Local Order and Dynamics of Nanoconstrained Ethylene-Butylene Chain Segments in SEBS




Abstract
:
1. Introduction
2. Materials and Methods
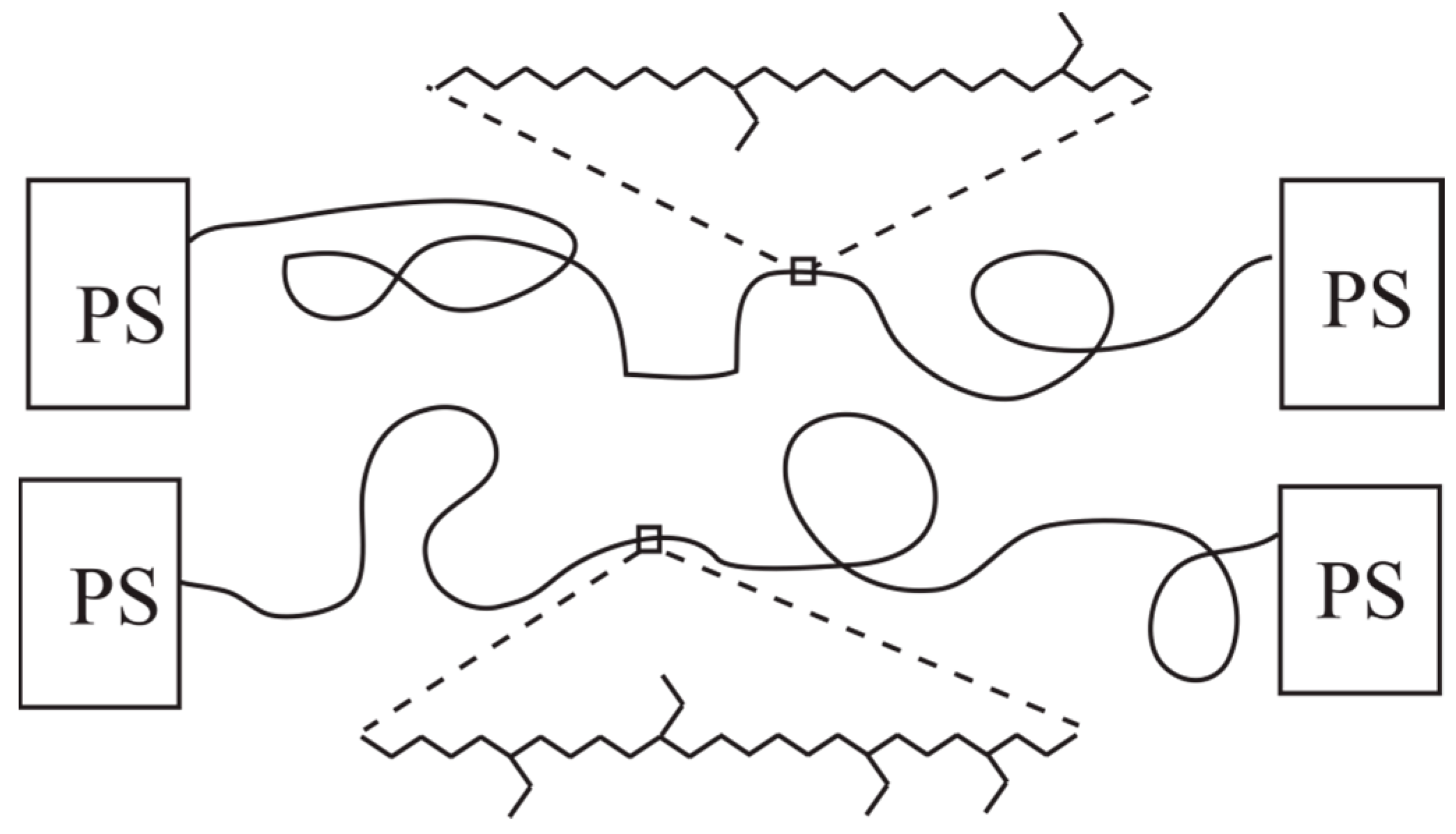
2.1. Materials
2.2. X-ray Diffraction
2.3. Differential Scanning Calorimetry
2.4. Size Exclusion Chromatography
2.5. High Resolution Liquid State NMR
2.6. Magic Angle Spinning NMR
2.7. Dielectric Spectroscopy
2.8. Time Domain NMR
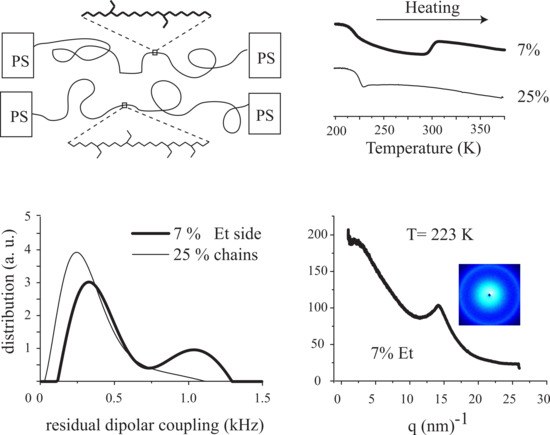
3. Results
3.1. Polymer Characterization
3.2. Film Characterization
3.3. Magic Angle Spinning NMR
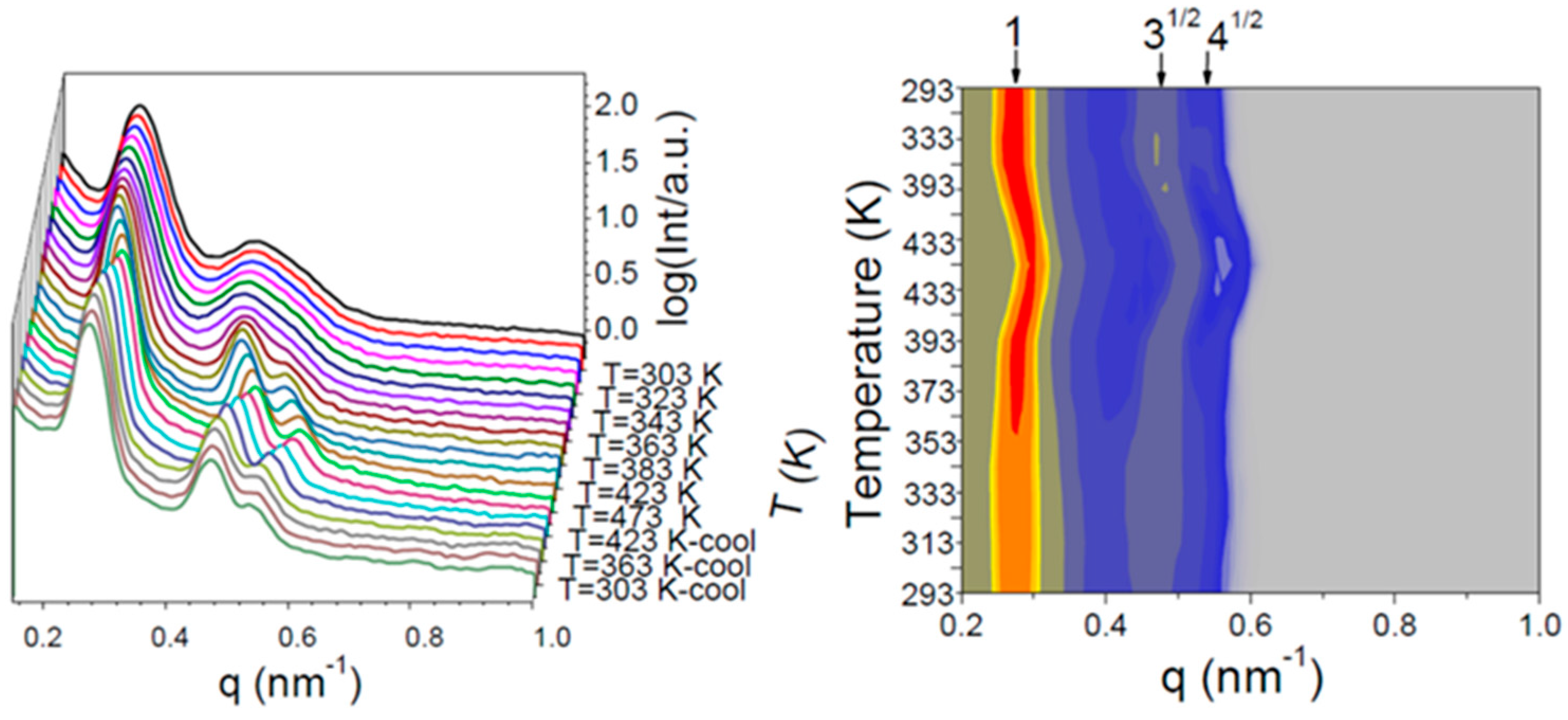
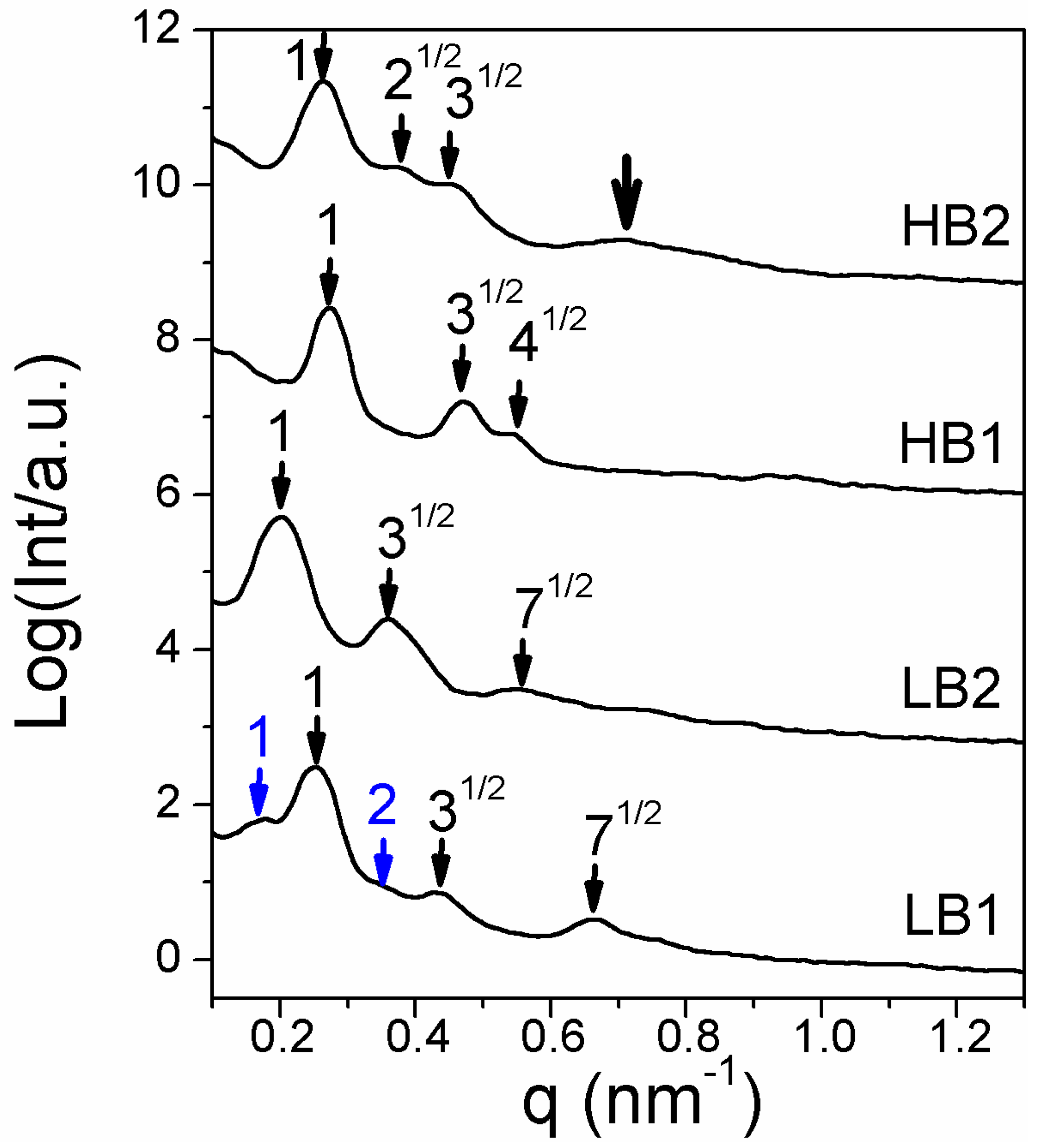
3.4. Wide-Angle X-ray Diffraction (WAXD)
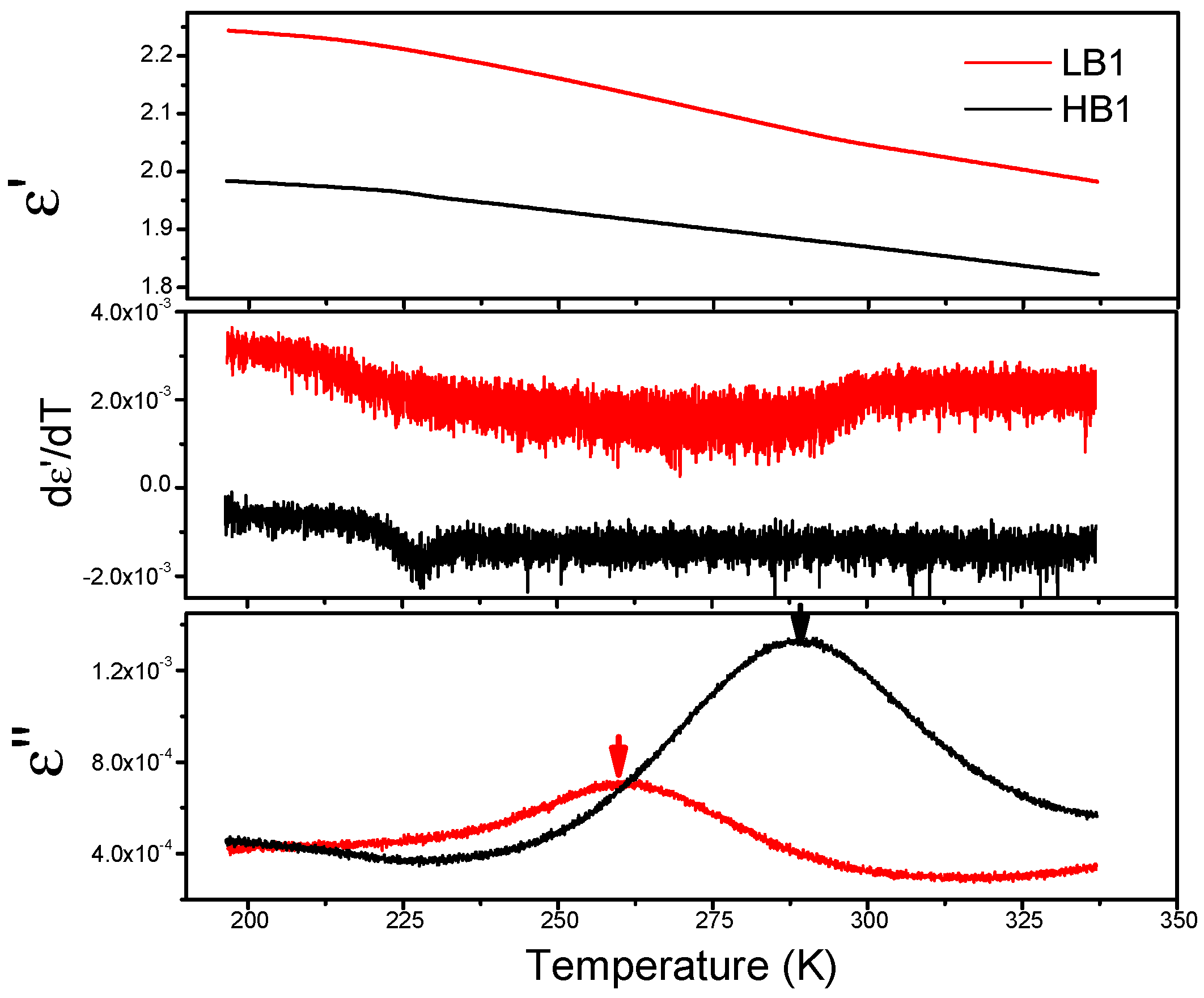
3.5. Dielectric Spectroscopy
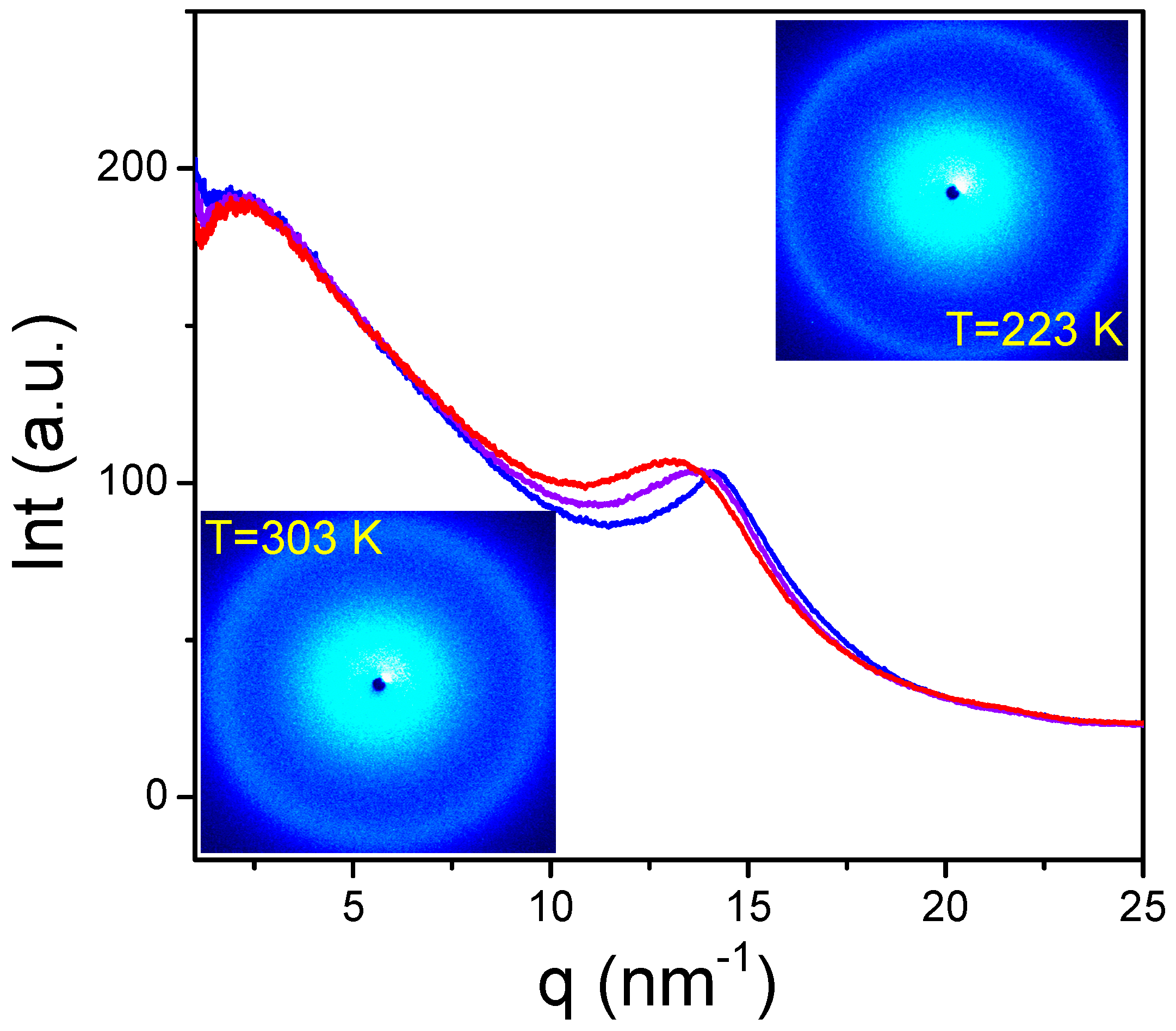
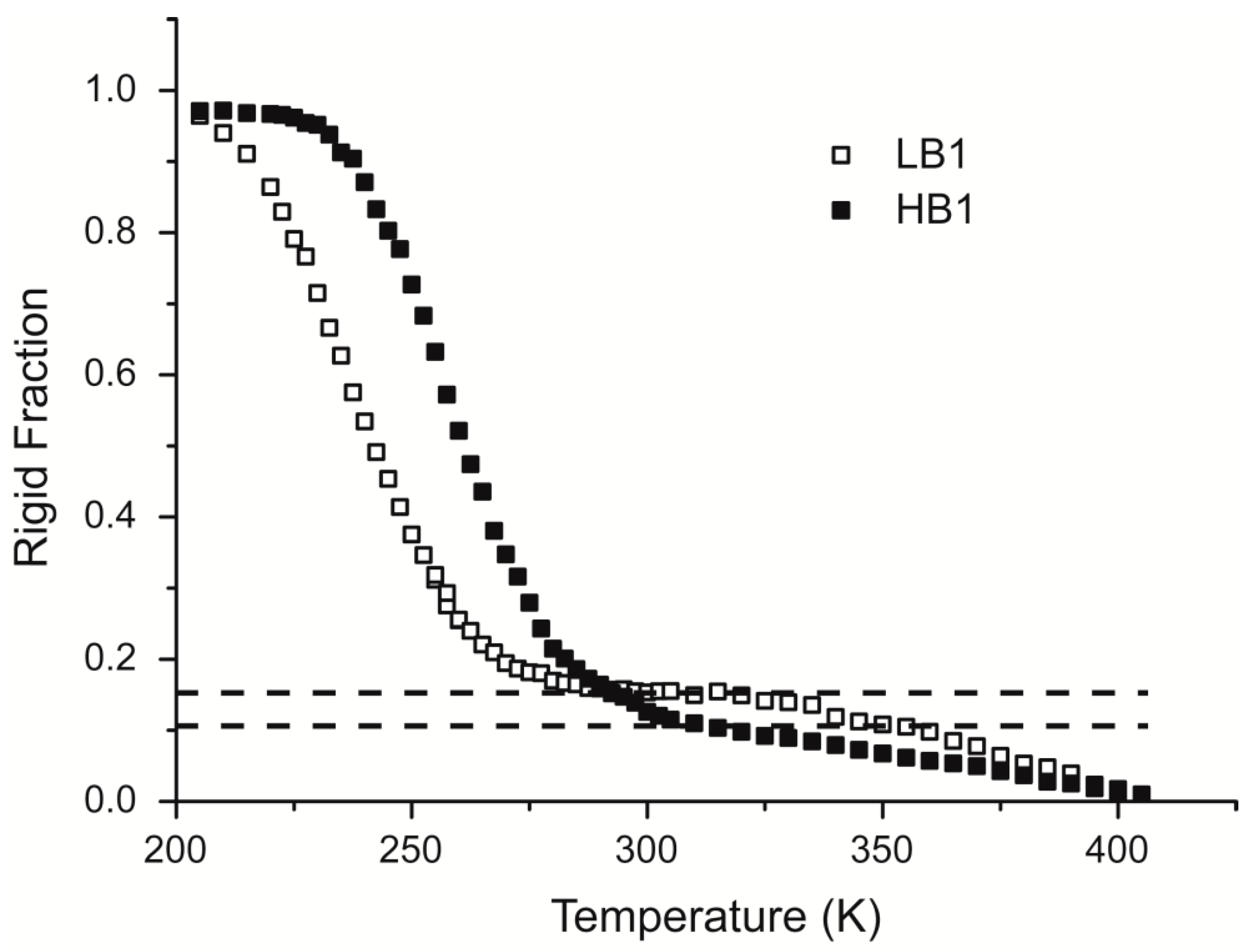
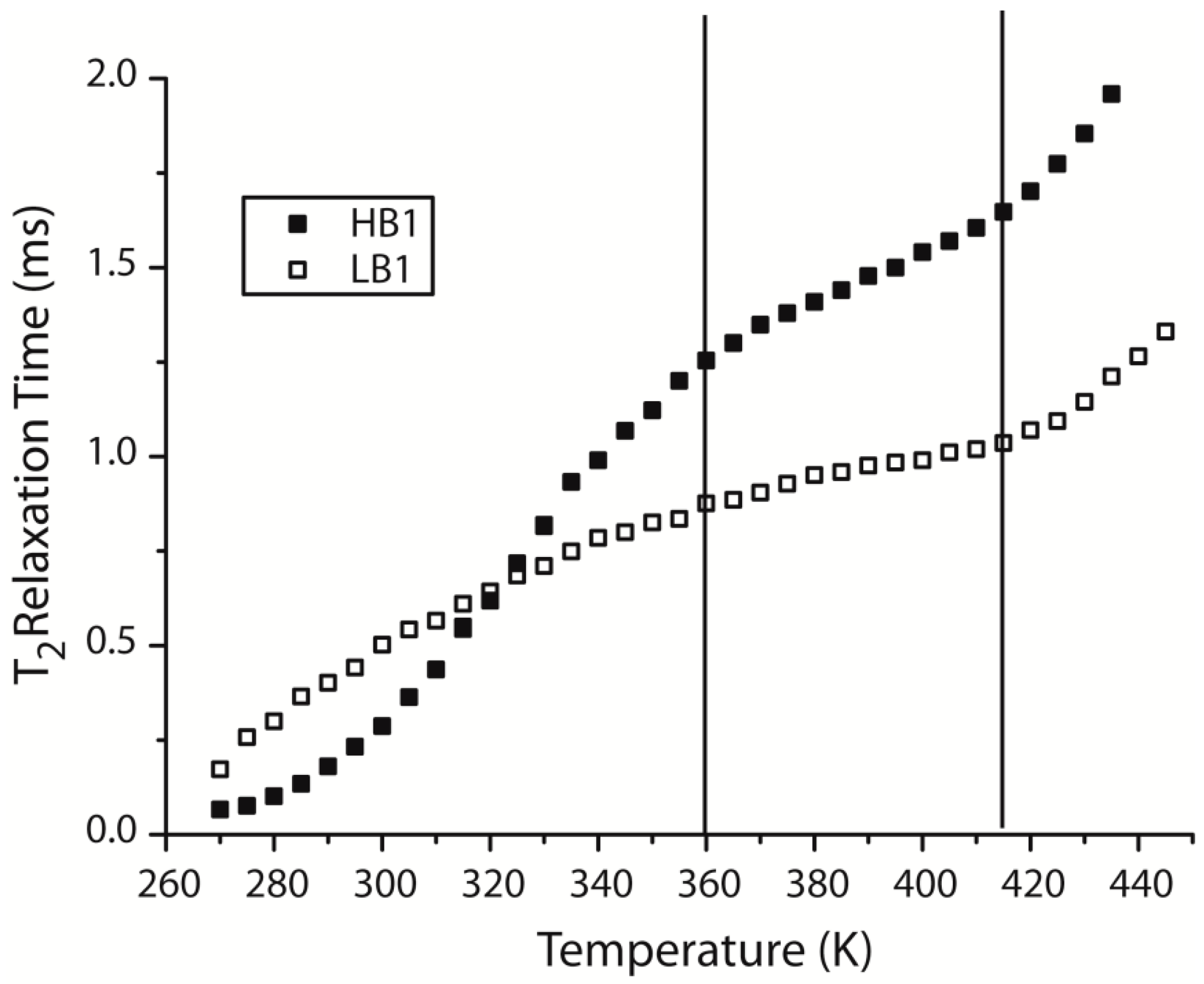
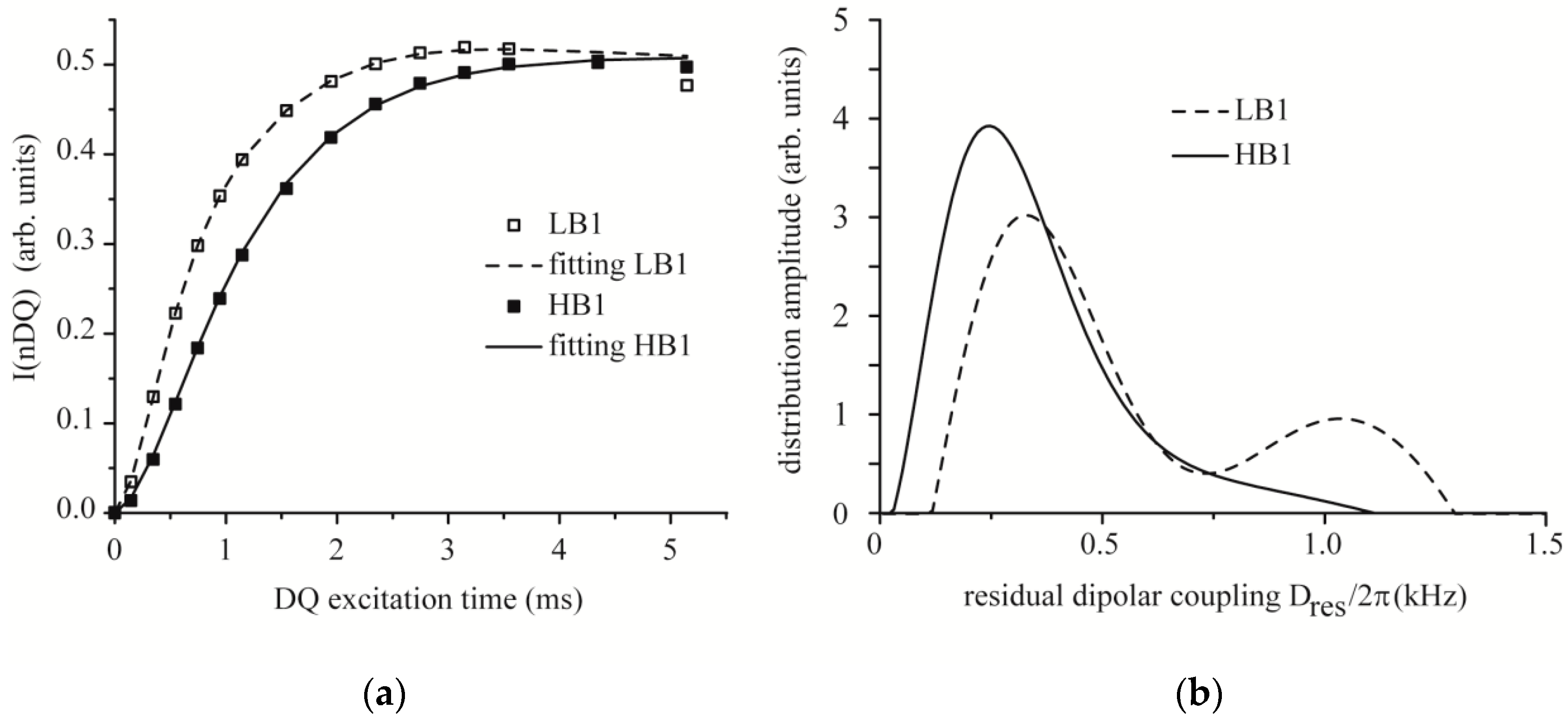
3.6. Time Domain NMR
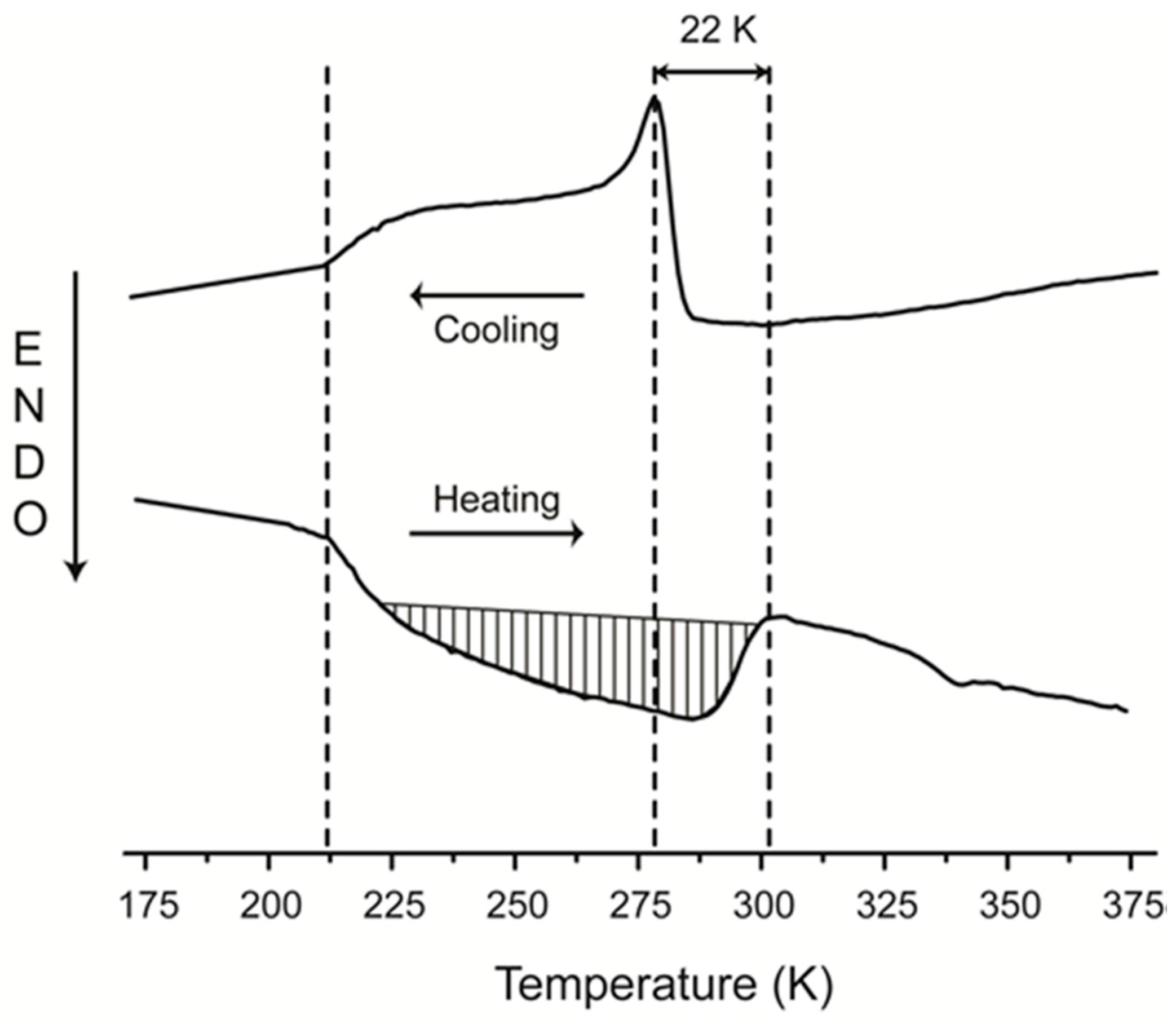
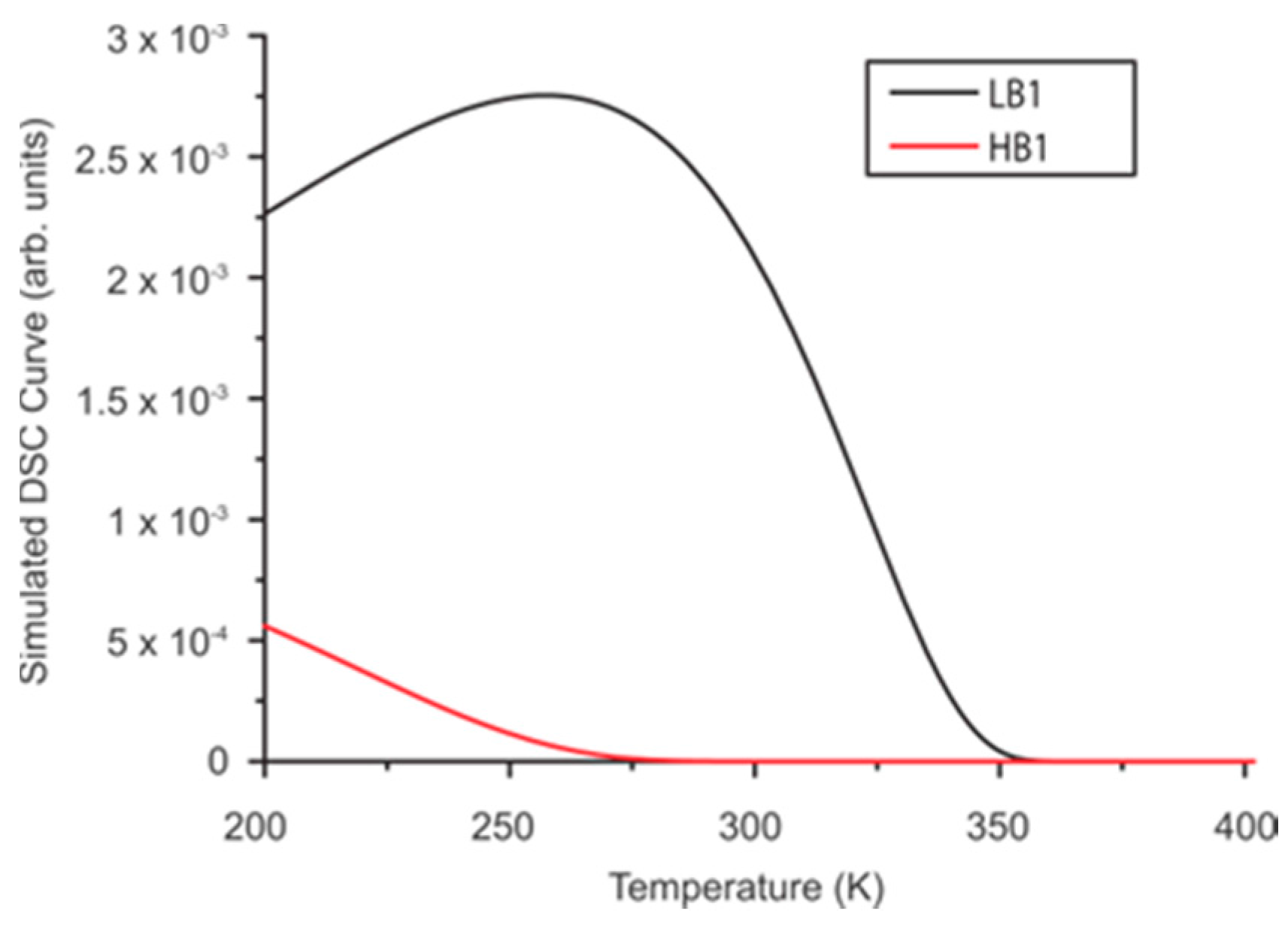
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Drobny, J.G. Handbook of Thermoplastic Elastomers; Elsevier Science: New York, NY, USA, 2007. [Google Scholar]
- Matsen, M.W.; Bates, F.S. Unifying weak- and strong-segregation block copolymer theories. Macromolecules 1996, 29, 1091–1098. [Google Scholar] [CrossRef]
- Fredrickson, G.H.; Bates, F.S. Dynamics of block copolymers: Theory and experiment. Annu. Rev. Mater. Sci. 1996, 26, 501–550. [Google Scholar] [CrossRef]
- Bates, F.S.; Fredrickson, G.H. Block copolymers—Designer soft materials. Phys. Today 1999, 52, 32. [Google Scholar] [CrossRef]
- Heck, B.; Arends, P.; Ganter, M.; Kressler, J.; Stuhn, B. Saxs and tem studies on poly(styrene)-block-poly(ethene-co-but-1-ene)-block-poly(styrene) in bulk and at various interfaces. Macromolecules 1997, 30, 4559–4566. [Google Scholar] [CrossRef]
