FA Polymerization Disruption by Protic Polar Solvents



Abstract
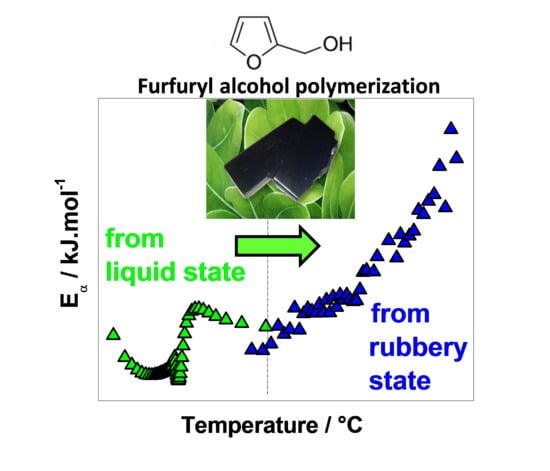
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Liquid FA/solvent Mixtures
2.3. Preparation of Cured PFA Materials
2.4. Analytical Techniques
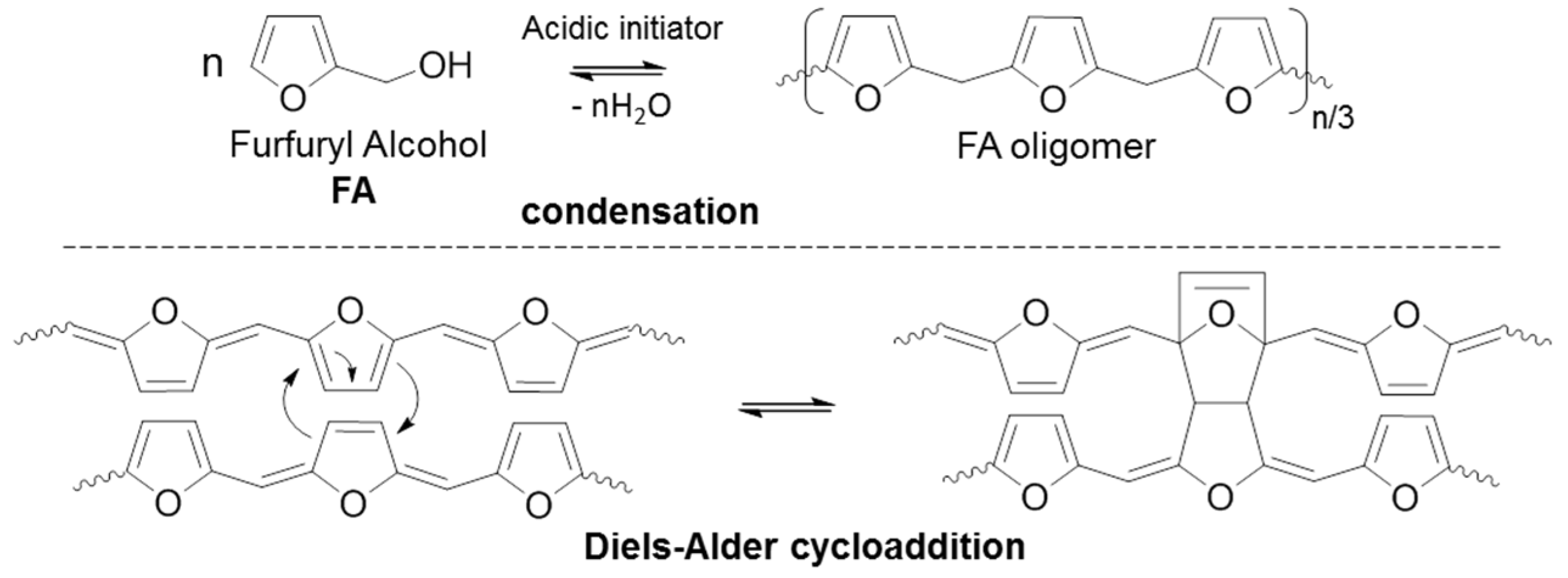
2.5. Theoretical Approaches
3. Results and Discussion
3.1. FA Polymerization Evolution from the Liquid State
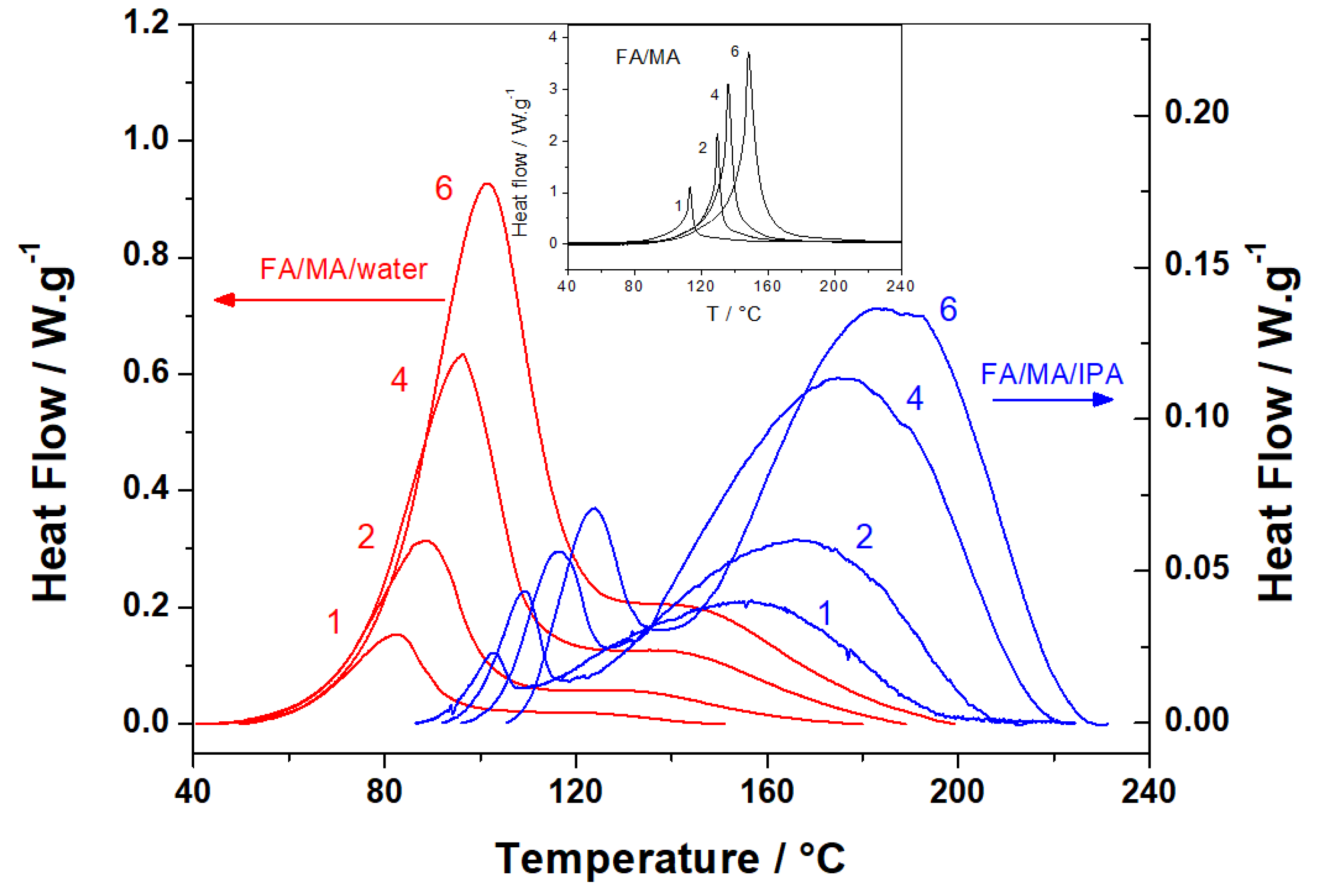
3.1.1. Non-Isothermal DSC Investigation
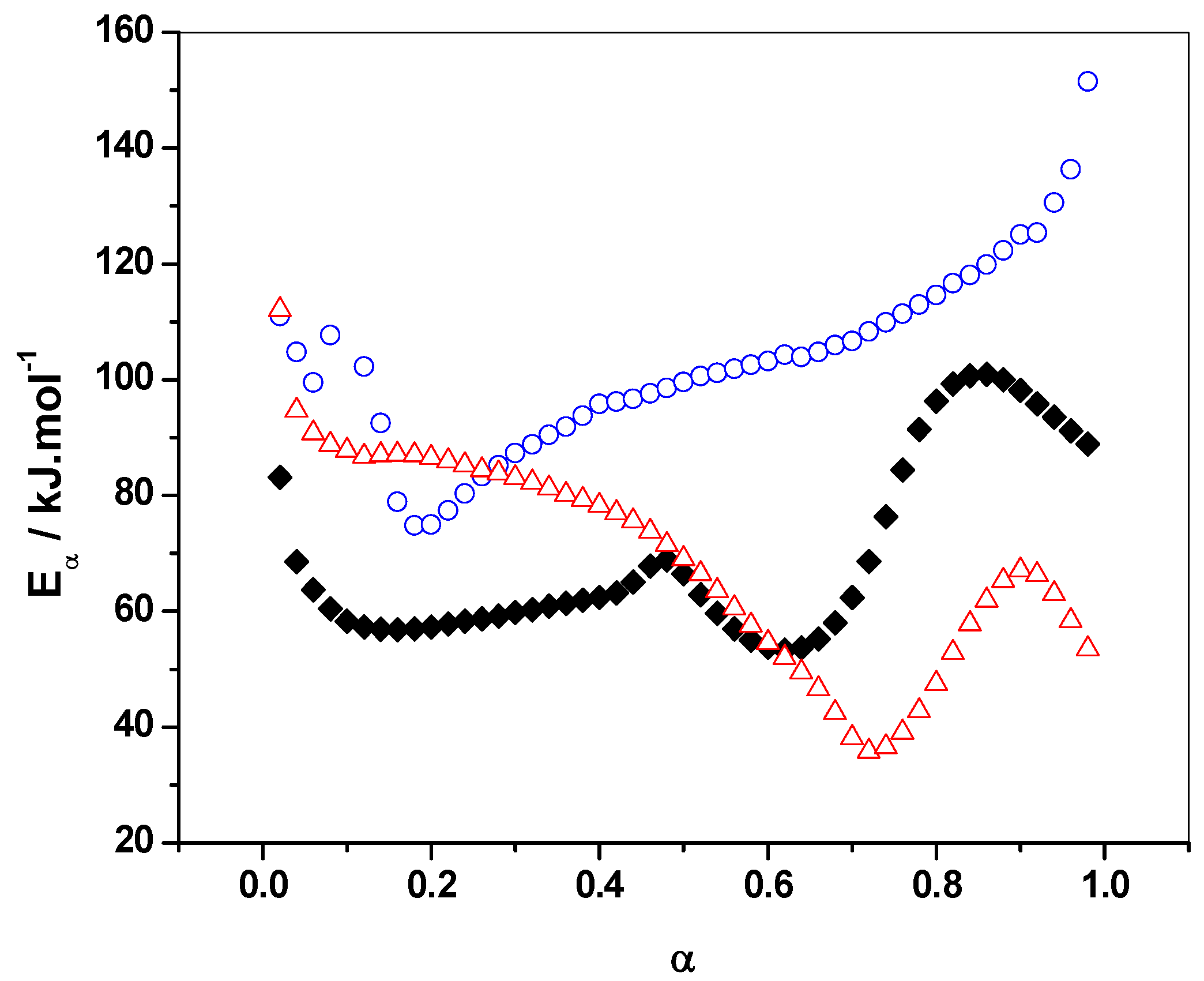
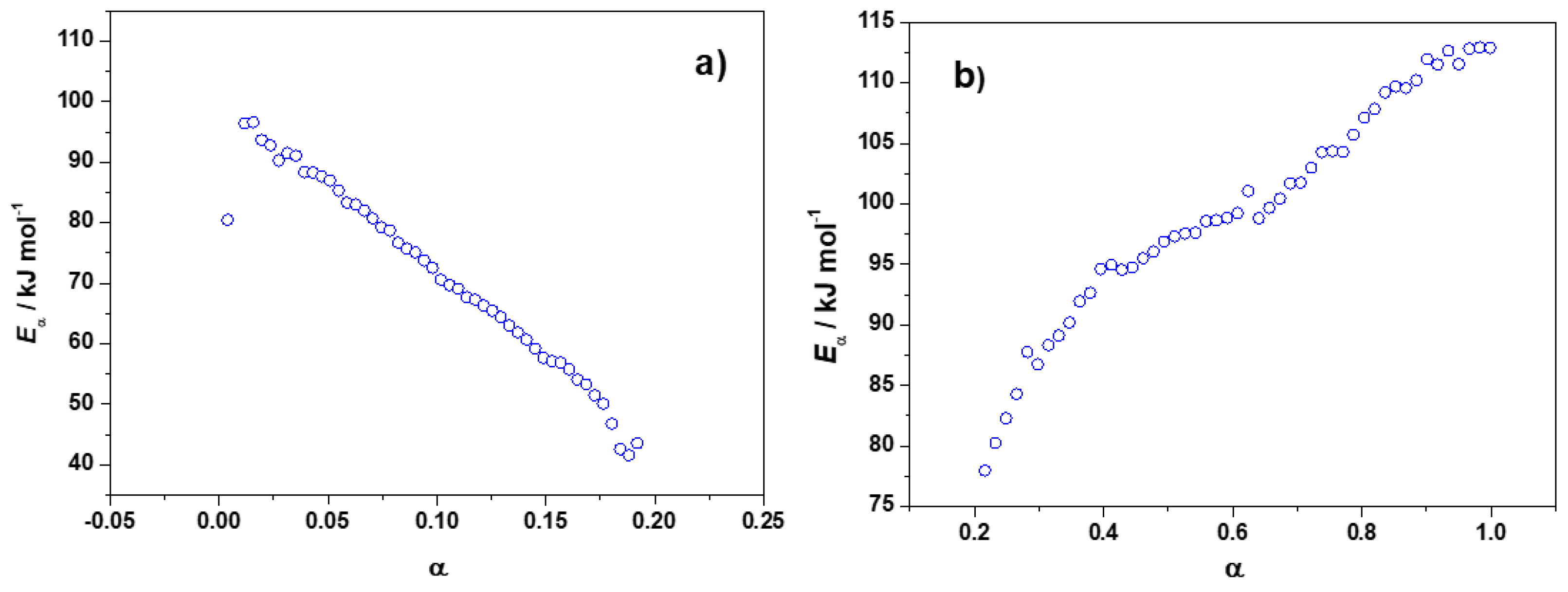
3.1.2. Eα vs. α-Dependence
3.1.3. Additionnal Kinetic Computation of FA/MA/IPA
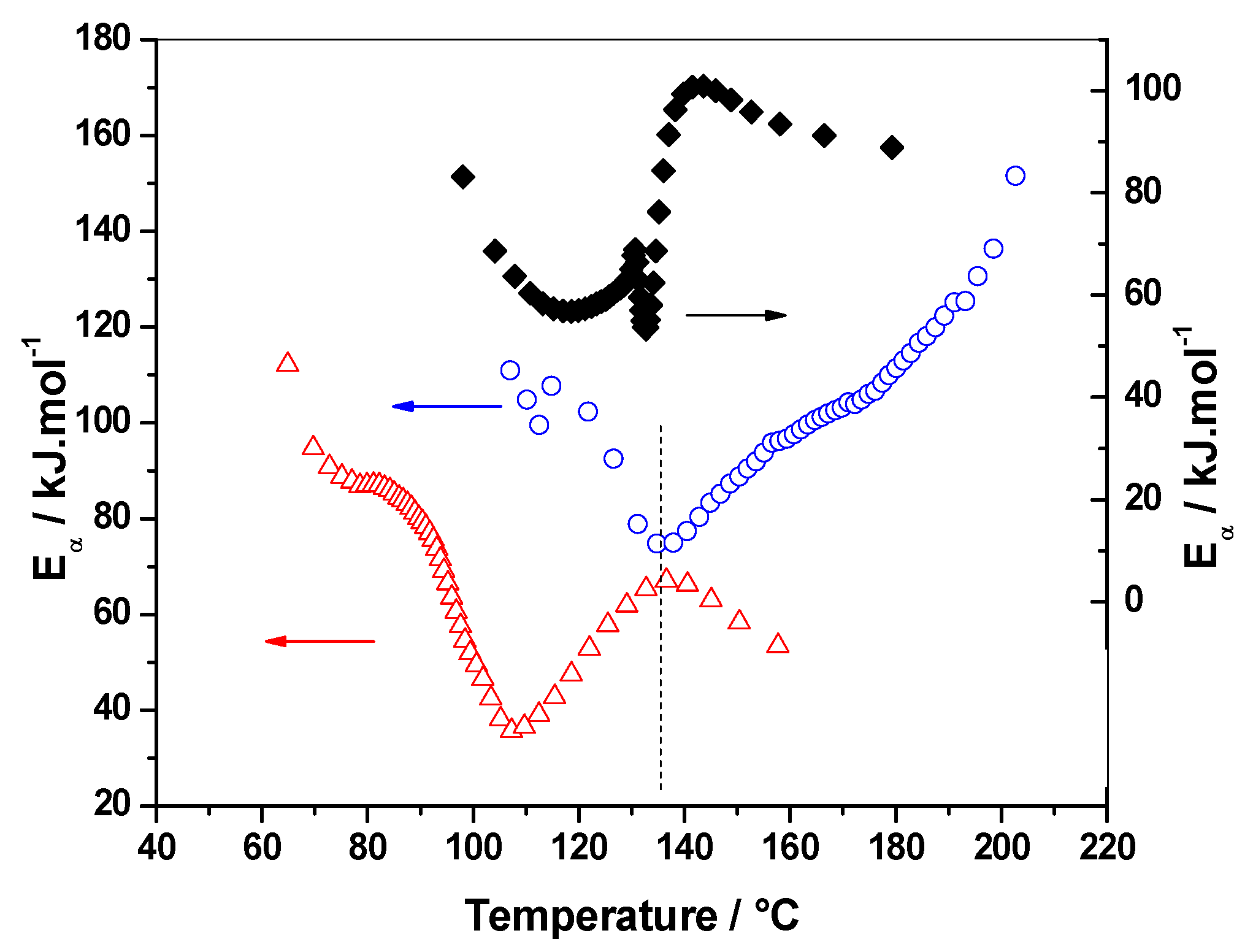
3.1.4. Eα vs. T-Dependence
3.2. Residual Cross-Linking Reactions in Rubbery State
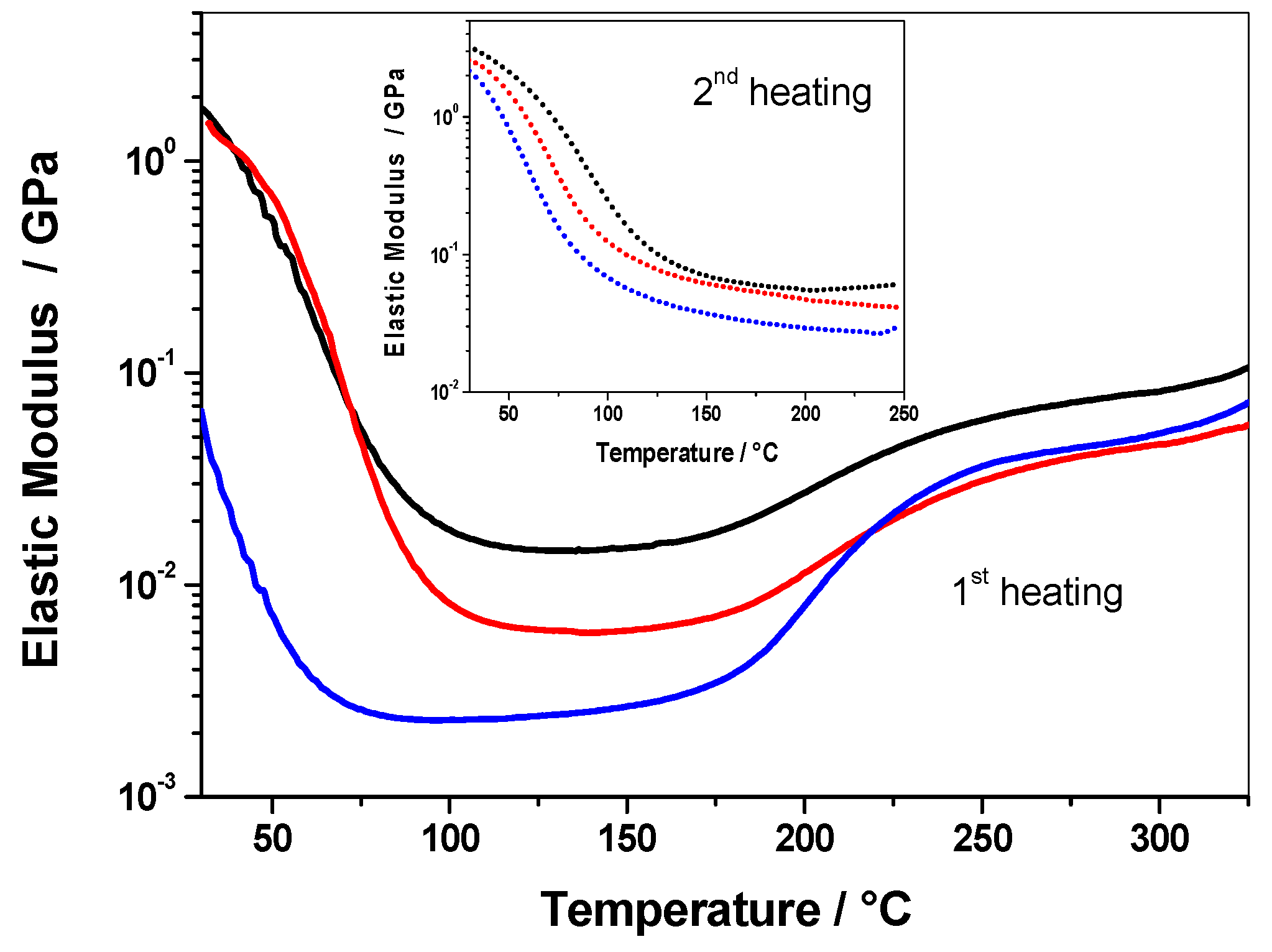
3.2.1. Non-Isothermal DMA Investigation
3.2.2. Eα vs. T-Dependence
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Kamm, B.; Kamm, M.; Schmidt, M.; Hirth, T.; Schulze, M. Lignocellulose-based chemical products and product family trees. In Biorefineries–Industrial Processes and Products; Kamm, B., Gruber, P.R., Kamm, M., Eds.; WILEY-VCH Verlag GmbH & Co.: Weinheim, Germany, 2006. [Google Scholar]
