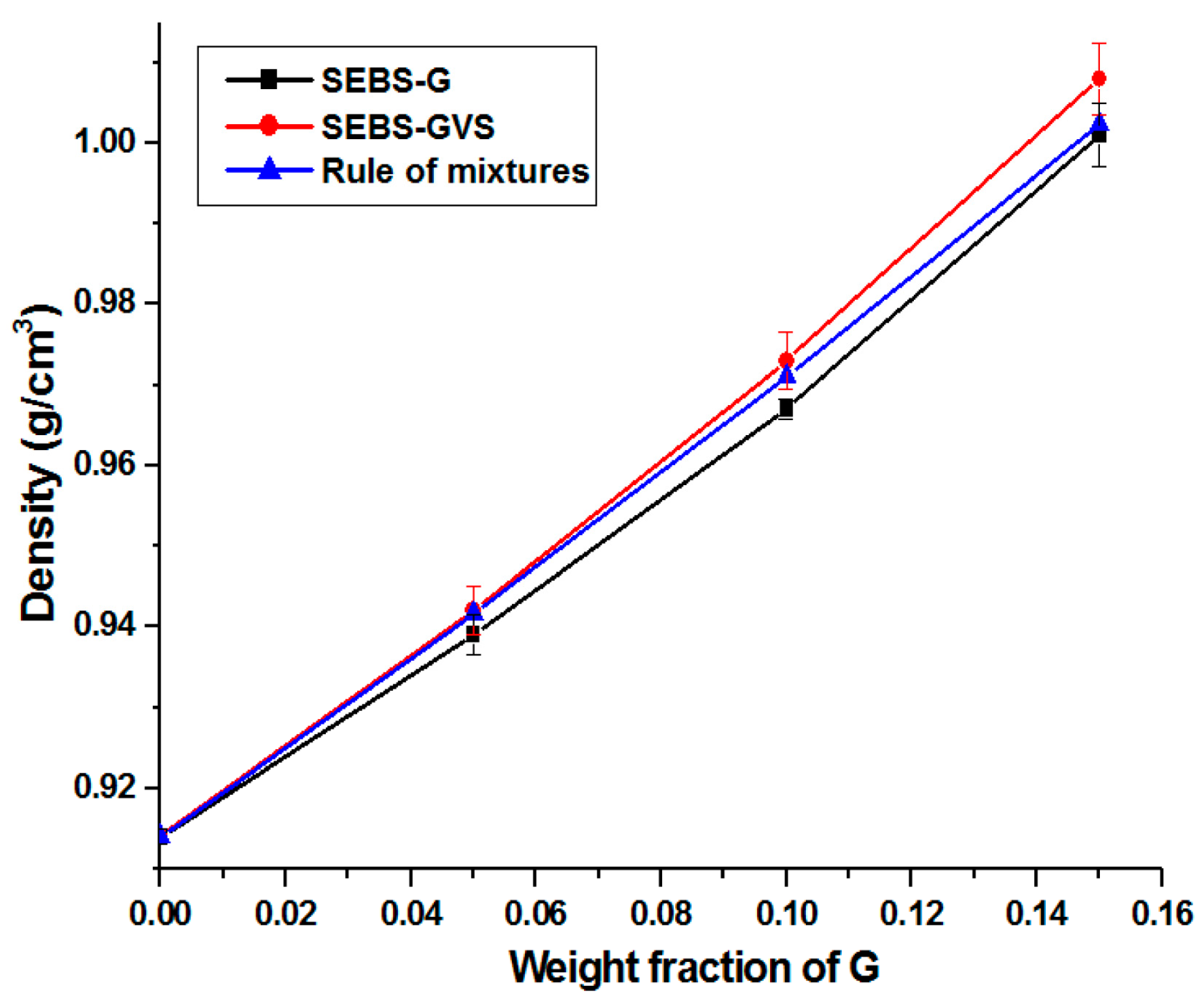
3.4. Density and Morphology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sadasivuni, K.K.; Ponnamma, D.; Thomas, S.; Grohens, Y. Evolution from graphite to graphene elastomer composites. Prog. Polym. Sci. 2014, 39, 749–780. [Google Scholar] [CrossRef]
- Zhang, L.; Jiao, H.; Jiu, H.; Chang, J.; Zhang, S.; Zhao, Y. Thermal, mechanical and electrical properties of polyurethane/(3-aminopropyl) triethoxysilane functionalized graphene/epoxy resin interpenetrating shape memory polymer composites. Compos. Part A Appl. Sci. Manuf. 2016, 90, 286–295. [Google Scholar] [CrossRef]
- Rath, T.; Li, Y. Nanocomposites based on polystyrene-b-poly(ethylene-r-butylene)-b-polystyrene and exfoliated graphite nanoplates: Effect of nanoplatelet loading on morphology and mechanical properties. Compos. Part A Appl. Sci. Manuf. 2011, 42, 1995–2002. [Google Scholar] [CrossRef]
- Ramezanzadeh, B.; Ghasemi, E.; Mahdavian, M.; Changizi, E.; Mohamadzadeh Moghadam, M.H. Characterization of covalently-grafted polyisocyanate chains onto graphene oxide for polyurethane composites with improved mechanical properties. Chem. Eng. J. 2015, 281, 869–883. [Google Scholar] [CrossRef]
- Galimberti, M.; Kumar, V.; Coombs, M.; Cipolletti, V.; Agnelli, S.; Pandini, S.; Conzatti, L. Filler networking of a nanographite with a high shape anisotropyand synergism with carbon black in poly(1,4-cisisoprene)-based nanocomposites. Rubber Chem. Technol. 2014, 87, 197–218. [Google Scholar] [CrossRef]
- Chattopadhyay, P.K.; Chattopadhyay, S.; Das, N.C.; Bandyopadhyay, P.P. Impact of carbon black substitution with nanoclay on microstructure and tribological properties of ternary elastomeric composites. Mater. Des. 2011, 32, 4696–4704. [Google Scholar] [CrossRef]
- Zhao, W.; Fang, M.; Wu, F.; Wu, H.; Wang, L.; Chen, G. Preparation of graphene by exfoliation of graphite using wet ball milling. J. Mater. Chem. 2010, 20, 5817–5819. [Google Scholar] [CrossRef]