- Lin, G.; Aucoin, D.; Giotto, M.; Canfield, A.; Wen, W.Y.; Jones, A.A. Lattice model simulation of penetrant diffusion along hexagonally packed rods in a barrier matrix as determined by pulse-field-gradient nuclear magnetic resonance. Macromolecules 2007, 40, 1521–1528. [Google Scholar] [CrossRef]
- Mamodia, M.; Indukuri, K.; Atkins, E.T.; Jeu, W.H.; Lesser, A.J. Hierarchical description of deformation in block copolymer tpes. J. Mater. Sci. 2008, 43, 7035–7046. [Google Scholar] [CrossRef]
- Tan, X.; Li, J.; Guo, S. Temperature-dependent order-to-order transition of polystyrene-block-poly(ethylene-co-butylene)-block-polystyrene triblock copolymer under multilayered confinement. Macromolecules 2018, 51, 2099–2109. [Google Scholar] [CrossRef]
- Sierra, C.A.; Galán, C.; Fatou, J.G.; Parellada, M.D.; Barrio, J.A. Thermal and mechanical properties of poly (styrene-b-ethylene-co-butylene-b-styrene) triblock copolymers. Polymer 1997, 38, 4325–4335. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, A.; Zhao, C.; Liang, H.; Wu, Z.; Xia, J. Molecular chain movements and transitions of sebs above room temperature studied by moving-window two-dimensional correlation infrared spectroscopy. Macromolecules 2007, 40, 9009–9017. [Google Scholar] [CrossRef]
- Indukuri, K.K.; Lesser, A.J. Comparative deformational characteristics of poly(styrene-b-ethylene-co-butylene-b-styrene) thermoplastic elastomers and cross-linked natural rubber. Polymer 2005, 46, 7218–7229. [Google Scholar] [CrossRef]
- Mamodia, M.; Panday, A.; Gido, S.P.; Lesser, A.J. Effect of microdomain structure and process conditions on the mechanical behavior of cylindrical block copolymer systems. Macromolecules 2007, 40, 7320–7328. [Google Scholar] [CrossRef]