- Chheda, J.N.; Román-Leshkov, Y.; Dumesic, J.A. Production of 5-hydroxymethylfurfural and furfural by dehydration of biomass-derived mono- and poly-saccharides. Green Chem. 2007, 9, 342–350. [Google Scholar] [CrossRef]
- Rosatella, A.A.; Simeonov, S.P.; Frade, R.F.M.; Afonso, C.A.M. 5-hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754–793. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sadaba, I.; Lopez Granados, M. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Gandini, A. The irruption of polymers from renewable resources on the scene of macromolecular science and technology. Green Chem. 2011, 13, 1061–1083. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—the us department of energy’s “top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.J.F.; Trovatti, E. Progress of polymers from renewable resources: Furans, vegetable oils, and polysaccharides. Chem. Rev. 2016, 116, 1637–1669. [Google Scholar] [CrossRef] [PubMed]
- Choura, M.; Belgacem, N.M.; Gandini, A. Acid-catalyzed polycondensation of furfuryl alcohol: Mechanisms of chromophore formation and cross-linking. Macromolecules 1996, 29, 3839–3850. [Google Scholar] [CrossRef]
- Oliva-Teles, M.T.; Delerue-Matos, C.; Alvim-Ferraz, M.C.M. Determination of free furfuryl alcohol in foundry resins by chromatographic techniques. Anal. Chim. Acta 2005, 537, 47–51. [Google Scholar] [CrossRef]
- Lande, S.; Westin, M.; Schneider, M.H. Eco-efficient wood protection: Furfurylated wood as alternative to traditional wood preservation. Manag. Environ. Qual. Int. J. 2004, 15, 529–540. [Google Scholar] [CrossRef]
- Ju, M.; Zeng, C.; Wang, C.; Zhang, L. Preparation of ultrafine carbon spheres by controlled polymerization of furfuryl alcohol in microdroplets. Ind. Eng. Chem. Res. 2014, 53, 3084–3090. [Google Scholar] [CrossRef]
- Spange, S.; Grund, S. Nanostructured organic–inorganic composite materials by twin polymerization of hybrid monomers. Adv. Mater. 2009, 21, 2111–2116. [Google Scholar] [CrossRef]
- Bosq, N.; Guigo, N.; Falco, G.; Persello, J.; Sbirrazzuoli, N. Impact of silica nanoclusters on furfuryl alcohol polymerization and molecular mobility. J. Phys. Chem. C 2017, 121, 7485–7494. [Google Scholar] [CrossRef]
- Deka, H.; Misra, M.; Mohanty, A. Renewable resource based “all green composites” from kenaf biofiber and poly(furfuryl alcohol) bioresin. Ind. Crops Prod. 2013, 41, 94–101. [Google Scholar] [CrossRef]