- Leon, V.; Rodriguez, A.M.; Prieto, P.; Prato, M.; Vazquez, E. Exfoliation of graphite with triazine derivatives under ball-milling conditions: Preparation of few-layer graphene via selective noncovalent interactions. ACS Nano 2014, 8, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Grigorescu, R.M.; Ciuprina, F.; Ghioca, P.; Ghiurea, M.; Iancu, L.; Spurcaciu, B.; Panaitescu, D.M. Mechanical and dielectric properties of SEBS modified by graphite inclusion and composite interface. J. Phys. Chem. Solids 2016, 89, 97–106. [Google Scholar] [CrossRef]
- Rungswang, W.; Kotaki, M.; Shimojima, T.; Kimura, G.; Sakurai, S.; Chirachanchai, S. Existence of microdomain orientation in thermoplastic elastomer through a case study of SEBS electrospun fibers. Polymer 2011, 52, 844–853. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Gabor, R.A.; Nicolae, C.A.; Ghiurea, M.; Mihailescu, M.; Grigorescu, R.M. Influence of melt processing induced orientation on the morphology and mechanical properties of poly(styrene-b-ethylene/butylene-b-styrene) block copolymers and their composites with graphite. Mater. Des. 2014, 64, 694–705. [Google Scholar] [CrossRef]
- Weidisch, R.; Gido, S.P.; Uhrig, D.; Iatrou, H.; Mays, J.; Hadjichristidis, N. Tetrafunctional Multigraft Copolymers as Novel Thermoplastic Elastomers. Macromolecules 2001, 34, 6333–6337. [Google Scholar] [CrossRef]
- Feng, H.; Lu, X.; Wang, W.; Kang, N.-G.; Mays, J.W. Block Copolymers: Synthesis, Self-Assembly, and Applications. Polymers 2017, 9, 494. [Google Scholar] [CrossRef]
- Helal, E.; Amurin, L.G.; Carastan, D.J.; de Sousa, R.R., Jr.; David, E.; Frechette, M.; Demarquette, N.R. Interfacial molecular dynamics of styrenic block copolymer-based nanocomposites with controlled spatial distribution. Polymer 2017, 113, 9–26. [Google Scholar] [CrossRef]