- Ho, R.-M.; Adedeji, A.; Giles, D.W.; Hajduk, D.A.; Macosko, C.W.; Bates, F.S. Microstructure of triblock copolymers in asphalt oligomers. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 2857–2877. [Google Scholar] [CrossRef]
- Beiner, M. Nanoconfinement as a tool to study early stages of polymer crystallization. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 1556–1561. [Google Scholar] [CrossRef]
- Bracco, S.; Comotti, A.; Simonutti, R.; Camurati, I.; Sozzani, P. Low-temperature crystallization of ethylene-ran-propylene copolymers: Conformational rearrangement of sequences during the formation of the aggregates. Macromolecules 2002, 35, 1677–1684. [Google Scholar] [CrossRef]
- Kuwabara, K.; Horii, F. Solid-state 13c nmr analyses of the orthorhombic-to-hexagonal phase transition for constrained ultradrawn polyethylene fibers. Macromolecules 1999, 32, 5600–5605. [Google Scholar] [CrossRef]
- Vittoria, V.; Guadagno, L.; Comotti, A.; Simonutti, R.; Auriemma, F.; De Rosa, C. Mesomorphic form of syndiotactic polypropylene. Macromolecules 2000, 33, 6200–6204. [Google Scholar] [CrossRef]
- Sozzani, P.; Simonutti, R.; Galimberti, M. Mas nmr characterization of syndiotactic polypropylene: Crystal structure and amorphous phase conformation. Macromolecules 1993, 26, 5782–5789. [Google Scholar] [CrossRef]
- Sozzani, P.; Bracco, S.; Comotti, A.; Simonutti, R. Motional phase disorder of polymer chains as crystallized to hexagonal lattices. In Interphases and Mesophases in Polymer Crystallization II; Springer: Berlin/Heidelberg, Germany, 2005; Volume 181, pp. 153–177. [Google Scholar]
- Litvinov, V.M.; Steeman, P.A.M. Epdm-carbon black interactions and the reinforcement mechanisms, as studied by low-resolution 1 h nmr. Macromolecules 1999, 32, 8476–8490. [Google Scholar] [CrossRef]