- Guigo, N.; Mija, A.; Vincent, L.; Sbirrazzuoli, N. Eco-friendly composite resins based on renewable biomass resources: Polyfurfuryl alcohol/lignin thermosets. Eur. Pol. J. 2010, 46, 1016–1023. [Google Scholar] [CrossRef]
- Roudsari, G.M.; Misra, M.; Mohanty, A.K. A study of mechanical properties of biobased epoxy network: Effect of addition of epoxidized soybean oil and poly(furfuryl alcohol). J. Appl. Polym. Sci. 2017, 134. [Google Scholar] [CrossRef]
- Pin, J.M.; Guigo, N.; Vincent, L.; Sbirrazzuoli, N.; Mija, A. Copolymerization as a strategy to combine epoxidized linseed oil and furfuryl alcohol: The design of a fully bio-based thermoset. ChemSusChem 2015, 8, 4149–4161. [Google Scholar] [CrossRef] [PubMed]
- Conley, T.; Metil, I. An investigation of the structure of furfuryl alcohol polycondensatee with infrared spectroscopy. J. Appl. Polym. Sci. 1963, 7, 37–52. [Google Scholar] [CrossRef]
- Dunlop, A.; Peters, F. The Furans; Reinhold Publishing Corporation: New York, NY, USA, 1953. [Google Scholar]
- Montero, A.L.; Montero, L.A.; Martínez, R.; Spange, S. Ab initio modelling of crosslinking in polymers. A case of chains with furan rings. J. Mol. Struct. THEOCHEM 2006, 770, 99–106. [Google Scholar] [CrossRef]