- He, Y.; Li, W.; Yang, G.; Liu, H.; Lu, J.; Zheng, T.; Li, X. A novel method for fabricating wearable, piezoresistive, and pressure sensors based on modified-graphite/polyurethane composite films. Materials 2017, 10, 684. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Xu, Z.; Wu, Y.; Cheng, Q.; Guo, Q.; Wang, H. Super-tough artificial nacre based on graphene oxide via synergistic interface interactions of π-π stacking and hydrogen bonding. Carbon 2017, 111, 807–812. [Google Scholar] [CrossRef]
- Szeluga, U.; Kumanek, B.; Trzebicka, B. Synergy in hybrid polymer/nanocarbon composites. A review. Compos. Part A Appl. Sci. Manuf. 2015, 73, 204–231. [Google Scholar] [CrossRef]
- Rohini, R.; Katti, P.; Bose, S. Tailoring the interface in graphene/thermoset polymer composites: A critical review. Polymer 2015, 70, A17–A34. [Google Scholar] [CrossRef]
- Chipara, M.; Hui, D.; Notingher, P.V.; Chipara, M.D.; Lau, K.T.; Sankar, J.; Panaitescu, D. On polyethylene–polyaniline composites. Compos. Part B Appl. Sci. Manuf. 2003, 34, 637–645. [Google Scholar] [CrossRef]
- Katbab, A.A.; Hrymak, A.N.; Kasmadjian, K. Preparation of interfacially compatibilized PP-EPDM thermoplastic vulcanizate/graphite nanocomposites: Effects of graphite microstructure upon morphology, electrical conductivity, and melt rheology. J. Appl. Polym. Sci. 2008, 107, 3425–3433. [Google Scholar] [CrossRef]
- Tavman, I.; Krupa, I.; Omastova, M.; Sarikanat, M.; Novak, I.; Sever, K.; Ozdemir, I.; Seki, Y.; Podhradska, S.; Moskova, D.; et al. Effects of conductive graphite filler loading on physical properties of high-density polyethylene, Composite. Polym. Compos. 2012, 33, 1071–1076. [Google Scholar] [CrossRef]
- Kalaitzidou, K.; Fukushima, H.; Drzal, L.T. A route for polymer nanocomposites with engineered electrical conductivity and percolation threshold. Materials 2010, 3, 1089–1103. [Google Scholar] [CrossRef]
- Planes, E.; Chazeau, L.; Vigier, G.; Stuhldreier, T. Influence of silica fillers on the ageing by gamma radiation of EDPM nanocomposites. Compos. Sci. Technol. 2010, 70, 1530–1536. [Google Scholar] [CrossRef]
- Xie, Y.; Hill, C.A.S.; Xiao, Z.; Militz, H.; Mai, C. Silane coupling agents used for natural fiber/polymer composites: A review. Compos. Part A Appl. Sci. Manuf. 2010, 41, 806–819. [Google Scholar] [CrossRef]