- Saalwachter, K. Proton multiple-quantum nmr for the study of chain dynamics and structural constraints in polymeric soft materials. Prog. Nuclear Magn. Resonan. Spectrosc. 2007, 51, 1–35. [Google Scholar] [CrossRef]
- Bonetti, S.; Farina, M.; Mauri, M.; Koynov, K.; Butt, H.J.; Kappl, M.; Simonutti, R. Core@shell poly(N-butylacrylate)@polystyrene nanoparticles: Baroplastic force-responsiveness in presence of strong phase separation. Macromol. Rapid Commun. 2016, 37, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Kalinowski, H.O.; Berger, S. 150 and More Basic nmr Experiments: A Practical Course; Wiley-VCH: Weinheim, Germany, 1998. [Google Scholar]
- Saalwächter, K. Artifacts in transverse proton nmr relaxation studies of elastomers. Macromolecules 2005, 38, 1508–1512. [Google Scholar] [CrossRef]
- Vaca Chávez, F.; Saalwächter, K. Nmr observation of entangled polymer dynamics: Tube model predictions and constraint release. Phys. Rev. Lett. 2010, 104, 198305. [Google Scholar] [CrossRef] [PubMed]
- Valentín, J.L.; Posadas, P.; Fernández-Torres, A.; Malmierca, M.A.; González, L.; Chassé, W.; Saalwächter, K. Inhomogeneities and chain dynamics in diene rubbers vulcanized with different cure systems†. Macromolecules 2010, 43, 4210–4222. [Google Scholar] [CrossRef]
- Dibbanti, M.K.; Mauri, M.; Mauri, L.; Medaglia, G.; Simonutti, R. Probing small network differences in sulfur-cured rubber compounds by combining nuclear magnetic resonance and swelling methods. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Saalwächter, K.; Gottlieb, M.; Liu, R.; Oppermann, W. Gelation as studied by proton multiple-quantum nmr. Macromolecules 2007, 40, 1555–1561. [Google Scholar] [CrossRef]