- Wang, S.; Vorotnikov, V.; Vlachos, D.G. A DFT study of furan hydrogenation and ring opening on Pd (111). Green Chem. 2014, 16, 736–747. [Google Scholar] [CrossRef]
- Kim, T.; Jeong, J.; Rahman, M.; Zhu, E.; Mahajan, D. Characterizations of furfuryl alcohol oligomer/polymerization catalyzed by homogeneous and heterogeneous acid catalysts. Korean J. Chem. Eng. 2014, 31, 2124–2129. [Google Scholar] [CrossRef]
- Kim, T.; Assary, R.S.; Marshall, C.L.; Gosztola, D.J.; Curtiss, L.A.; Stair, P.C. Acid-catalyzed furfuryl alcohol polymerization: Characterizations of molecular structure and thermodynamic properties. ChemCatChem 2011, 3, 1451–1458. [Google Scholar] [CrossRef]
- Falco, G.; Guigo, N.; Vincent, L.; Sbirrazzuoli, N. Opening furan for tailoring properties of biobased poly(furfuryl alcohol) thermoset. ChemSusChem 2018. [Google Scholar] [CrossRef] [PubMed]
- Sbirrazzuoli, N. Determination of pre-exponential factors and of the mathematical functions f(α) or g(α) that describe the reaction mechanism in a model-free way. Thermochim. Acta 2013, 564, 59–69. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N. Is the friedman method applicable to transformations with temperature dependent reaction heat? Macromol. Chem. Phys. 2007, 208, 1592–1597. [Google Scholar] [CrossRef]