- Yang, Y.; Murali, R. Binding mechanisms of molecular oxygen and moisture to grapheme. Appl. Phys. Lett. 2011, 98, 093116. [Google Scholar] [CrossRef]
- Yu, B.; Wang, X.; Xing, W.; Yang, H.; Wang, X.; Song, L.; Hua, Y.; Lo, S. Enhanced thermal and mechanical properties of functionalized graphene/thiol-ene systems by photopolymerization technology. Chem. Eng. J. 2013, 228, 318–326. [Google Scholar] [CrossRef]
- Lan, S.; Lia, L.; Xu, D.; Zhu, D.; Liu, Z.; Nie, F. Surface modification of magnesium hydroxide using vinyltriethoxysilane by dry process. Appl. Surf. Sci. 2016, 382, 56–62. [Google Scholar] [CrossRef]
- Akbari, A.; Yegani, R.; Pourabbas, B. Synthesis of high dispersible hydrophilic poly(ethylene glycol)/vinyl silane grafted silica nanoparticles to fabricate protein repellent polyethylene nanocomposite. Eur. Polym. J. 2016, 81, 86–97. [Google Scholar] [CrossRef]
- Kim, H.; Jang, J. Infrared spectroscopic study of SiOx film formation and decomposition of vinyl silane derivative by heat treatment. I. On KBr and gold surface. J. Appl. Polym. Sci. 1998, 68, 775–784. [Google Scholar] [CrossRef]
- Penso, I.; Cechinatto, E.A.; Machado, G.; Luvison, C.; Wanke, C.H.; Bianchi, O.; Soares, M.R.F. Preparation and characterization of polyhedral oligomeric silsesquioxane (POSS) using domestic microwave oven. J. Non-Cryst. Solids 2015, 428, 82–89. [Google Scholar] [CrossRef]
- Meloa, R.P.; Aguiara, V.O.; Marques, M.F.V. Silane crosslinked polyethylene from different commercial PE’s: Influence of comonomer, catalyst type and evaluation of hlpb as crosslinking coagent. Mater. Res. 2015, 18, 313–319. [Google Scholar] [CrossRef]
- Ma, P.C.; Kim, J.K.; Tang, B.Z. Effects of silane functionalization on the properties of carbon nanotube/epoxy nanocomposites. Compos. Sci. Technol. 2007, 67, 2965–2972. [Google Scholar] [CrossRef]
- Lopez, J.F.; Pelaez, G.J.; Perez, L.D. Monitoring the formation of polystyrene/silica nanocomposites from vinyl triethoxysilane containing copolymers. Colloid Polym. Sci. 2013, 291, 1143–1153. [Google Scholar] [CrossRef]