- Dollase, T.; Graf, R.; Heuer, A.; Spiess, H.W. Local order and chain dynamics in molten polymer blocks revealed by proton double-quantum nmr. Macromolecules 2001, 34, 298–309. [Google Scholar] [CrossRef]
- Metz, G.; Wu, X.L.; Smith, S.O. Ramped-amplitude cross polarization in magic-angle-spinning nmr. J. Magn. Resonan. Ser. A 1994, 110, 219–227. [Google Scholar] [CrossRef]
- Maus, A.; Hertlein, C.; Saalwächter, K. A robust proton nmr method to investigate hard/soft ratios, crystallinity, and component mobility in polymers. Macromol. Chem. Phys. 2006, 207, 1150–1158. [Google Scholar] [CrossRef]
- Mauri, M.; Thomann, Y.; Schneider, H.; Saalwächter, K. Spin-diffusion nmr at low field for the study of multiphase solids. Solid State Nuclear Magn. Resonan. 2008, 34, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Mauri, M.; Dibbanti, M.K.; Calzavara, M.; Mauri, L.; Simonutti, R.; Causin, V. Time domain nuclear magnetic resonance: A key complementary technique for the forensic differentiation of foam traces. Anal. Methods 2013, 5, 4336–4344. [Google Scholar] [CrossRef]
- Chassé, W.; Valentín, J.L.; Genesky, G.D.; Cohen, C.; Saalwächter, K. Precise dipolar coupling constant distribution analysis in proton multiple-quantum NMR of elastomers. J. Chem. Phys. 2011, 134, 044907. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Zhang, T.; Reddy, D.V.; Rinaldi, P.L.; McIntosh, L.H.; Quirk, R.P. Multidimensional nmr studies of poly(ethylene-co-1-butene) microstructures. Macromolecules 2003, 36, 4017–4028. [Google Scholar] [CrossRef]
- Wunderlich, B.; Czornyj, G. A study of equilibrium melting of polyethylene. Macromolecules 1977, 10, 906–913. [Google Scholar] [CrossRef]