- Vyazovkin, S. Isoconversional Kinetics of Thermally Stimulated Processes; Springer: Berlin, Germany, 2015. [Google Scholar]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. Ictac kinetics committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Sbirrazzuoli, N. Isoconversional kinetic analysis of thermally stimulated processes in polymers. Macromol. Rapid Commun. 2006, 27, 1515–1532. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Achilias, D.S.; Karayannidis, G.P. Estimation of thermal transitions in poly(ethylene naphthalate): Experiments and modeling using isoconversional methods. Polymer 2010, 51, 2565–2575. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Vincent, L.; Vyazovkin, S. Comparison of several computational procedures for evaluating the kinetics of thermally stimulated condensed phase reactions. Chemom. Intell. Lab. Syst. 2000, 54, 53–60. [Google Scholar] [CrossRef]
- Atkins, P.; Paula, J.D. Physical Chemistry, 9th ed.; W.H Freeman: New York, NY, USA, 2010. [Google Scholar]
- Vyazovkin, S. Evaluation of activation energy of thermally stimulated solid-state reactions under arbitrary variation of temperature. J. Comput. Chem. 1997, 18, 393–402. [Google Scholar] [CrossRef]
- Vyazovkin, S. Modification of the integral isoconversional method to account for variation in the activation energy. J. Comput. Chem. 2001, 22, 178–183. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Brunel, D.; Elegant, L. Different kinetic equations analysis. J. Therm. Anal. Calorim. 1992, 38, 1509–1524. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Girault, Y.; Elegant, L. Simulations for evaluation of kinetic methods in differential scanning calorimetry. Part 3—Peak maximum evolution methods and isoconversional methods. Thermochim. Acta 1997, 293, 25–37. [Google Scholar] [CrossRef]
- Kim, T.; Assary, R.S.; Kim, H.; Marshall, C.L.; Gosztola, D.J.; Curtiss, L.A.; Stair, P.C. Effects of solvent on the furfuryl alcohol polymerization reaction: Uv raman spectroscopy study. Catal. Today 2013, 205, 60–66. [Google Scholar] [CrossRef]
- Guigo, N.; Mija, A.; Vincent, L.; Sbirrazzuoli, N. Chemorheological analysis and model-free kinetics of acid catalysed furfuryl alcohol polymerization. Phys. Chem. Chem. Phys. 2007, 9, 5359–5366. [Google Scholar] [CrossRef] [PubMed]