- Mauritz, K.A.; Blackwell, R.I.; Beyer, F.L. Viscoelastic properties and morphology of sulfonated poly(styrene-bethylene/butylene-b-styrene) block copolymers (sBCP), and sBCP/[silicate] nanostructured materials. Polymer 2004, 45, 3001–3016. [Google Scholar] [CrossRef]
- Cho, D.; Lee, S.; Yang, G.; Fukushima, H.; Drzal, L.T. Dynamic mechanical and thermal properties of phenylethynyl-terminated polyimide composites reinforced with expanded graphite nanoplatelets. Macromol. Mater. Eng. 2005, 290, 179–187. [Google Scholar] [CrossRef]
- Ibrahim, H.; Farag, M.; Megahed, H.; Mehanny, S. Characteristics of starch-based biodegradable composites reinforced with date palm and flax fibers. Carbohydr. Polym. 2014, 101, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Habsuda, J.; Simon, G.P.; Cheng, Y.B.; Hewitt, D.G.; Diggins, D.R.; Toh, H.; Cser, F. Sol–gel derived composites from poly(silicic acid) and 2-hydroxyethylmethacrylate: Thermal, physical and morphological properties. Polymer 2002, 43, 4627–4638. [Google Scholar] [CrossRef]
- Panda, J.N.; Bijwe, J.; Pandey, R.K. Role of treatment to graphite particles to increase the thermal conductivity in controlling tribo-performance of polymer composites. Wear 2016, 360–361, 87–96. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
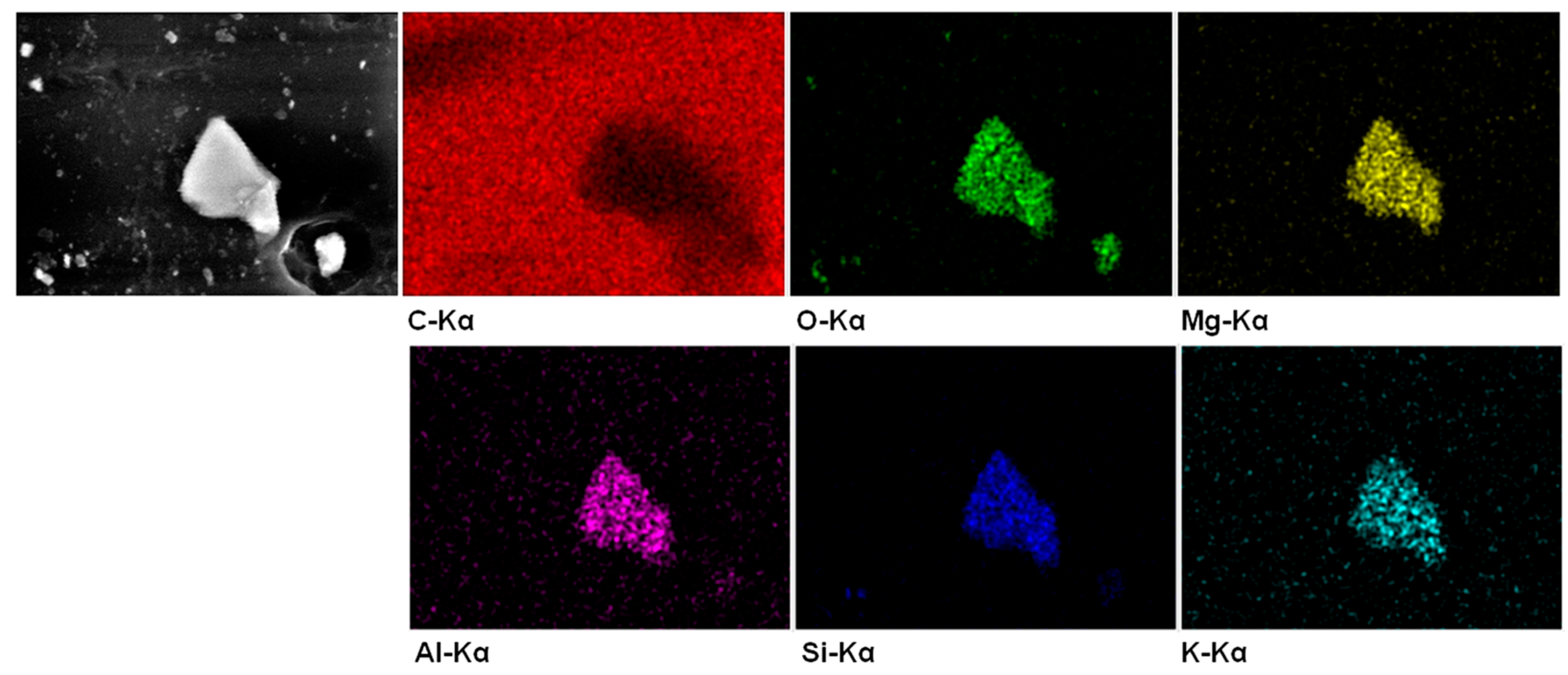
| Sample | Elemental Composition (wt %) * | ||
|---|---|---|---|
| C | O | Si | |
| SEBS | 95.9 | 4.1 | - |
| SEBS-5G | 96.3 | 3.7 | - |
| SEBS-5GVS | 96.1 | 3.9 | - |
| SEBS-10G | 96.9 | 3.1 | - |
| SEBS-10GVS | 95.2 | 4.2 | 0.6 |
| SEBS-15G | 97.3 | 2.7 | - |
| SEBS-15GVS | 93.6 | 5.2 | 1.2 |
| Sample | T20%, °C | WL450 °C, % | Residue at 700 °C, % |
|---|---|---|---|
| SEBS | 422.4 | 77.6 | 0.11 |
| SEBS-5G | 426.7 | 67.1 | 5.15 |
| SEBS-5GVS | 426.4 | 67.7 | 5.49 |
| SEBS-10G | 428.1 | 64.0 | 9.62 |
| SEBS-10GVS | 427.7 | 63.0 | 11.32 |
| SEBS-15G | 429.7 | 58.6 | 14.75 |