- Brandolini, A.J.; Hills, D.D. Nmr Spectra of Polymers and Polymer Additives; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Floudas, G.; Placke, P.; Stepanek, P.; Brown, W.; Fytas, G.; Ngai, K.L. Dynamics of the “strong” polymer of n-lauryl methacrylate below and above the glass transition. Macromolecules 1995, 28, 6799–6807. [Google Scholar] [CrossRef]
- Floudas, G.; Štepánek, P. Structure and dynamics of poly(N-decyl methacrylate) below and above the glass transition. Macromolecules 1998, 31, 6951–6957. [Google Scholar] [CrossRef]
- Hu, W.; Srinivas, S.; Sirota, E.B. Crystalline structure and properties of ep and eb copolymers by solid-state nmr, dsc, and waxs. Macromolecules 2002, 35, 5013–5024. [Google Scholar] [CrossRef]
- Grigoriadis, C.; Duran, H.; Steinhart, M.; Kappl, M.; Butt, H.-J.; Floudas, G. Suppression of phase transitions in a confined rodlike liquid crystal. ACS Nano 2011, 5, 9208–9215. [Google Scholar] [CrossRef] [PubMed]
- Selevou, A.; Papamokos, G.; Steinhart, M.; Floudas, G. 8ocb and 8cb liquid crystals confined in nanoporous alumina: Effect of confinement on the structure and dynamics. J. Phys. Chem. B 2017, 121, 7382–7394. [Google Scholar] [CrossRef] [PubMed]
- Saalwächter, K.; Thomann, Y.; Hasenhindl, A.; Schneider, H. Direct observation of interphase composition in block copolymers. Macromolecules 2008, 41, 9187–9191. [Google Scholar] [CrossRef]
- Li, K.L.; Inglefield, P.T.; Jones, A.A.; Bendler, J.T.; English, A.D. Heterogeneous microscopic mobility near the glass transition from nmr line shapes. Macromolecules 1988, 21, 2940–2944. [Google Scholar] [CrossRef]
- Simha, R.; Somcynsky, T. On the statistical thermodynamics of spherical and chain molecule fluids. Macromolecules 1969, 2, 342–350. [Google Scholar] [CrossRef]
- Utracki, L.A.; Simha, R. Analytical representation of solutions to lattice-hole theory. Macromol. Theory Simul. 2001, 10, 17–24. [Google Scholar] [CrossRef]