- Vyazovkin, S.; Sbirrazzuoli, N. Mechanism and kinetics of epoxy−amine cure studied by differential scanning calorimetry. Macromolecules 1996, 29, 1867–1873. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
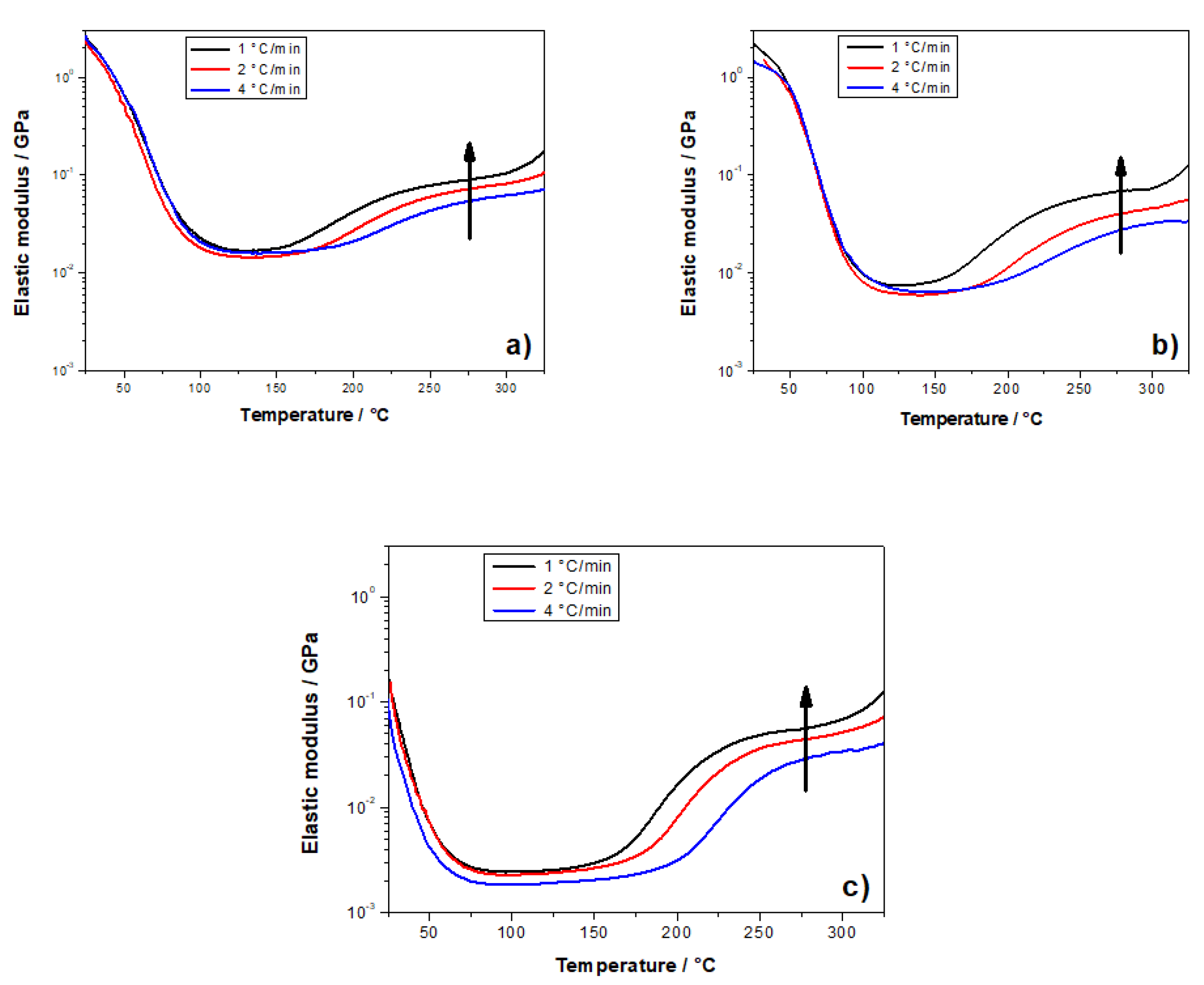{kind=link}
{kind=link}
| FA/MA/Water | FA/MA/IPA | FA/MA (Reference) | |
|---|---|---|---|
| β/°C·min−1 | Q/J·g−1 of FA | ||
| 1 | 490 ± 20 | 300 ± 30 | 709 ± 30 |
| 2 | 632 ± 30 | 230 ± 20 | 685 ± 30 |
| 4 | 678 ± 30 | 216 ± 20 | 620 ± 30 |
| 6 | 712 ± 30 | 168 ± 20 | 593 ± 30 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falco, G.; Guigo, N.; Vincent, L.; Sbirrazzuoli, N. FA Polymerization Disruption by Protic Polar Solvents. Polymers 2018, 10, 529. https://doi.org/10.3390/polym10050529
Falco G, Guigo N, Vincent L, Sbirrazzuoli N. FA Polymerization Disruption by Protic Polar Solvents. Polymers. 2018; 10(5):529. https://doi.org/10.3390/polym10050529
Chicago/Turabian StyleFalco, Guillaume, Nathanaël Guigo, Luc Vincent, and Nicolas Sbirrazzuoli. 2018. "FA Polymerization Disruption by Protic Polar Solvents" Polymers 10, no. 5: 529. https://doi.org/10.3390/polym10050529
APA StyleFalco, G., Guigo, N., Vincent, L., & Sbirrazzuoli, N. (2018). FA Polymerization Disruption by Protic Polar Solvents. Polymers, 10(5), 529. https://doi.org/10.3390/polym10050529