| SEBS-15GVS | 430.3 | 55.6 | 19.07 |
| Sample | E’ (⊥) 30 °C, MPa | E’ (II) 30 °C, MPa | E’ (⊥) 100 °C, MPa | E’ (II) 100 °C, MPa | Tg (⊥) °C | Tg (II) °C |
|---|---|---|---|---|---|---|
| SEBS | 11.0 ± 0.2 | 166.3 ± 1.3 | 4.0 ± 0.1 | 10.0 ± 0.1 | 99 | 101 |
| SEBS-5G | 21.1 ± 0.2 | 171.2 ± 1.4 | 5.8 ± 0.1 | 14.3 ± 0.3 | 102 | 99 |
| SEBS-10G | 33.1 ± 0.3 | 182.9 ± 1.4 | 9.3 ± 0.0 | 21.8 ± 0.2 | 101 | 100 |
| SEBS-15G | 40.7 ± 0.9 | 197.8 ± 0.6 | 12.4 ± 0.1 | 21.2 ± 0.2 | 100 | 100 |
| SEBS-5GVS | 20.4 ± 0.1 | 164.1 ± 0.9 | 5.6 ± 0.2 | 10.2 ± 0.1 | 97 | 97 |
| SEBS-10GVS | 36.5 ± 0.3 | 164.8 ± 0.6 | 10.2 ± 0.2 | 15.8 ± 0.1 | 99 | 101 |
| SEBS-15GVS | 54.5 ± 1.1 | 160.9 ± 1.1 | 15.4 ± 0.2 | 30.7 ± 0.6 | 103 | 97 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panaitescu, D.M.; Gabor, R.A.; Nicolae, C.A.; Parau, A.C.; Vitelaru, C.; Raditoiu, V.; Chipara, M. Block Copolymer Elastomer with Graphite Filler: Effect of Processing Conditions and Silane Coupling Agent on the Composite Properties. Polymers 2018, 10, 46. https://doi.org/10.3390/polym10010046
Panaitescu DM, Gabor RA, Nicolae CA, Parau AC, Vitelaru C, Raditoiu V, Chipara M. Block Copolymer Elastomer with Graphite Filler: Effect of Processing Conditions and Silane Coupling Agent on the Composite Properties. Polymers. 2018; 10(1):46. https://doi.org/10.3390/polym10010046
Chicago/Turabian StylePanaitescu, Denis Mihaela, Raluca Augusta Gabor, Cristian Andi Nicolae, Anca Constantina Parau, Catalin Vitelaru, Valentin Raditoiu, and Mircea Chipara. 2018. "Block Copolymer Elastomer with Graphite Filler: Effect of Processing Conditions and Silane Coupling Agent on the Composite Properties" Polymers 10, no. 1: 46. https://doi.org/10.3390/polym10010046
APA StylePanaitescu, D. M., Gabor, R. A., Nicolae, C. A., Parau, A. C., Vitelaru, C., Raditoiu, V., & Chipara, M. (2018). Block Copolymer Elastomer with Graphite Filler: Effect of Processing Conditions and Silane Coupling Agent on the Composite Properties. Polymers, 10(1), 46. https://doi.org/10.3390/polym10010046