- Herrmann, V.; Unseld, K.; Fuchs, H.B.; Blumich, B. Molecular dynamics of elastomers investigated by dmta and the NMR-mouse (r). Colloid Polym. Sci. 2002, 280, 758–764. [Google Scholar] [CrossRef]
- Polgar, L.M.; Hagting, E.; Raffa, P.; Mauri, M.; Simonutti, R.; Picchioni, F.; van Duin, M. Effect of rubber polarity on cluster formation in rubbers cross-linked with diels–alder chemistry. Macromolecules 2017, 50, 8955–8964. [Google Scholar] [CrossRef] [PubMed]
- Baughman, T.W.; Sworen, J.C.; Wagener, K.B. Sequenced ethylene−propylene copolymers: Effects of short ethylene run lengths. Macromolecules 2006, 39, 5028–5036. [Google Scholar] [CrossRef]
- Crist, B.; Claudio, E.S. Isothermal crystallization of random ethylene−butene copolymers: Bimodal kinetics. Macromolecules 1999, 32, 8945–8951. [Google Scholar] [CrossRef]
- Crist, B.; Williams, D.N. Crystallization and melting of model ethylene-butene random copolymers: Thermal studies. J. Macromol. Sci. Phys. B 2000, 39, 1–13. [Google Scholar] [CrossRef]
- Carella, J.M.; Graessley, W.W.; Fetters, L.J. Effects of chain microstructure on the viscoelastic properties of linear polymer melts: Polybutadienes and hydrogenated polybutadienes. Macromolecules 1984, 17, 2775–2786. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Mathot, V.B.F. Partitioning of main and side-chain units between different phases: A solid-state 13c nmr inversion-recovery cross-polarization study on a homogeneous, metallocene-based, ethylene-1-octene copolymer. Solid State Nuclear Magn. Resonan. 2002, 22, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Rogunova, M.; Hiltner, A.; Baer, E.; Nowacki, R.; Galeski, A.; Piorkowska, E. Structure of polypropylene crystallized in confined nanolayers. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 3380–3396. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Styrene mol % | Butylene mol % | Ethylene mol * | Mn × 104 | PDI | PS units (per block) | EB units | |
|---|---|---|---|---|---|---|---|
| LB1 | 18.6 | 23.8 | 76.2 | 6.0 | 1.07 | 86 | 752 |
| LB2 | 18.6 | 23.0 | 77 | 7.9 | 1.08 | 113 | 990 |
| LBS | 27.7 § | 21.8 | 78.2 | 10.6 | 1.13 | 212 | 1105 |
| HB3 | 11.5 | 57.8 | 42.2 | 10.0 | 1.06 | 93 | 1439 |
| HB1 | 11.2 | 67.4 | 32.6 | 10.2 | 1.09 | 93 | 1476 |
| HB2 | 7.1 | 64.3 | 35.7 | 15.9 | 1.08 | 95 | 2486 |
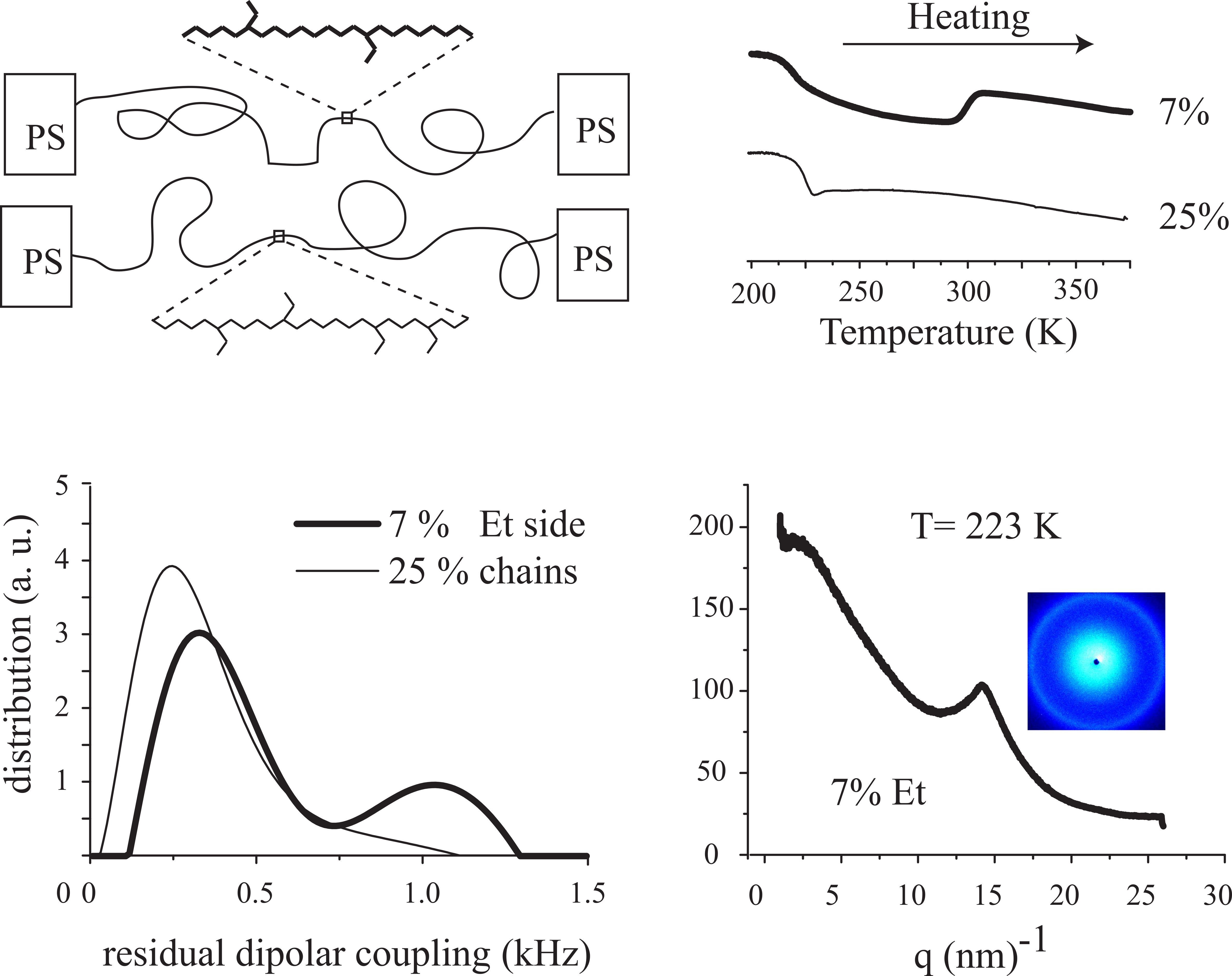
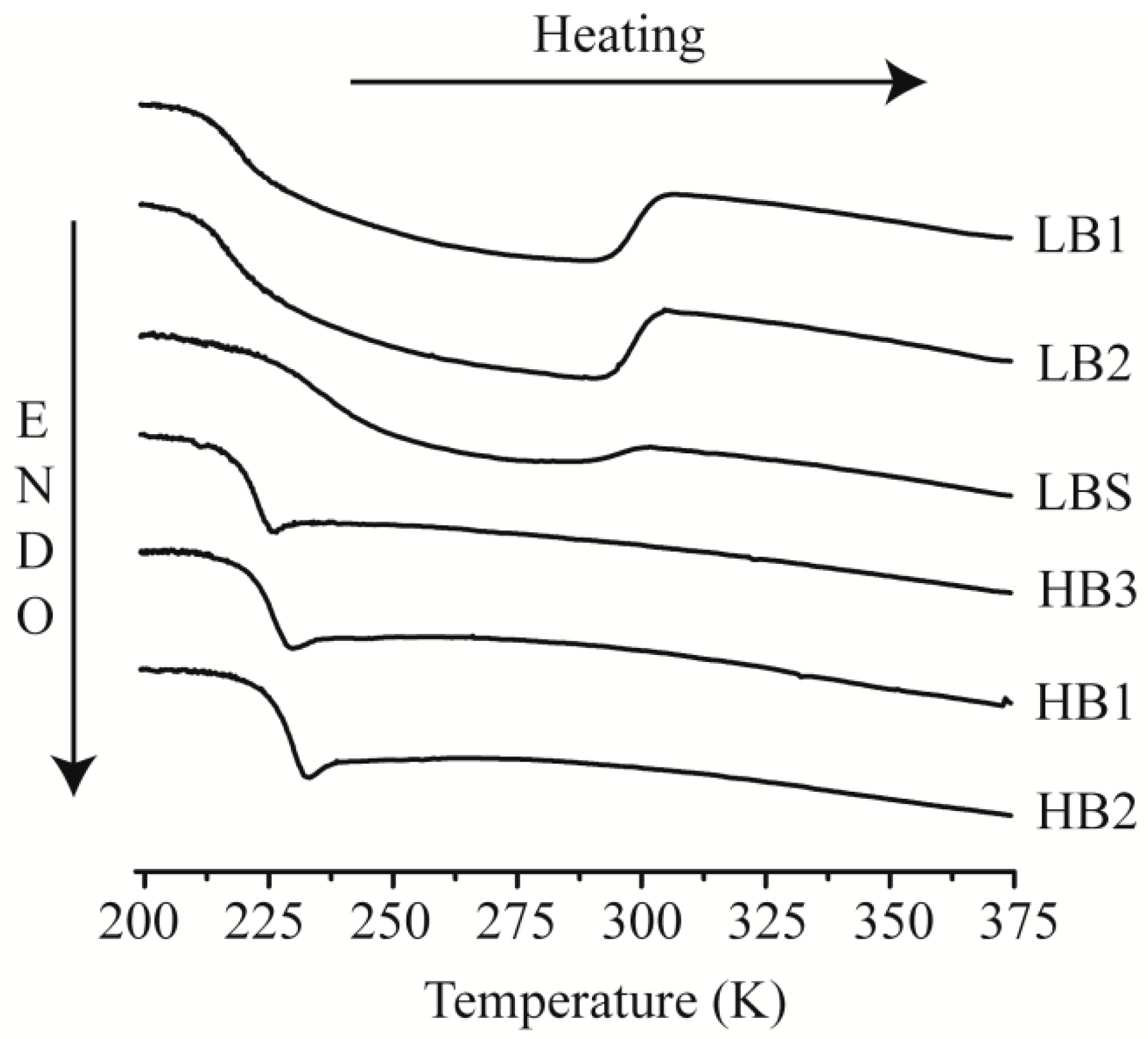
| Sample | Lower Tg (K) | Upper bound of transition (K) | Nanodomain morphology a | Characteristic spacing, d (nm) b |
|---|---|---|---|---|
| LB1 | 213 | 301 | lam/hcpc | 35.7 (lam) 25.2 (hcpc) |
| LB2 | 211 | 300 | hcpc | 34.5 |
| LBS | 226 | 298 | ||
| HB3 | 218 | - | ||
| HB1 | 221 | - | hcpc | 24.3 |
| HB2 | 225 | - | bcc | 23.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauri, M.; Floudas, G.; Simonutti, R. Local Order and Dynamics of Nanoconstrained Ethylene-Butylene Chain Segments in SEBS. Polymers 2018, 10, 655. https://doi.org/10.3390/polym10060655
Mauri M, Floudas G, Simonutti R. Local Order and Dynamics of Nanoconstrained Ethylene-Butylene Chain Segments in SEBS. Polymers. 2018; 10(6):655. https://doi.org/10.3390/polym10060655
Chicago/Turabian StyleMauri, Michele, George Floudas, and Roberto Simonutti. 2018. "Local Order and Dynamics of Nanoconstrained Ethylene-Butylene Chain Segments in SEBS" Polymers 10, no. 6: 655. https://doi.org/10.3390/polym10060655
APA StyleMauri, M., Floudas, G., & Simonutti, R. (2018). Local Order and Dynamics of Nanoconstrained Ethylene-Butylene Chain Segments in SEBS. Polymers, 10(6), 655. https://doi.org/10.3390/polym10060655